
Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin

Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
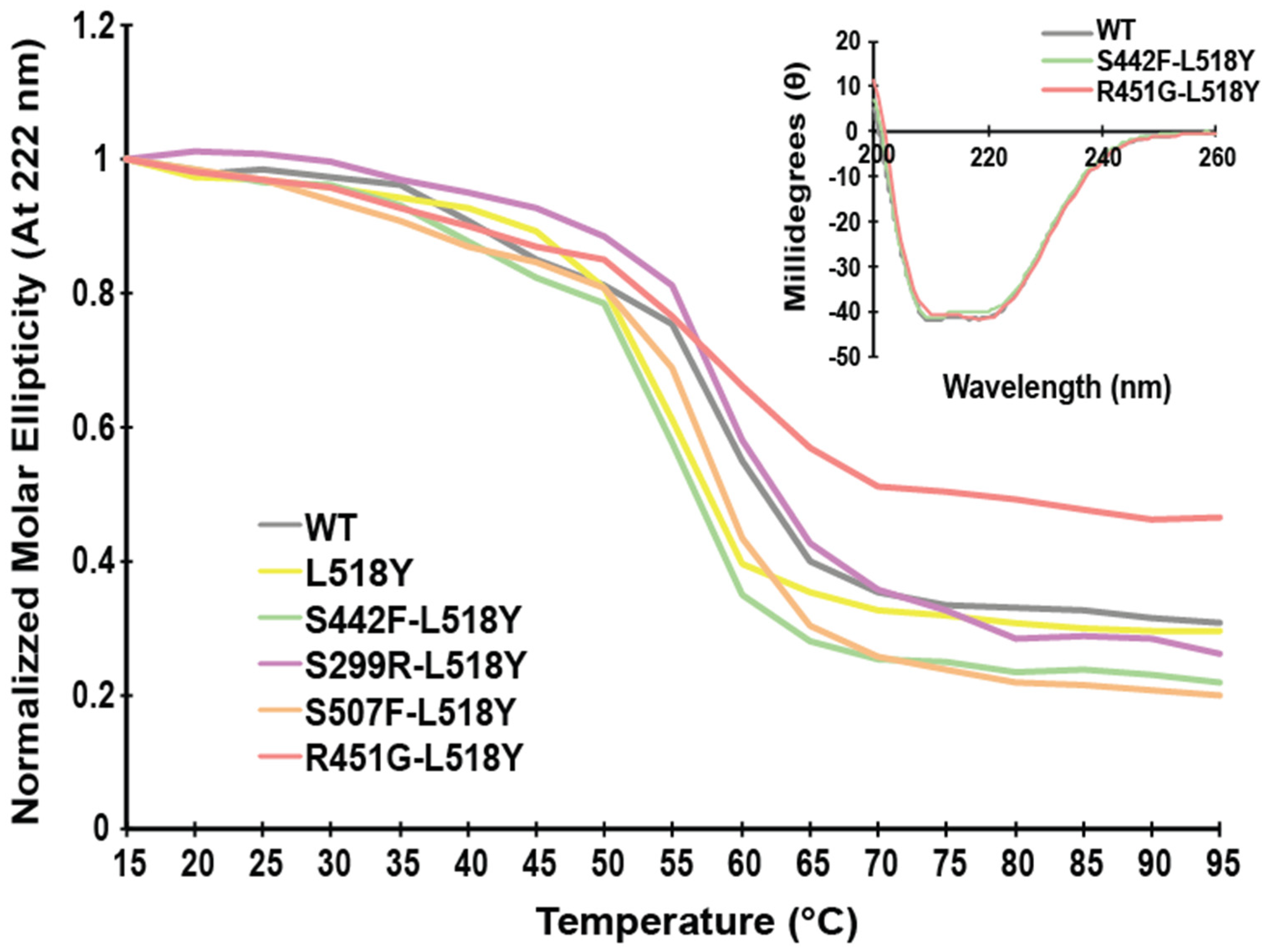
3.1. L518Y DSP Point Mutation Proximal to the Calpain Target Site Results in No Overt Structural Changes
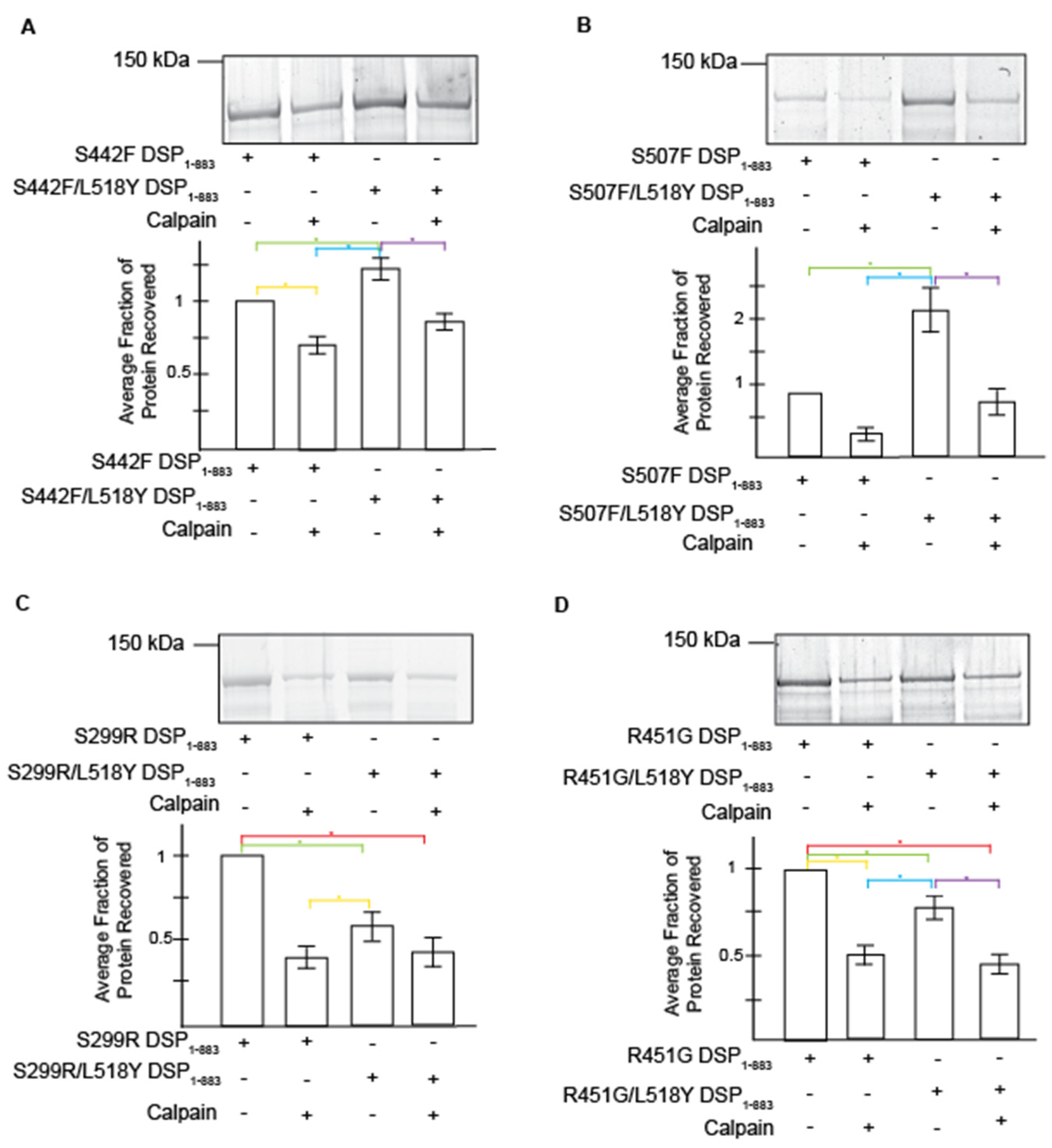
3.2. L518Y DSP Point Mutation Results in Moderate Protection from Degradation by Calpain
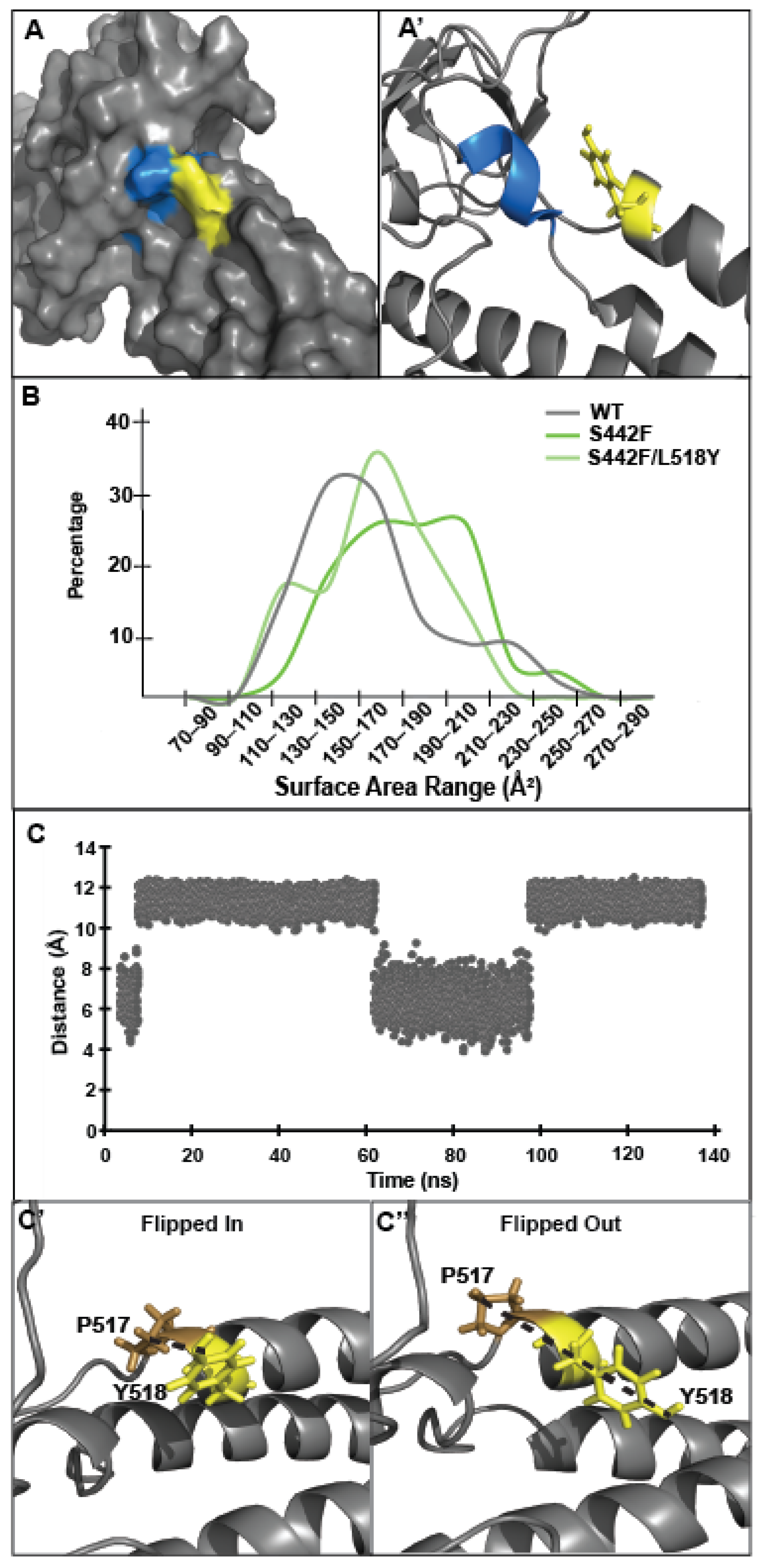
3.3. Tyrosine at Position 518 Is Insufficient to Robustly and Ubiquitously Protect DSP Clinical Variants from Calpain Targeting Due to Its Size, Position, and Mobility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm Electrophysiol. Rev. 2016, 5, 90–101. [Google Scholar] [CrossRef]
- Murray, B. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C): A Review of Molecular and Clinical Literature. J. Genet. Couns. 2012, 21, 494–504. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular Mechanisms of Arrhythmogenic Cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Morgan, R.D.; Chambers, J.C.; McKenna, W.J. Arrhythmogenic Cardiomyopathy: Etiology, Diagnosis, and Treatment. Annu. Rev. Med. 2010, 61, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.; Marchlinski, F.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Eur. Heart J. 2015, ehv162. [Google Scholar] [CrossRef] [PubMed]
