
Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias


 , , ,
, , ,  , , ,
, , ,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
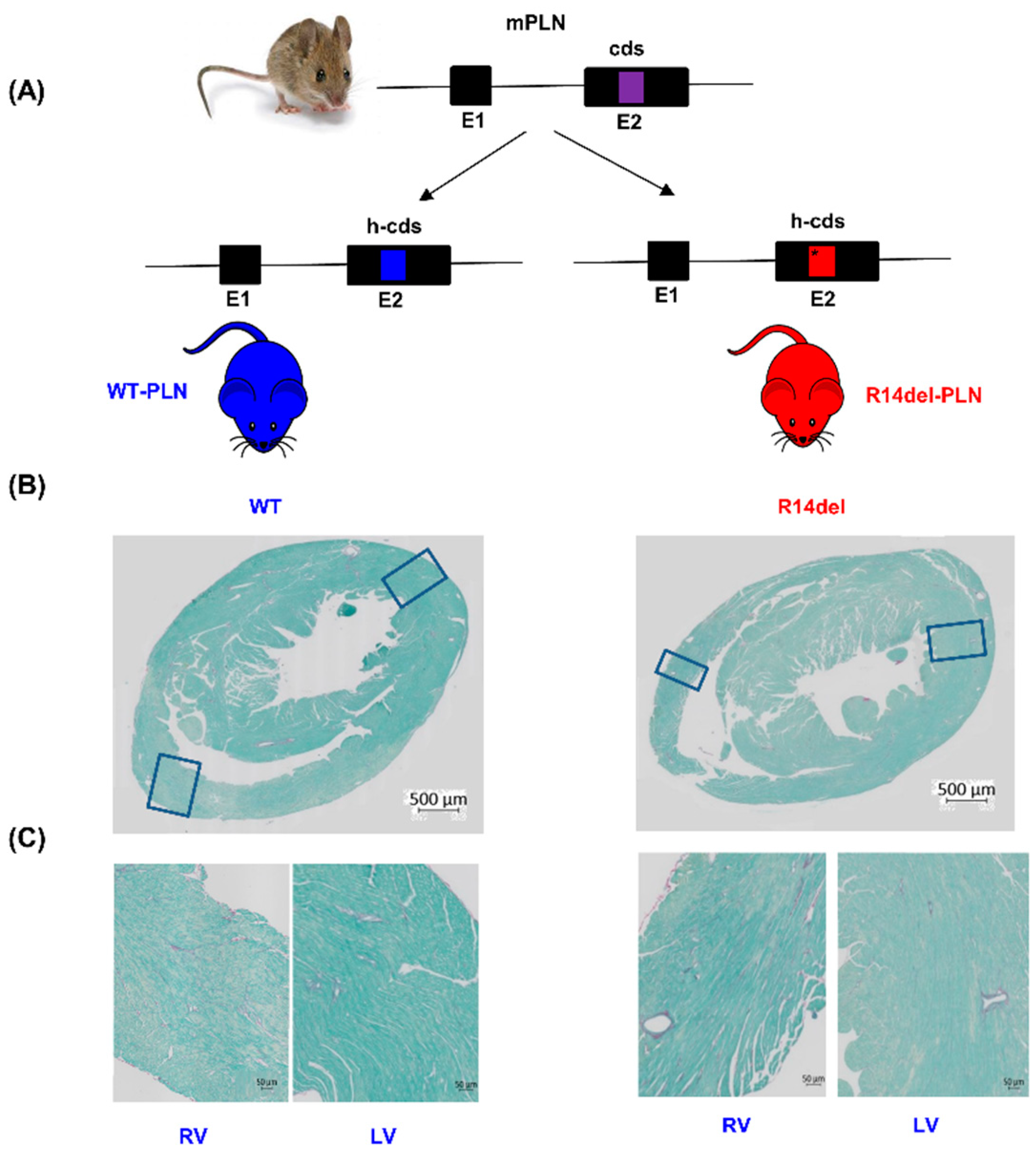
2.1. Humanized WT-PLN and R14del-PLN Knock-in Mice
2.2. Histology
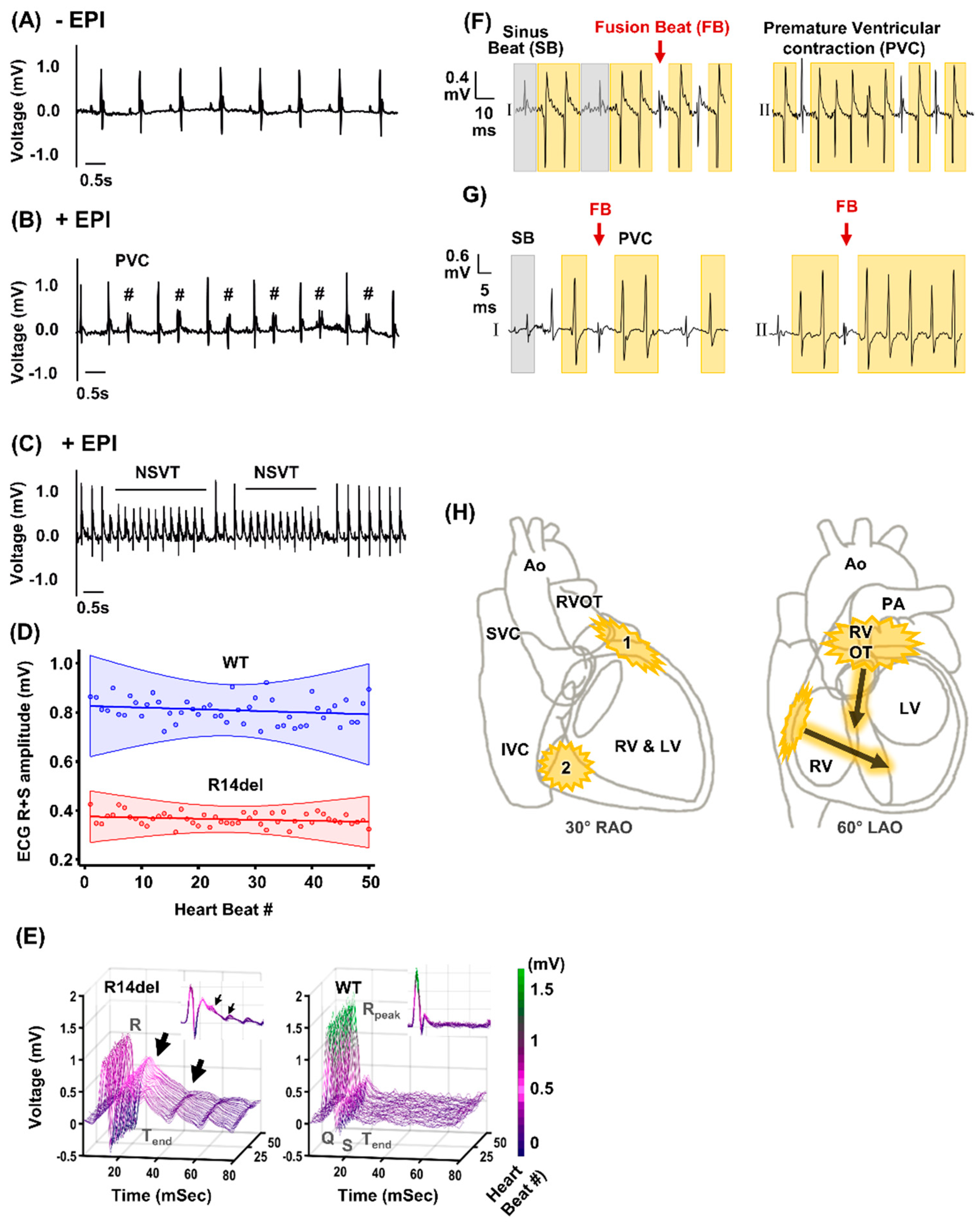
2.3. In Vivo Stress-Induced Arrhythmias
2.4. Human R14del-PLN Electrocardiogram Studies
2.5. Electrophysiological Recordings
2.6. Mouse Myocyte Mechanics, Ca Kinetics
2.7. Measurement of Calcium Sparks
2.8. Aftercontractions and CaMKII Inhibitor KN93 in Isolated Cardiomyocytes
2.9. Immunofluorescence Staining and Quantitative Immunoblotting
2.10. RNA Sequencing Analysis
2.11. Statistical Analysis
3. Results
3.1. Ventricular Ectopy Originating in the Right Ventricle of Humanized Mice Harboring the R14del-PLN Mutation
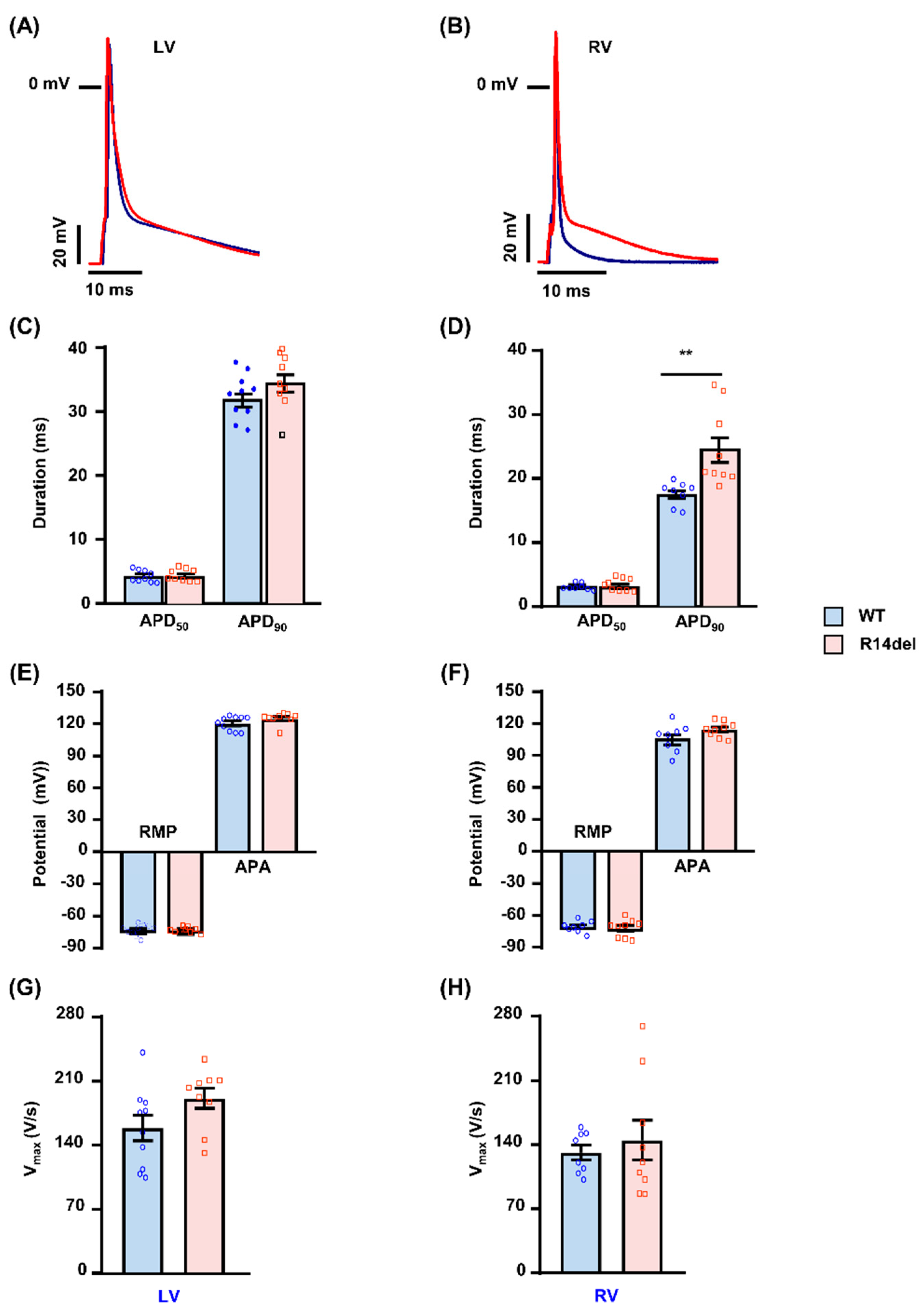
3.2. R14del-PLN Associates with Prolongation of Action Potential in RV Myocytes
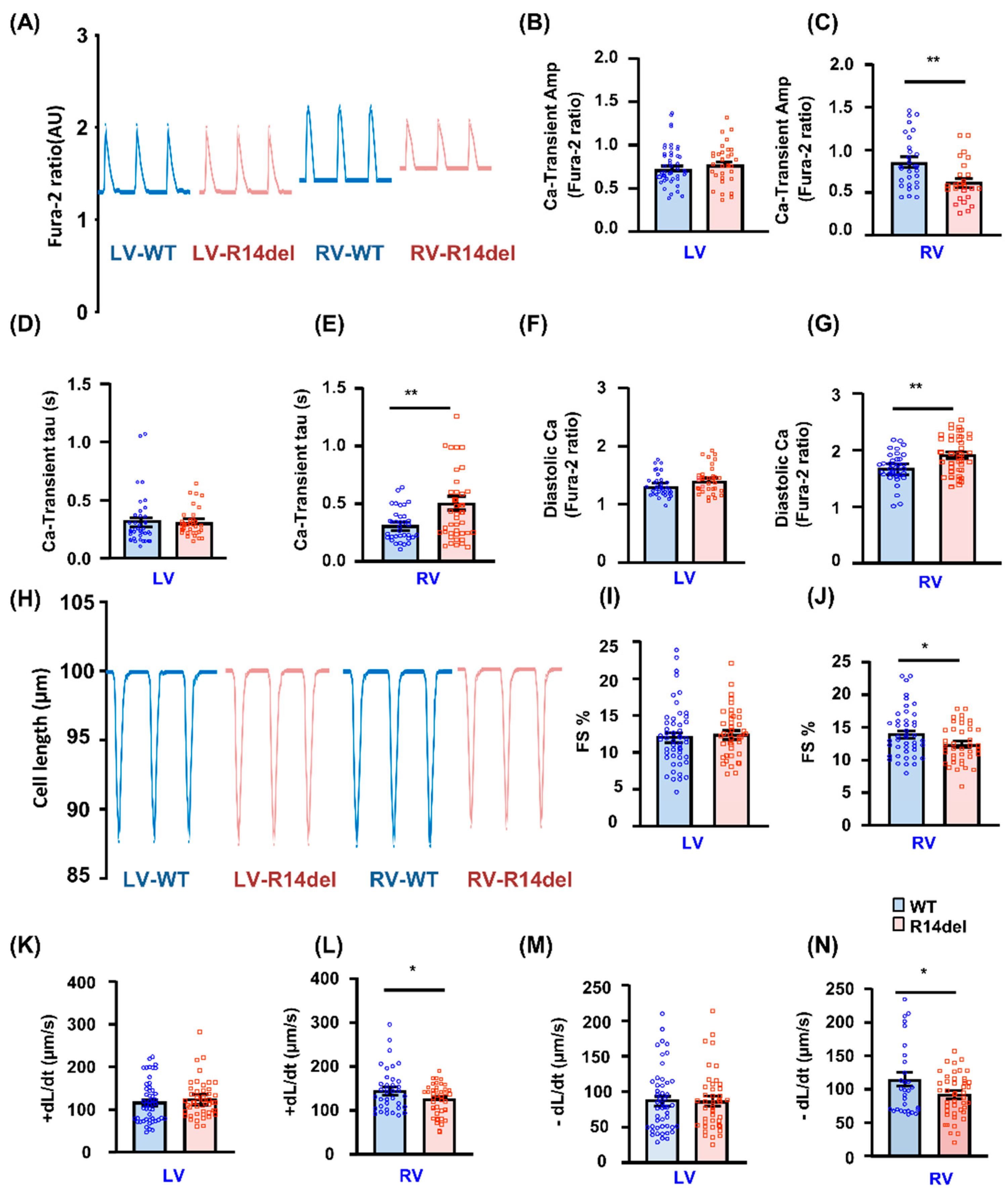
3.3. Calcium Kinetics and Contractile Parameters Are Depressed in RV Myocytes
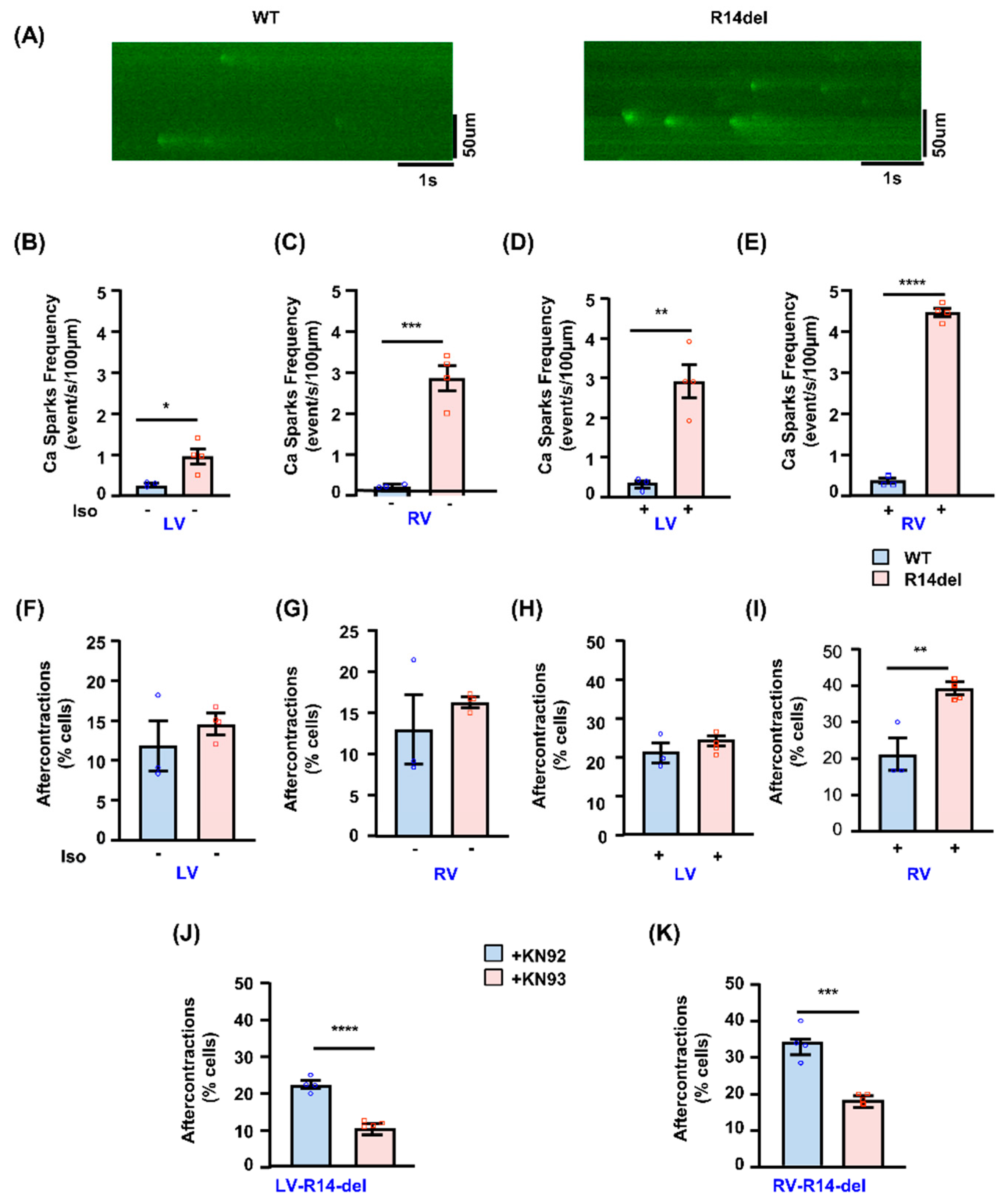
3.4. Calcium Sparks and After-Contractions Are Increased in RV Myocytes from R14del-PLN Hearts
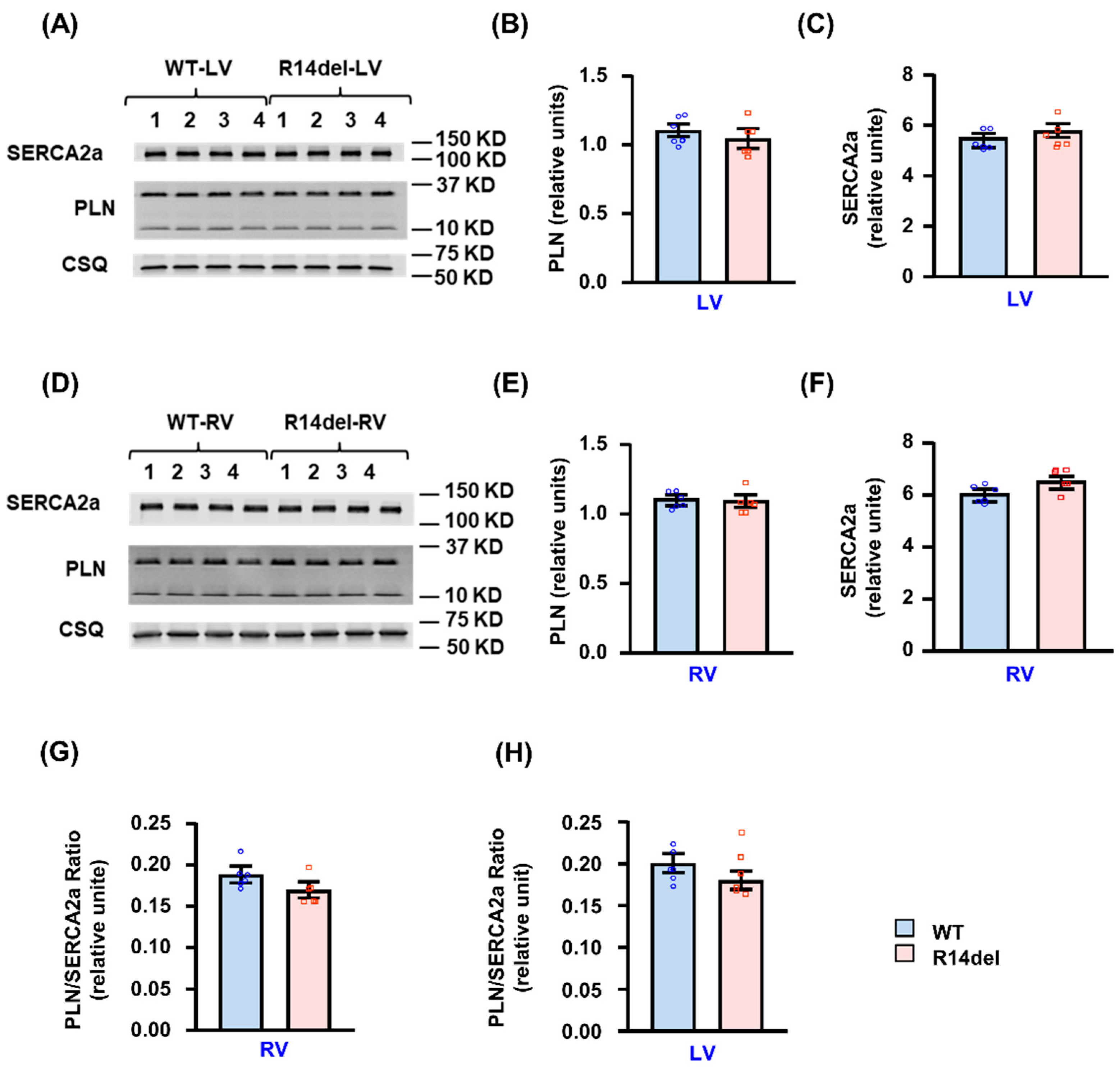
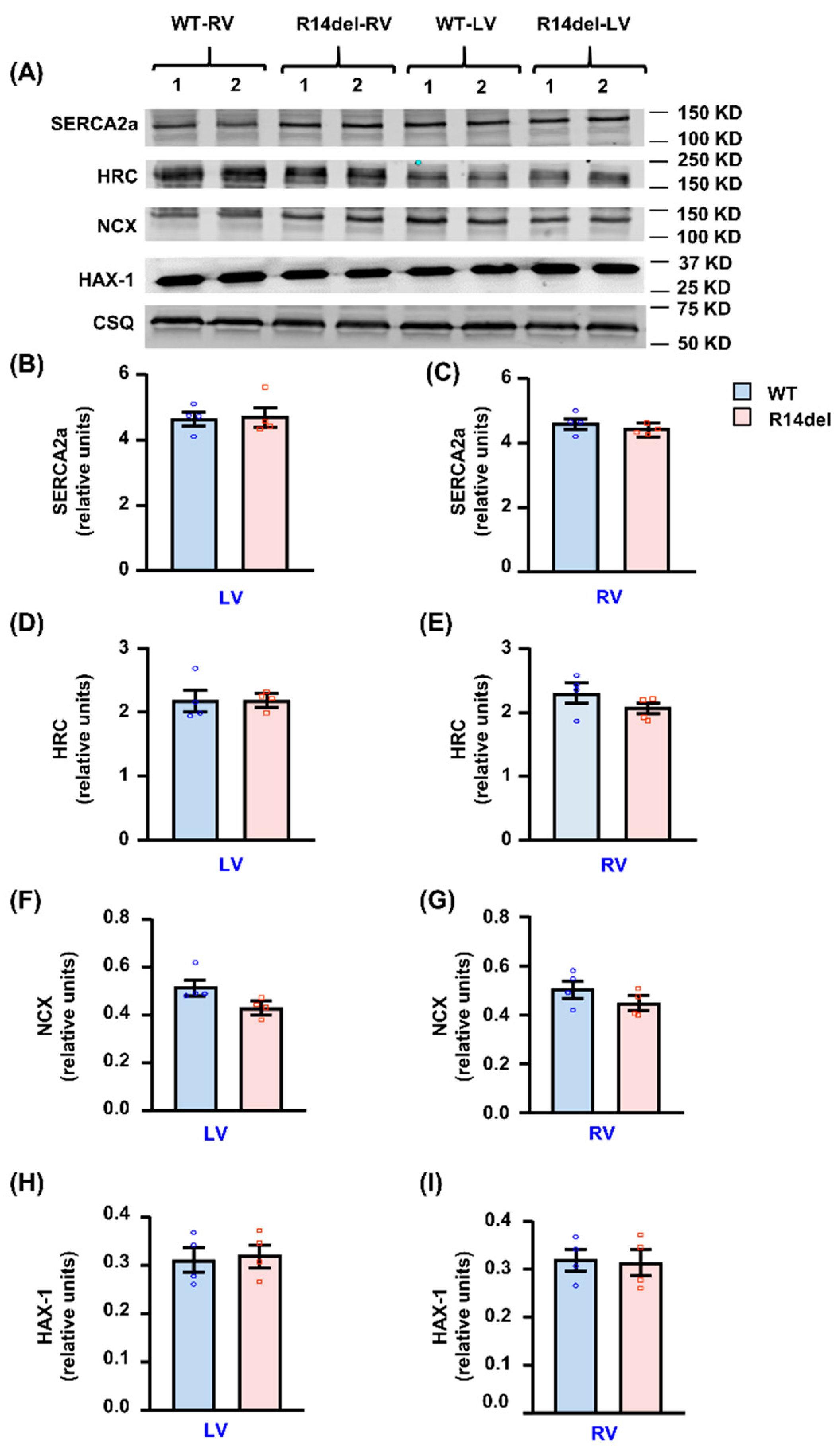
3.5. Calcium Handling Protein Levels
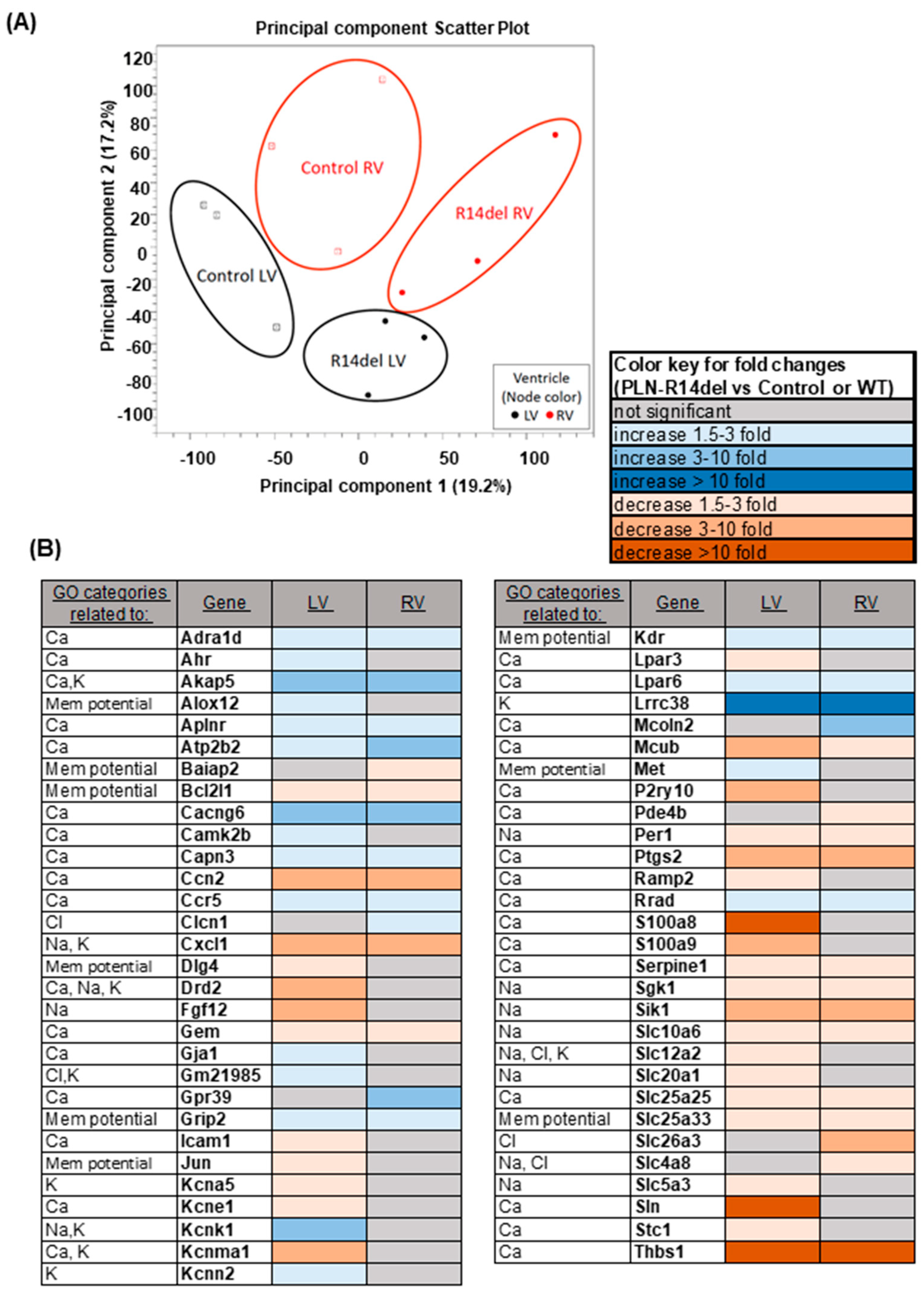
3.6. Gene Expression Profiles
4. Discussion
4.1. Ventricular Tachycardia and Its Origin in RV
