
Saliva as a Blood Alternative for Genome-Wide DNA Methylation Profiling by Methylated DNA Immunoprecipitation (MeDIP) Sequencing

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
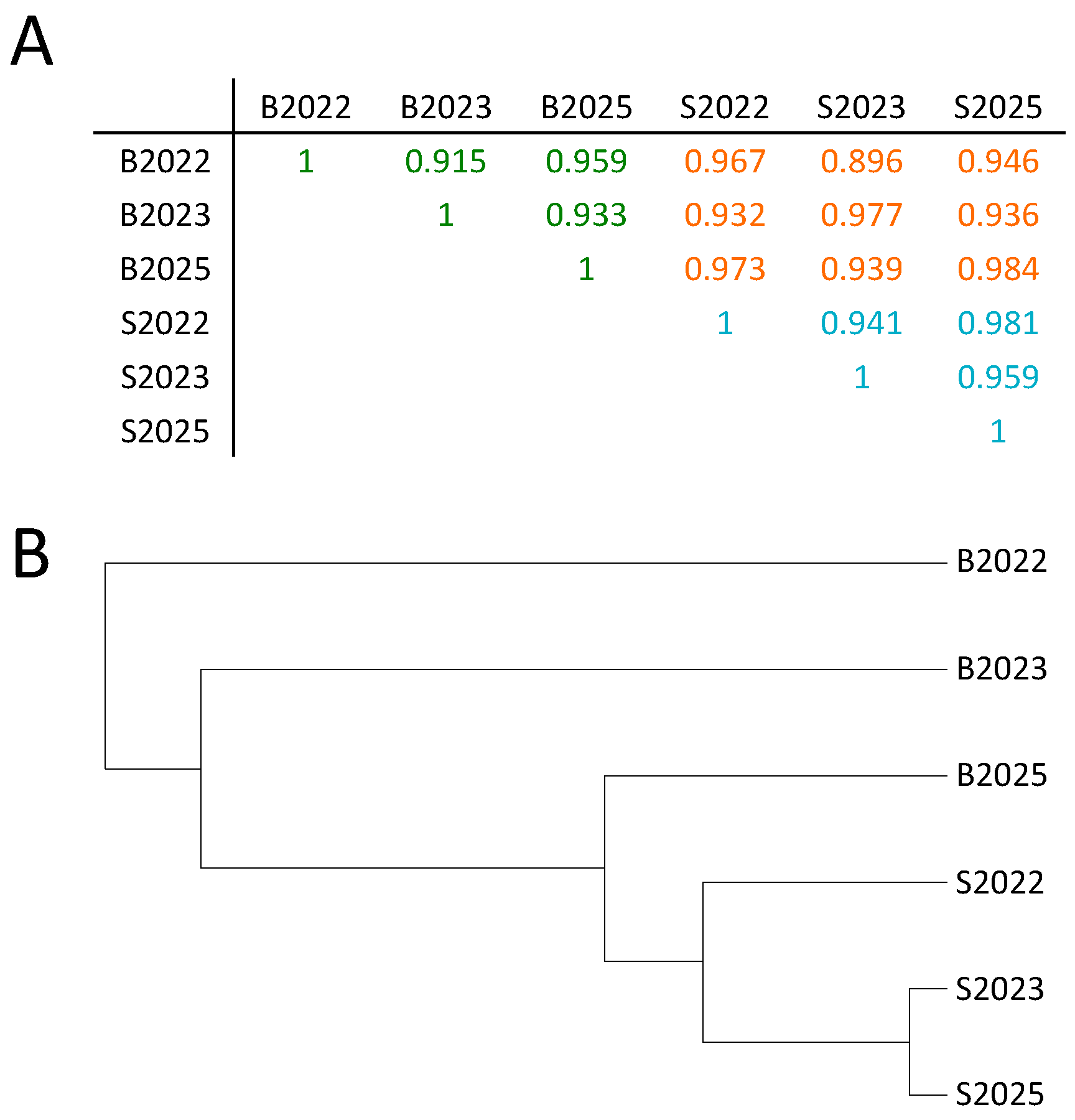
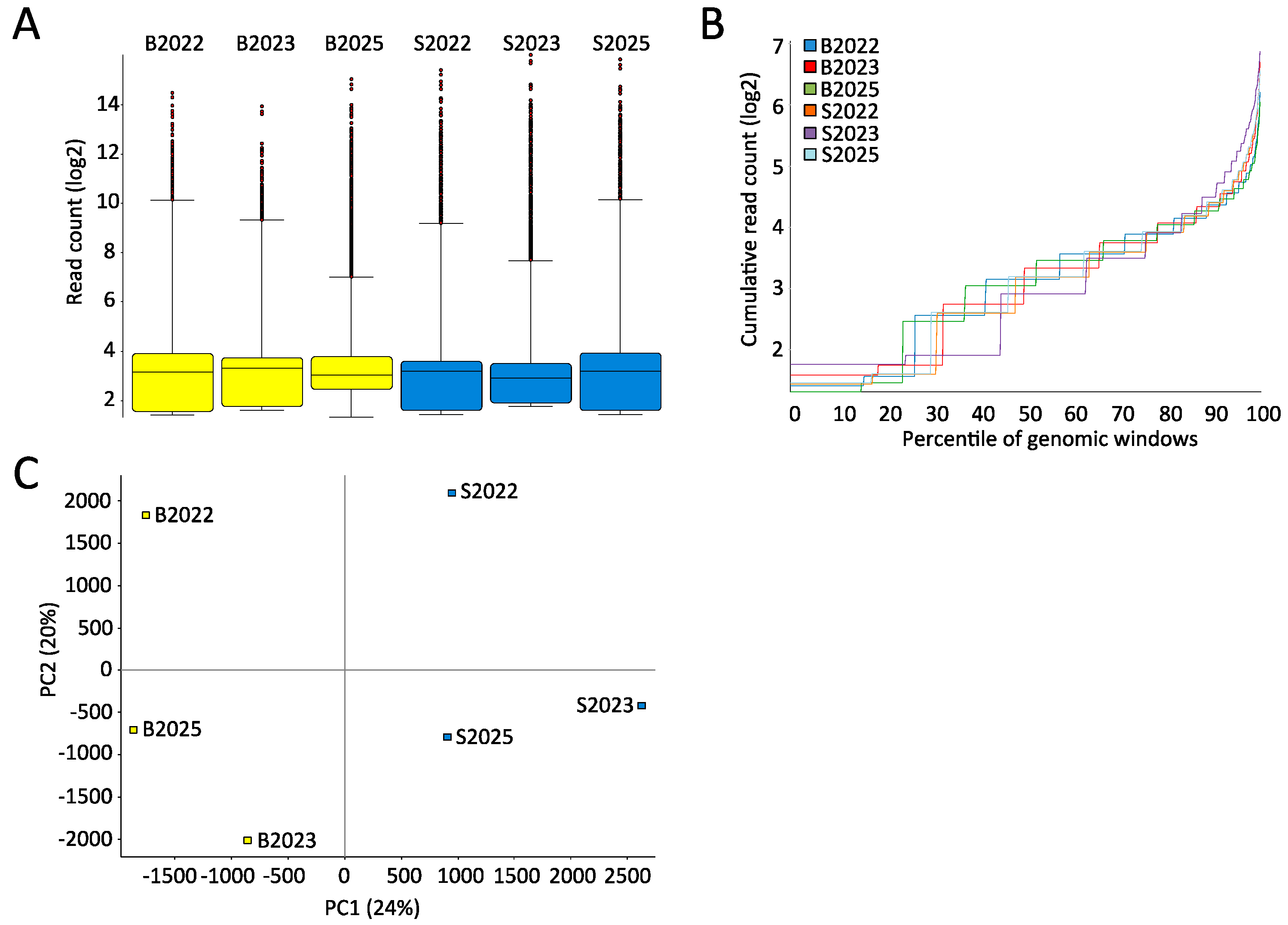
2.1. High-Quality Sequencing Data Generated by MeDIP-seq Using Saliva-Extracted DNA
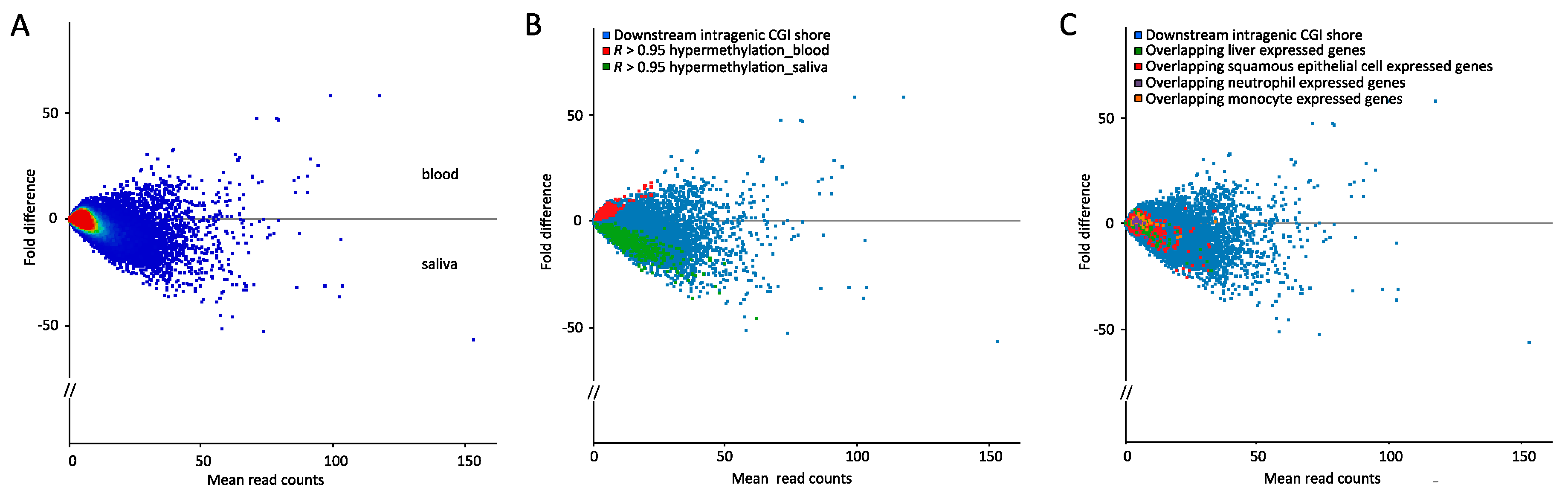
2.2. Few Differently-Methylated Windows are Detected between Saliva and Blood
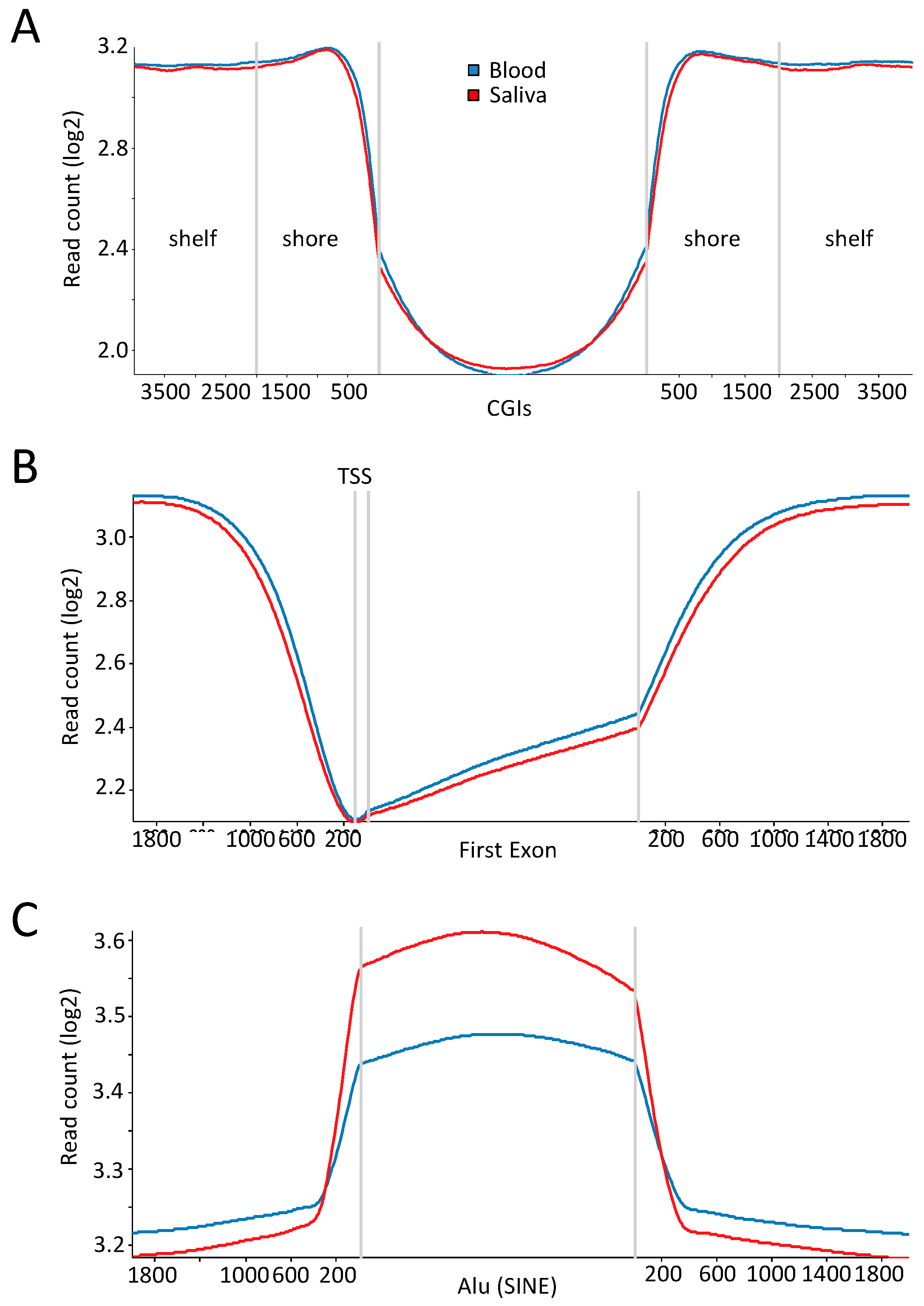
2.3. Comparable DNA Methylation Patterns between Blood and Saliva at Defined Genomic Regions
3. Materials and Methods
3.1. Biological Samples
3.2. MeDIP-seq
3.3. Sequencing and Bioinformatics
3.4. Ethics Statement
3.5. Availability of Data and Materials
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bjornsson, H.T.; Cui, H.; Gius, D.; Fallin, M.D.; Feinberg, A.P. The new field of epigenomics: Implications for cancer and other common disease research. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. Epigenetics at the epicenter of modern medicine. JAMA 2008, 299, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppede, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [PubMed]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2014, 122, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.A.; McClay, J.L.; Nerella, S.; Clark, S.; Kumar, G.; Chen, W.; Khachane, A.N.; Xie, L.; Hudson, A.; Gao, G.; et al. Methylome-wide association study of schizophrenia: Identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 2014, 71, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Dempster, E.L.; Pidsley, R.; Schalkwyk, L.C.; Owens, S.; Georgiades, A.; Kane, F.; Kalidindi, S.; Picchioni, M.; Kravariti, E.; Toulopoulou, T.; et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum. Mol. Genet. 2011, 20, 4786–4796. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Numata, S.; Tajima, A.; Ohi, K.; Hashimoto, R.; Shimodera, S.; Imoto, I.; Takeda, M.; Ohmori, T. Aberrant DNA methylation of blood in schizophrenia by adjusting for estimated cellular proportions. Neuromol. Med. 2014, 16, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, J.; Ehrlich, S.; Walton, E.; White, T.; Perrone-Bizzozero, N.; Bustillo, J.; Turner, J.A.; Calhoun, V.D. Methylation patterns in