- Garrod, D.; Chidgey, M. Desmosome Structure, Composition and Function. Biochim. Biophys. Acta (BBA) Biomembr. 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic Cardiomyopathy: Pathology, Genetics, and Concepts in Pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Basso, C. Ultrastructural Evidence of Intercalated Disc Remodelling in Arrhythmogenic Right Ventricular Cardiomyopathy: An Electron Microscopy Investigation on Endomyocardial Biopsies. Eur. Heart J. 2006, 27, 1847–1854. [Google Scholar] [CrossRef]
- Ronny, A.; Shulamit, M.; Shimon, R.; Vardiella, M.; Tova, C.-S. A Recessive Mutation in Desmoplakin Causes Arrhythmogenic Right Ventricular Dysplasia, Skin Disorder, and Woolly Hair. J. Am. Coll. Cardiol. 2003, 42, 319–327. [Google Scholar] [CrossRef]
- Uzumcu, A.; Norgett, E.E.; Dindar, A.; Uyguner, O.; Nisli, K.; Kayserili, H.; Sahin, S.E.; Dupont, E.; Severs, N.J.; Leigh, I.M.; et al. Loss of Desmoplakin Isoform I Causes Early Onset Cardiomyopathy and Heart Failure in a Naxos-like Syndrome. J. Med. Genet. 2006, 43, e5. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in Human Desmoplakin Domain Binding to Plakoglobin Causes a Dominant Form of Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Stevens, T.L.; Wallace, M.J.; El Refaey, M.; Roberts, J.D.; Koenig, S.N.; Mohler, P.J. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. J. Cardiovasc. Dev. Dis. 2020, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Huen, A.C.; Park, J.K.; Godsel, L.M.; Chen, X.; Bannon, L.J.; Amargo, E.V.; Hudson, T.Y.; Mongiu, A.K.; Leigh, I.M.; Kelsell, D.P.; et