4.2. Right Ventricular Specificity of R14del-PLN
4.3. Decreases in ER Stress and Apoptosis Pathways
4.4. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Roux-Buisson, N.; Gandjbakhch, E.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; Cosnay, P.; et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Results of a systematic screening. Heart Rhythm 2014, 11, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq Transatlantic Network of Excellence to Cure Phospholamban-Induced Cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Doevendans, P.A.; Glijnis, P.C.; Hajjar, R.J. PLN Foundation. Circ. Res. 2018, 123, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef]
- Haghighi, K.; Pritchard, T.; Bossuyt, J.; Waggoner, J.R.; Yuan, Q.; Fan, G.C.; Osinska, H.; Anjak, A.; Rubinstein, J.; Robbins, J.; et al. The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase. J. Mol. Cell. Cardiol. 2012, 52, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Eijgenraam, T.R.; Boukens, B.J.; Boogerd, C.J.; Schouten, E.M.; van de Kolk, C.W.A.; Stege, N.M.; Te Rijdt, W.P.; Hoorntje, E.T.; van der Zwaag, P.A.; van Rooij, E.; et al. The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy. Sci. Rep. 2020, 10, 9819. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yuan, Q.; Qian, J.; Waggoner, J.R.; Pathak, A.; Chu, G.; Mitton, B.; Sun, X.; Jin, J.; Braz, J.C.; et al. The presence of Lys27 instead of Asn27 in human phospholamban promotes sarcoplasmic reticulum Ca2+-ATPase superinhibition and cardiac remodeling. Circulation 2006, 113, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Chadda, K.R.; Ahmad, S.; Valli, H.; den Uijl, I.; Al-Hadithi, A.B.; Salvage, S.C.; Grace, A.A.; Huang, C.L.; Jeevaratnam, K. The effects of ageing and adrenergic challenge on electrocardiographic phenotypes in a murine model of long QT syndrome type 3. Sci. Rep. 2017, 7, 11070. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.; Zhao, X.; Gao, S.; Hong, C.; Vatner, D.E.; Vatner, S.F. Heart Rate and Electrocardiography Monitoring in Mice. Curr Protoc Mouse Biol. 2011, 1, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Zhang, X.; Cheng, L.; Lammers, W.J.; Grace, A.A.; Fraser, J.A.; Zhang, H.; Huang, C.L.; Lei, M. Altered sinoatrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/DeltaKPQ hearts suggest an overlap syndrome. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1510–H1523. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Rubinstein, J.; Arvanitis, D.A.; Ren, X.; Gao, X.; Haghighi, K.; Gilbert, M.; Iyer, V.R.; Kim, D.H.; Cho, C.; et al. Abnormal calcium cycling and cardiac arrhythmias associated with the human Ser96Ala genetic variant of histidine-rich calcium-binding protein. J. Am. Heart Assoc. 2013, 2, e000460. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Pritchard, T.J.; Liu, G.S.; Singh, V.P.; Bidwell, P.; Lam, C.K.; Vafiadaki, E.; Das, P.; Ma, J.; Kunduri, S.; et al. Human G109E-inhibitor-1 impairs cardiac function and promotes arrhythmias. J. Mol. Cell. Cardiol. 2015, 89, 349–359. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Green, L.C.; Anthony, S.R.; Slone, S.; Lanzillotta, L.; Nieman, M.L.; Wu, X.; Robbins, N.; Jones, S.M.; Roy, S.; Owens, A.P., 3rd; et al. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. JCI Insight 2019, 4, e121541. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Morales, A.; Vafiadaki, E.; Lam, C.K.; Cai, W.F.; Haghighi, K.; Adly, G.; Hershberger, R.E.; Kranias, E.G. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc. Res. 2015, 107, 164–174. [Google Scholar] [CrossRef] [PubMed]
- DeMazumder, D.; Limpitikul, W.B.; Dorante, M.; Dey, S.; Mukhopadhyay, B.; Zhang, Y.; Moorman, J.R.; Cheng, A.; Berger, R.D.; Guallar, E.; et al. Entropy of cardiac repolarization predicts ventricular arrhythmias and mortality in patients receiving an implantable cardioverter-defibrillator for primary prevention of sudden death. Europace 2016, 18, 818–1828. [Google Scholar] [CrossRef] [PubMed]
- Kleber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [PubMed]