whole blood correlate with symptoms in schizophrenia patients. Schizophr. Bull. 2014, 40, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A.; et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Montano, C.; Taub, M.A.; Jaffe, A.; Briem, E.; Feinberg, J.I.; Trygvadottir, R.; Idrizi, A.; Runarsson, A.; Berndsen, B.; Gur, R.C.; et al. Association of DNA Methylation Differences With Schizophrenia in an Epigenome-Wide Association Study. JAMA Psychiatry 2016, 73, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B.; et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, L.; Carli, V.; Sarchiapone, M.; George, D.K.; Goldman, D.; Roy, A.; Enoch, M.A. Interaction between FKBP5 and childhood trauma and risk of aggressive behavior. Arch. Gener. Psychiatry 2012, 69, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Bahlo, M.; Stankovich, J.; Danoy, P.; Hickey, P.F.; Taylor, B.V.; Browning, S.R.; The Australian and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene); Brown, M.A.; Rubio, J.P. Saliva-derived DNA performs well in large-scale, high-density single-nucleotide polymorphism microarray studies. Cancer Epidemiol. Biomark. Prev. 2010, 19, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, J.S.; Erdman, C.A.; Hamilton, S.P. Array-based whole-genome survey of dog saliva DNA yields high quality SNP data. PLoS ONE 2010, 5, e10809. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.E.; Maranian, M.J.; Spiteri, I.; Russell, R.; Ingle, S.; Luccarini, C.; Earl, H.M.; Pharoah, P.P.; Dunning, A.M.; Caldas, C. Saliva samples are a viable alternative to blood samples as a source of DNA for high throughput genotyping. BMC Med. Genom. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.K.; Kilaru, V.; Klengel, T.; Mercer, K.B.; Bradley, B.; Conneely, K.N.; Ressler, K.J.; Binder, E.B. DNA extracted from saliva for methylation studies of psychiatric traits: Evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168B, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.M.; Sharfi, D.; Lee, M.; Yrigollen, C.M.; Naumova, O.Y.; Grigorenko, E.L. Comparison of whole-genome DNA methylation patterns in whole blood, saliva, and lymphoblastoid cell lines. Behav. Genet. 