al. Intermediate Filament–Membrane Attachments Function Synergistically with Actin-Dependent Contacts to Regulate Intercellular Adhesive Strength. J. Cell Biol. 2002, 159, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Manring, H.R.; Dorn, L.E.; Ex-Willey, A.; Accornero, F.; Ackermann, M.A. At the Heart of Inter- and Intracellular Signaling: The Intercalated Disc. Biophys. Rev. 2018, 10, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Bauce, B.; Rampazzo, A.; Basso, C.; Mazzotti, E.; Rigato, I.; Steriotis, A.; Beffagna, G.; Lorenzon, A.; De Bortoli, M.; Pilichou, K.; et al. Clinical Phenotype and Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy in Pediatric Patients Carrying Desmosomal Gene Mutations. Heart Rhythm 2011, 8, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient Mutations Linked to Arrhythmogenic Cardiomyopathy Enhance Calpain-Mediated Desmoplakin Degradation. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical Profile of Four Families with Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Dominant Desmoplakin Mutations. Eur. Hear. J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- López-Ayala, J.M.; Gómez-Milanés, I.; Sánchez Muñoz, J.J.; Ruiz-Espejo, F.; Ortíz, M.; González-Carrillo, J.; López-Cuenca, D.; Oliva-Sandoval, M.J.; Monserrat, L.; Valdés, M.; et al. Desmoplakin Truncations and Arrhythmogenic Left Ventricular Cardiomyopathy: Characterizing a Phenotype. EP Eur. 2014, 16, 1838–1846. [Google Scholar] [CrossRef]