- van de Leur, R.R.; Taha, K.; Bos, M.N.; van der Heijden, J.F.; Gupta, D.; Cramer, M.J.; Hassink, R.J.; van der Harst, P.; Doevendans, P.A.; Asselbergs, F.W.; et al. Discovering and Visualizing Disease-Specific Electrocardiogram Features Using Deep Learning: Proof-of-Concept in Phospholamban Gene Mutation Carriers. Circ. Arrhythm. Electrophysiol. 2021, 14, e009056. [Google Scholar] [CrossRef] [PubMed]
- Keramati, A.R.; DeMazumder, D.; Misra, S.; Chrispin, J.; Assis, F.R.; Raghuram, C.; Dey, S.; Calkins, H.; Tandri, H. Anterior pericardial access to facilitate electrophysiology study and catheter ablation of ventricular arrhythmias: A single tertiary center experience. J. Cardiovasc. Electrophysiol. 2017, 28, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Wang, P.; Liu, X.; Lim, E.L.; Wang, Z.; Yeager, M.; Wong, M.P.; Sham, P.C.; Chanock, S.J.; Wang, J. GWASdb: A database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2012, 40, D1047–D1054. [Google Scholar] [CrossRef]
- Anderson, D.M.; Makarewich, C.A.; Anderson, K.M.; Shelton, J.M.; Bezprozvannaya, S.; Bassel-Duby, R.; Olson, E.N. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci. Signal. 2016, 9, ra119. [Google Scholar] [CrossRef] [PubMed]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Schoenberg, M. Strength-duration curves in cardiac Purkinje fibres: Effects of liminal length and charge distribution. J. Physiol. 1972, 226, 593–618. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Nguyen, T.P.; Olcese, R.; Chen, P.S.; Qu, Z. Perspective: A dynamics-based classification of ventricular arrhythmias. J. Mol. Cell. Cardiol. 