2013, 43, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.; Tochigi, M.; Jia, P.; Pal, M.; Mill, J.; Kwan, A.; Ioshikhes, I.; Vincent, J.B.; Kennedy, J.L.; Strauss, J.; et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol. Psychiatry 2012, 17, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.C.; O’Reilly, R.L.; Singh, S.M. DNA methylation and mRNA expression of SYN III, a candidate gene for schizophrenia. BMC Med. Genet. 2008, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Evans, J.; Chae, H.; Pilrose, J.; Kim, S.; Yan, P.; Huang, R.L.; Lai, H.C.; Lin, H.; Liu, Y.; et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene 2013, 32, 4519–4528. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Prange-Krex, G.; Freiberg-Richter, J.; Bornhauser, M.; Ehninger, G. Buccal swabs but not mouthwash samples can be used to obtain pretransplant DNA fingerprints from recipients of allogeneic bone marrow transplants. Bone Marrow Transpl. 2000, 25, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, A.; Vidovic Juras, D.; Vucicevic Boras, V.; Lukac, J.; Grubisic-Ilic, M.; Rak, D.; Sabioncello, A. Determination of leucocyte subsets in human saliva by flow cytometry. Arch. Oral. Biol. 2012, 57, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Webb, S.; Kerr, A.R.; Illingworth, R.S.; Guy, J.; Andrews, R.; Bird, A. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011, 21, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schubeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Essex, M.J.; Boyce, W.T.; Hertzman, C.; Lam, L.L.; Armstrong, J.M.; Neumann, S.M.; Kobor, M.S. Epigenetic vestiges of early developmental adversity: Childhood stress exposure and DNA methylation in adolescence. Child Dev. 2013, 84, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F.; et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Z.; Zhang, H.; Ge, W.; Weder, N.; Douglas-Palumberi, H.; Perepletchikova, F.; Gelernter, J.; Kaufman, J. Child abuse and epigenetic mechanisms of disease risk. Am. J. Prev. Med. 2013, 44, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.; Palta, P.; Joyce, C.J.; Scott, C.; Grundberg, E.; Deloukas, P.; Palotie, A.; Coffey, A.J. A comparison of the whole genome approach of MeDIP-seq to the targeted approach of the Infinium HumanMethylation450 BeadChip(®) for methylome profiling. PLoS ONE 2012, 7, e50233. [Google Scholar] [CrossRef] [PubMed]