- Sorimachi, H.; Ishiura, S.; Suzuki, K. Structure and Physiological Function of Calpains. Biochem. J. 1997, 328, 721–732. [Google Scholar] [CrossRef]
- Patterson, C.; Portbury, A.L.; Schisler, J.C.; Willis, M.S. Tear Me Down: Role of Calpain in the Development of Cardiac Ventricular Hypertrophy. Circ. Res. 2011, 109, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Lendeckel, U.; Bode-Böger, S.M.; Goette, A. Physiologic and Pathophysiologic Role of Calpain: Implications for the Occurrence of Atrial Fibrillation. Cardiovasc. Ther. 2012, 30, e115–e127. [Google Scholar] [CrossRef]
- Potz, B.A.; Abid, M.R.; Sellke, F.W. Role of Calpain in Pathogenesis of Human Disease Processes. J. Nat. Appl. Sci. 2016, 2, 2. [Google Scholar]
- Kudo-Sakamoto, Y.; Akazawa, H.; Ito, K.; Takano, J.; Yano, M.; Yabumoto, C.; Naito, A.T.; Oka, T.; Lee, J.-K.; Sakata, Y.; et al. Calpain-Dependent Cleavage of N-Cadherin Is Involved in the Progression of Post-Myocardial Infarction Remodeling. J. Biol. Chem. 2014, 289, 19408–19419. [Google Scholar] [CrossRef]
- Tompa, P.; Buzder-Lantos, P.; Tantos, A.; Farkas, A.; Szilágyi, A.; Bánóczi, Z.; Hudecz, F.; Friedrich, P. On the Sequential Determinants of Calpain Cleavage. J. Biol. Chem. 2004, 279, 20775–20785. [Google Scholar] [CrossRef]
- Cuerrier, D.; Moldoveanu, T.; Davies, P.L. Determination of Peptide Substrate Specificity for μ-Calpain by a Peptide Library-Based Approach. J. Biol. Chem. 2005, 280, 40632–40641. [Google Scholar] [CrossRef]