2015, 82, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Denis, A.; Sacher, F.; Derval, N.; Martin, R.; Lim, H.S.; Pambrun, T.; Massoullie, G.; Duchateau, J.; Cochet, H.; Pillois, X.; et al. Arrhythmogenic response to isoproterenol testing vs. exercise testing in arrhythmogenic right ventricular cardiomyopathy patients. Europace 2018, 20, f30–f36. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.A.; Goldhaber, J.I.; So, J.M.; Han, T.; Motter, C.; Ngo, A.; Chantawansri, C.; Ritter, M.R.; Friedlander, M.; Nicoll, D.A.; et al. Functional adult myocardium in the absence of Na+-Ca2+ exchange: Cardiac-specific knockout of NCX1. Circ. Res. 2004, 95, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Falcon, D.; Galeano-Otero, I.; Calderon-Sanchez, E.; Del Toro, R.; Martin-Bornez, M.; Rosado, J.A.; Hmadcha, A.; Smani, T. TRP Channels: Current Perspectives in the Adverse Cardiac Remodeling. Front. Physiol. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Lev, S.; Zeevi, D.A.; Frumkin, A.; Offen-Glasner, V.; Bach, G.; Minke, B. Constitutive activity of the human TRPML2 channel induces cell degeneration. J. Biol. Chem. 2010, 285, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Babu, G.J.; Bhupathy, P.; Timofeyev, V.; Petrashevskaya, N.N.; Reiser, P.J.; Chiamvimonvat, N.; Periasamy, M. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc. Natl. Acad. Sci. USA 2007, 104, 17867–17872. [Google Scholar] [CrossRef] [PubMed]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.M.; Maillet, M.; Vanhoutte, D.; Schloemer, A.; Sargent, M.A.; Blair, N.S.; Lynch, K.A.; Okada, T.; Aronow, B.J.; Osinska, H.; et al. A thrombospondin-dependent pathway for a protective ER stress response. Cell 2012, 149, 1257–1268. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT-PLN | R14del-PLN | |
|---|---|---|
| Heart rate (bpm) | 498 ± 28 | 483 ± 15 |
| PR interval (ms) | 41.6 ± 1.7 | 50.3 ± 1.9 * |
| QRS interval (ms) | 9.6 ± 0.2 | 10.8 ± 0.3 * |
| Peak QRS voltage (mV) | 1.53 ± 0.05 | 0.42 ± 0.09 ** |
| JT interval (ms) | 12.2 ± 2.3 | 49.2 ± 2.2 ** |
| QT interval (ms) | 21.8 ± 2.2 | 60.0 ± 2.5 ** |
| QTc interval (ms) | 19.7 ± 1.6 | 53.7 ± 1.9 ** |
| R wave amplitude (mV) | 1.29 ± 0.09 | 0.86 ± 0.09 * |
| Human | |||
|---|---|---|---|
| Controls | Pre-Symptomatic R14del-PLN | Symptomatic R14del-PLN | |
| N | 272 | 15 | 53 |
| Age, years | 48 ± 0.8 | 46 ± 3.3 | 48 ± 1.7 |
| Heart rate, bpm | 71 ± 0.9 | 62 ± 3.5 * | 74 ± 2.5 |
| PR interval, ms | 153 ± 1.3 | 162 ± 7.6 | 180 ± 7.1 ** |
| QRS interval, ms | 95 ± 1.0 | 89 ± 2.2 * | 98 ± 3.5 * |
| Peak QRS voltage, mV | 1.1 ± 0.02 | 0.7 ± 0.08 ** | 0.6 ± 0.04 ** |
| QT interval, ms | 387 ± 1.9 | 419 ± 11.9 ** | 398 ± 6.5 * |
| QTc interval, ms | 418 ± 1.6 | 417 ± 5.5 | 433 ± 6.0 ** |