- Beyan, H.; Down, T.A.; Ramagopalan, S.V.; Uvebrant, K.; Nilsson, A.; Holland, M.L.; Gemma, C.; Giovannoni, G.; Boehm, B.O.; Ebers, G.C.; et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012, 22, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yu, D.; Chen, J.; Cao, R.; Yang, J.; Wang, H.; Ji, X.; Ning, B.; Shi, T. Re-annotation of presumed noncoding disease/trait-associated genetic variants by integrative analyses. Sci. Rep. 2015, 5, 9453. [Google Scholar] [CrossRef] [PubMed]
- Feber, A.; Wilson, G.A.; Zhang, L.; Presneau, N.; Idowu, B.; Down, T.A.; Rakyan, V.K.; Noon, L.A.; Lloyd, A.C.; Stupka, E.; et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res. 2011, 21, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Rajendiran, S.; Gibbs, L.D.; Van Treuren, T.; Klinkebiel, D.L.; Vishwanatha, J.K. MIEN1 is tightly regulated by SINE Alu methylation in its promoter. Oncotarget 2016, 7, 65307–65319. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ye, M.; Li, Y.; Yan, Z.; Butcher, L.M.; Sun, J.; Han, X.; Chen, Q.; Zhang, X.; Wang, J. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 2010, 52, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Chavez, L.; Jozefczuk, J.; Grimm, C.; Dietrich, J.; Timmermann, B.; Lehrach, H.; Herwig, R.; Adjaye, J. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010, 20, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Neary, J.L.; Perez, S.M.; Peterson, K.; Lodge, D.J.; Carless, M.A. Comparative analysis of MBD-seq and MeDIP-seq and estimation of gene expression changes in a rodent model of schizophrenia. Genomics 2017, 109, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Staunstrup, N.H.; Starnawska, A.; Nyegaard, M.; Christiansen, L.; Nielsen, A.L.; Borglum, A.; Mors, O. Genome-wide DNA methylation profiling with MeDIP-seq using archived dried blood spots. Clin. Epigenet. 2016, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.; Neeleman, M.; Luykx, J.J.; ten Berg, M.J.; Strengman, E.; Den Breeijen, H.H.; Stijvers, L.C.; Buizer-Voskamp, J.E.; Bakker, S.C.; Kahn, R.S.; et al. Seasonal changes in gene expression represent cell-type composition in whole blood. Hum. Mol. Genet. 2014, 23, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Whitney, A.R.; Diehn, M.; Popper, S.J.; Alizadeh, A.A.; Boldrick, J.C.; Relman, D.A.; Brown, P.O. Individuality and variation in gene expression patterns in human blood. Proc. Natl. Acad. Sci. USA 2003, 100, 1896–1901. [Google Scholar] [CrossRef] [PubMed]