- Sorimachi, H.; Mamitsuka, H.; Ono, Y. Understanding the Substrate Specificity of Conventional Calpains. Biol. Chem. 2012, 393, 853–871. [Google Scholar] [CrossRef]
- Ono, Y.; Saido, T.C.; Sorimachi, H. Calpain Research for Drug Discovery: Challenges and Potential. Nat. Rev. Drug Discov. 2016, 15, 854–876. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Ono, Y. Regulation and Physiological Roles of the Calpain System in Muscular Disorders. Cardiovasc. Res. 2012, 96, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Vainzof, M. The Effect of Calpain 3 Deficiency on the Pattern of Muscle Degeneration in the Earliest Stages of LGMD2A. J. Clin. Pathol. 2003, 56, 624–626. [Google Scholar] [CrossRef]
- Ono, Y.; Shimada, H.; Sorimachi, H.; Richard, I.; Saido, T.C.; Beckmann, J.S.; Ishiura, S.; Suzuki, K. Functional Defects of a Muscle-Specific Calpain, P94, Caused by Mutations Associated with Limb-Girdle Muscular Dystrophy Type 2A. J. Biol. Chem. 1998, 273, 17073–17078. [Google Scholar] [CrossRef]
- Barefield, D.Y.; McNamara, J.W.; Lynch, T.L.; Kuster, D.W.D.; Govindan, S.; Haar, L.; Wang, Y.; Taylor, E.N.; Lorenz, J.N.; Nieman, M.L.; et al. Ablation of the Calpain-Targeted Site in Cardiac Myosin Binding Protein-C Is Cardioprotective during Ischemia-Reperfusion Injury. J. Mol. Cell. Cardiol. 2019, 129, 236–246. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Schrödinger, LLC.: New York, NY, USA, 2021.