| R wave amplitude, mV | 1.0 ± 0.02 | 0.6 ± 0.07 ** | 0.4 ± 0.04 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haghighi, K.; Gardner, G.; Vafiadaki, E.; Kumar, M.; Green, L.C.; Ma, J.; Crocker, J.S.; Koch, S.; Arvanitis, D.A.; Bidwell, P.; et al. Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias. J. Pers. Med. 2021, 11, 502. https://doi.org/10.3390/jpm11060502
Haghighi K, Gardner G, Vafiadaki E, Kumar M, Green LC, Ma J, Crocker JS, Koch S, Arvanitis DA, Bidwell P, et al. Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias. Journal of Personalized Medicine. 2021; 11(6):502. https://doi.org/10.3390/jpm11060502
Chicago/Turabian StyleHaghighi, Kobra, George Gardner, Elizabeth Vafiadaki, Mohit Kumar, Lisa C. Green, Jianyong Ma, Jeffrey S. Crocker, Sheryl Koch, Demetrios A. Arvanitis, Phillip Bidwell, and et al. 2021. "Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias" Journal of Personalized Medicine 11, no. 6: 502. https://doi.org/10.3390/jpm11060502
APA StyleHaghighi, K., Gardner, G., Vafiadaki, E., Kumar, M., Green, L. C., Ma, J., Crocker, J. S., Koch, S., Arvanitis, D. A., Bidwell, P., Rubinstein, J., van de Leur, R., Doevendans, P. A., Akar, F. G., Tranter, M., Wang, H.-S., Sadayappan, S., DeMazumder, D., Sanoudou, D., ... Kranias, E. G. (2021). Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias. Journal of Personalized Medicine, 11(6), 502. https://doi.org/10.3390/jpm11060502