- Eady, J.J.; Wortley, G.M.; Wormstone, Y.M.; Hughes, J.C.; Astley, S.B.; Foxall, R.J.; Doleman, J.F.; Elliott, R.M. Variation in gene expression profiles of peripheral blood mononuclear cells from healthy volunteers. Physiol. Genom. 2005, 22, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Shao, X.; Caron, M.; Kwan, T.; Allum, F.; Cheung, W.A.; Ge, B.; Westfall, S.; Simon, M.M.; The Multiple Tissue Human Expression Resource; et al. Population whole-genome bisulfite sequencing across two tissues highlights the environment as the principal source of human methylome variation. Genome Biol. 2015, 16, 290. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013, 45, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Muller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Nivard, M.G.; Willemsen, G.; Hottenga, J.J.; Helmer, Q.; Dolan, C.V.; Ehli, E.A.; Davies, G.E.; van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.A.; Szarc Vel Szic, K.; Declerck, K.; Traen, S.; Koppen, G.; Van Camp, G.; Schoeters, G.; Vanden Berghe, W.; De Boever, P. Whole-Genome Saliva and Blood DNA Methylation Profiling in Individuals with a Respiratory Allergy. PLoS ONE 2016, 11, e0151109. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Qiu, C.; Li, J.; Tian, Q.; Deng, H.W. Characterization of the DNA methylome and its interindividual variation in human peripheral blood monocytes. Epigenomics 2013, 5, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Godderis, L.; Schouteden, C.; Tabish, A.; Poels, K.; Hoet, P.; Baccarelli, A.A.; Van Landuyt, K. Global Methylation and Hydroxymethylation in DNA from Blood and Saliva in Healthy Volunteers. BioMed Res. Int. 2015, 2015, 845041. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Wang, Q.; Chung, W.K.; Andrulis, I.L.; Daly, M.B.; John, E.M.; Keegan, T.H.; Knight, J.; Bradbury, A.R.; Kappil, M.A.; et al. Correlation of DNA methylation levels in blood and saliva DNA in young girls of the LEGACY Girls study. Epigenetics 2014, 9, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Grover, D.; Mukerji, M.; Bhatnagar, P.; Kannan, K.; Brahmachari, S.K. Alu repeat analysis