- Mitternacht, S. FreeSASA: An Open Source C Library for Solvent Accessible Surface Area Calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Choi, H.-J.; Weis, W.I. Crystal Structure of a Rigid Four-Spectrin-Repeat Fragment of the Human Desmoplakin Plakin Domain. J. Mol. Biol. 2011, 409, 800–812. [Google Scholar] [CrossRef]
- Choi, H.-J.; Park-Snyder, S.; Pascoe, L.T.; Green, K.J.; Weis, W.I. Structures of Two Intermediate Filament-Binding Fragments of Desmoplakin Reveal a Unique Repeat Motif Structure. Nat. Struct. Biol. 2002, 9, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins: Struct. Funct. Bioinform. 2000, 40, 389–408. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2015, 428, 720–725. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DSP Protein | Avg. Recovery without Calpain | L158Y Performance without Stress | Avg. Protein Recovery with Calpain | L518Y Performance under Stress | Avg. Exposed Surface Area of Target Site (Å2) |
|---|---|---|---|---|---|
| Proximal Variants | |||||
| S299R | 1 | 0.411 ± 0.063 * | 148.6 ± 2.9 | ||
| S299R/L518Y | 0.596 ± 0.082 * | ↓ 40.1% | 0.422 ± 0.085 * | ↑ 2.7% | 150.6 ± 3.2 |
| R451G | 1 | 0.485 ± 0.054 * | 176.4 ± 4.0 | ||
| R451G/L518Y | 0.734 ± 0.064 * | ↓ 26.6% | 0.482 ± 0.062 * | ↓ 0.6% | 135.7 ± 4.7 |
| Distal Variants | |||||
| S442F | 1 | 0.710 ± 0.060 * | 175.8 ± 3.0 | ||
| S442F/L518Y | 1.23 ± 0.07 * | ↑ 23% | 0.871 ± 0.056 | ↑ 22.7% | 160.7 ± 2.6 |
| S507F | 1 | 0.369 ± 0.064 | 173.7 ± 3.7 | ||
| S507F/L518Y | 2.31 ± 0.35 * | ↑ 131% | 0.849 ± 0.171 | ↑ 130% | 163.6 ± 3.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoover, C.A.; Ott, K.L.; Manring, H.R.; Dew, T.; Borzok, M.A.; Wright, N.T. Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin. J. Pers. Med. 2021, 11, 401. https://doi.org/10.3390/jpm11050401
Hoover CA, Ott KL, Manring HR, Dew T, Borzok MA, Wright NT. Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin. Journal of Personalized Medicine. 2021; 11(5):401. https://doi.org/10.3390/jpm11050401
Chicago/Turabian StyleHoover, Catherine A., Kendahl L. Ott, Heather R. Manring, Trevor Dew, Maegen A. Borzok, and Nathan T. Wright. 2021. "Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin" Journal of Personalized Medicine 11, no. 5: 401. https://doi.org/10.3390/jpm11050401
APA StyleHoover, C. A., Ott, K. L., Manring, H. R., Dew, T., Borzok, M. A., & Wright, N. T. (2021). Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin. Journal of Personalized Medicine, 11(5), 401. https://doi.org/10.3390/jpm11050401