in the complete human genome: Trends and variations with respect to genomic composition. Bioinformatics 2004, 20, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Buj, R.; Mallona, I.; Diez-Villanueva, A.; Barrera, V.; Mauricio, D.; Puig-Domingo, M.; Reverter, J.L.; Matias-Guiu, X.; Azuara, D.; Ramirez, J.L.; et al. Quantification of unmethylated Alu (QUAlu): A tool to assess global hypomethylation in routine clinical samples. Oncotarget 2016, 7, 10536–10546. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lu, X.; Xie, H. Dynamic Alu methylation during normal development, aging, and tumorigenesis. BioMed Res. Int. 2014, 2014, 784706. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.A.; Lesseur, C.; Conradt, E.; Lester, B.M.; Marsit, C.J. Global and gene-specific DNA methylation across multiple tissues in early infancy: Implications for children’s health research. FASEB J. 2014, 28, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, C.; Dell’Osso, B.; Palazzo, M.C.; Benatti, B.; Lietti, L.; Cattaneo, E.; Galimberti, D.; Fenoglio, C.; Cortini, F.; Scarpini, E.; et al. Selective DNA methylation of BDNF promoter in bipolar disorder: Differences among patients with BDI and BDII. Neuropsychopharmacology 2012, 37, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, H.; Iwamoto, K.; Bundo, M.; Ueda, J.; Miyauchi, T.; Komori, A.; Kazuno, A.; Adati, N.; Kusumi, I.; Okazaki, Y.; et al. Hypermethylation of serotonin transporter gene in bipolar disorder detected by epigenome analysis of discordant monozygotic twins. Transl. Psychiatry 2011, 1, e24. [Google Scholar] [CrossRef] [PubMed]
- Starnawska, A.; Demontis, D.; Pen, A.; Hedemand, A.; Nielsen, A.L.; Staunstrup, N.H.; Grove, J.; Als, T.D.; Jarram, A.; O’Brien, N.L.; et al. CACNA1C hypermethylation is associated with bipolar disorder. Transl. Psychiatry 2016, 6, e831. [Google Scholar] [CrossRef] [PubMed]
- Carrard, A.; Salzmann, A.; Malafosse, A.; Karege, F. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J. Affect. Disord. 2011, 132, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Kordi-Tamandani, D.M.; Sahranavard, R.; Torkamanzehi, A. DNA methylation and expression profiles of the brain-derived neurotrophic factor (BDNF) and dopamine transporter (DAT1) genes in patients with schizophrenia. Mol. Biol. Rep. 2012, 39, 10889–10893. [Google Scholar] [CrossRef] [PubMed]
- Melas, P.A.; Rogdaki, M.; Osby, U.; Schalling, M.; Lavebratt, C.; Ekstrom, T.J. Epigenetic aberrations in leukocytes of patients with schizophrenia: Association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012, 26, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J. Psychiatr. Res. 2011, 45, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, T.; Bundo, M.; Sunaga, F.; Asai, T.; Nishimura, F.; Yoshikawa, A.; Kawamura, Y.; Hibino, H.; Tochigi, M.; Kakiuchi, C.; et al. DNA methylation analysis of BDNF gene promoters in peripheral blood cells of schizophrenia patients. Neurosci. Res. 2013, 77, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Petronis, A.; Gottesman, II; Kan, P.; Kennedy, J.L.; Basile, V.S.; Paterson, A.D.; Popendikyte, V. Monozygotic twins exhibit numerous epigenetic differences: Clues to twin discordance? Schizophr. Bull. 2003, 29, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Martens, K.; Magi, R.; Kolde, R.; Koltsina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef] [PubMed]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Heath, S.; Bibikova, M.; Queiros, A.C.; Navarro, A.; Clot, G.; Martinez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Thorne, N.P.; Flicek, P.; Kulesha, E.; Graf, S.; Tomazou, E.M.; Backdahl, L.; Johnson, N.; Herberth, M.; et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res. 2008, 18, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Colot, V.; Jansen, R.C. Epigenome dynamics: A quantitative genetics perspective. Nat. Rev. Genet. 2008, 9, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Ozanne, S.E.; Constancia, M. Mechanisms of disease: The developmental origins of disease and the role of the epigenotype. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Reinius, L.E.; Acevedo, N.; Joerink, M.; Pershagen, G.; Dahlen, S.E.; Greco, D.; Soderhall, C.; Scheynius, A.; Kere, J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS ONE 2012, 7, e41361. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Morgan, M.; Hutchison, K.; Calhoun, V.D. A study of the influence of sex on genome wide methylation. PLoS ONE 2010, 5, e10028. [Google Scholar] [CrossRef] [PubMed]
- Jette, L.; Bissoon-Haqqani, S.; Le Francois, B.; Maroun, J.A.; Birnboim, H.C. Resistance of colorectal cancer cells to 5-FUdR and 5-FU caused by Mycoplasma infection. Anticancer Res. 2008, 28, 2175–2180. [Google Scholar] [PubMed]
- Speyer, H.; Norgaard, H.C.; Hjorthoj, C.; Madsen, T.A.; Drivsholm, S.; Pisinger, C.; Gluud, C.; Mors, O.; Krogh, J.; Nordentoft, M. Protocol for CHANGE: A randomized clinical trial assessing lifestyle coaching plus care coordination versus care coordination alone versus treatment as usual to reduce risks of cardiovascular disease in adults with schizophrenia and abdominal obesity. BMC Psychiatry 2015, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Enrichment Analysis | Number of Genes in Reference | Number of Genes in Input List | Fold Enrichment | Bonferroni Corrected p-Value |
|---|---|---|---|---|
| B-cell activation (P00010) | 72 | 9 | 4.54 | 3.4 × 10−2 |
| NFATC1, ITPR2, AC037459.4, MAPK8, CALM1 (+isoforms), NFATC2, MAP2K2 | ||||
| T-cell activation (P00053) | 96 | 11 | 4.16 | 1.5 × 10−2 |
| NFATC1, AC037459.4, MAPK8, CALM1 (+isoforms), NFATC2, MAP2K2, PIK3R1, PLCG1, CD247 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staunstrup, N.H.; Starnawska, A.; Nyegaard, M.; Nielsen, A.L.; Børglum, A.; Mors, O. Saliva as a Blood Alternative for Genome-Wide DNA Methylation Profiling by Methylated DNA Immunoprecipitation (MeDIP) Sequencing. Epigenomes 2017, 1, 14. https://doi.org/10.3390/epigenomes1030014
Staunstrup NH, Starnawska A, Nyegaard M, Nielsen AL, Børglum A, Mors O. Saliva as a Blood Alternative for Genome-Wide DNA Methylation Profiling by Methylated DNA Immunoprecipitation (MeDIP) Sequencing. Epigenomes. 2017; 1(3):14. https://doi.org/10.3390/epigenomes1030014
Chicago/Turabian StyleStaunstrup, Nicklas Heine, Anna Starnawska, Mette Nyegaard, Anders Lade Nielsen, Anders Børglum, and Ole Mors. 2017. "Saliva as a Blood Alternative for Genome-Wide DNA Methylation Profiling by Methylated DNA Immunoprecipitation (MeDIP) Sequencing" Epigenomes 1, no. 3: 14. https://doi.org/10.3390/epigenomes1030014
APA StyleStaunstrup, N. H., Starnawska, A., Nyegaard, M., Nielsen, A. L., Børglum, A., & Mors, O. (2017). Saliva as a Blood Alternative for Genome-Wide DNA Methylation Profiling by Methylated DNA Immunoprecipitation (MeDIP) Sequencing. Epigenomes, 1(3), 14. https://doi.org/10.3390/epigenomes1030014