
Animal Evidence for Synergistic Induction of Hepatic Injury by Dietary Fat and Alcohol Consumption and Its Potential Mechanisms


 ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Serum Biochemistry Analysis
2.3. Lipid Contents Determinations
2.4. Histopathological Analysis
2.5. Immunohistofluorescence (IHF) Analysis
2.6. Caspase-3/7 and Poly (ADP-Ribose) Polymerase (PARP) Activity Analysis
2.7. Western Blot Analysis
2.8. mRNA Expression Analysis Using qPCR
2.9. Statistical Analysis
3. Results
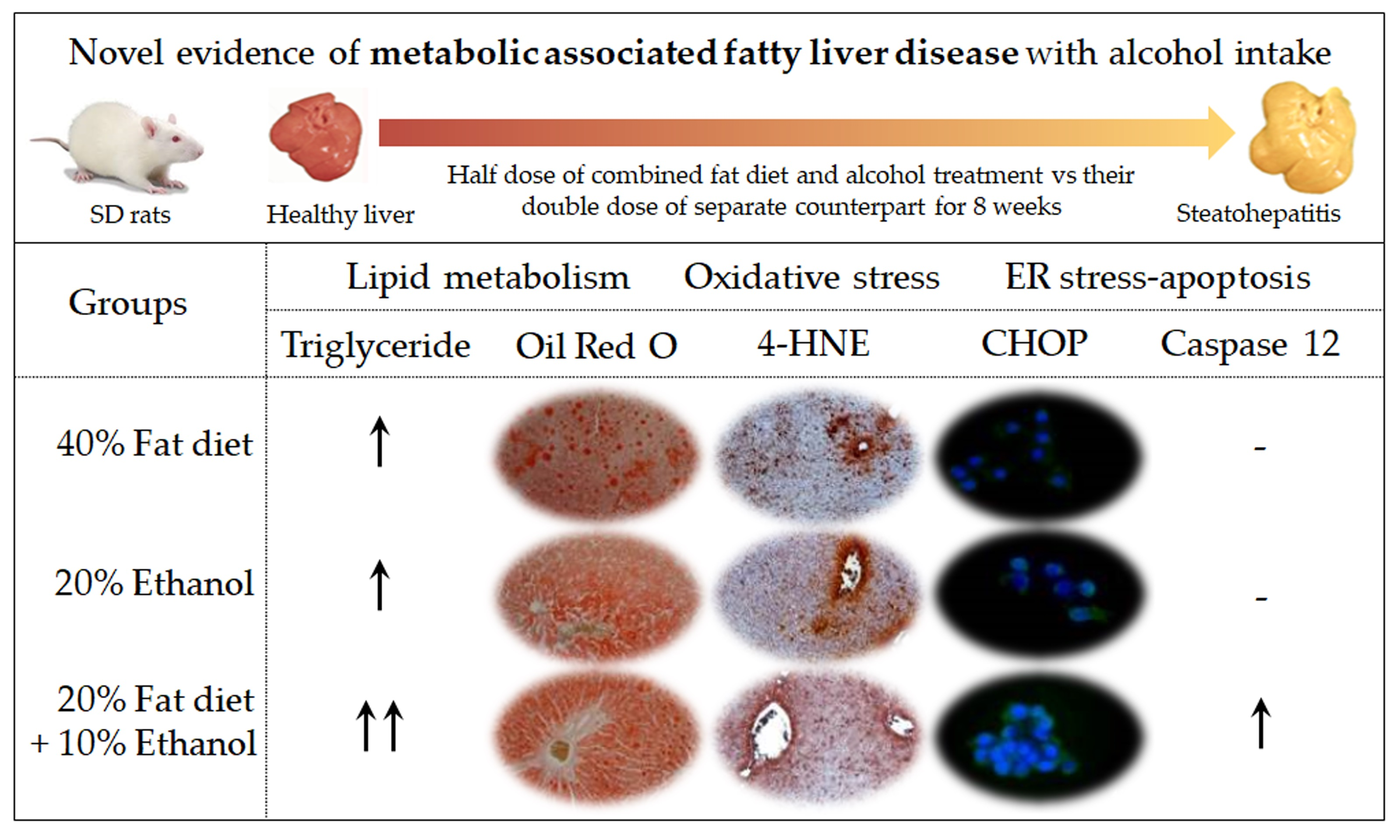
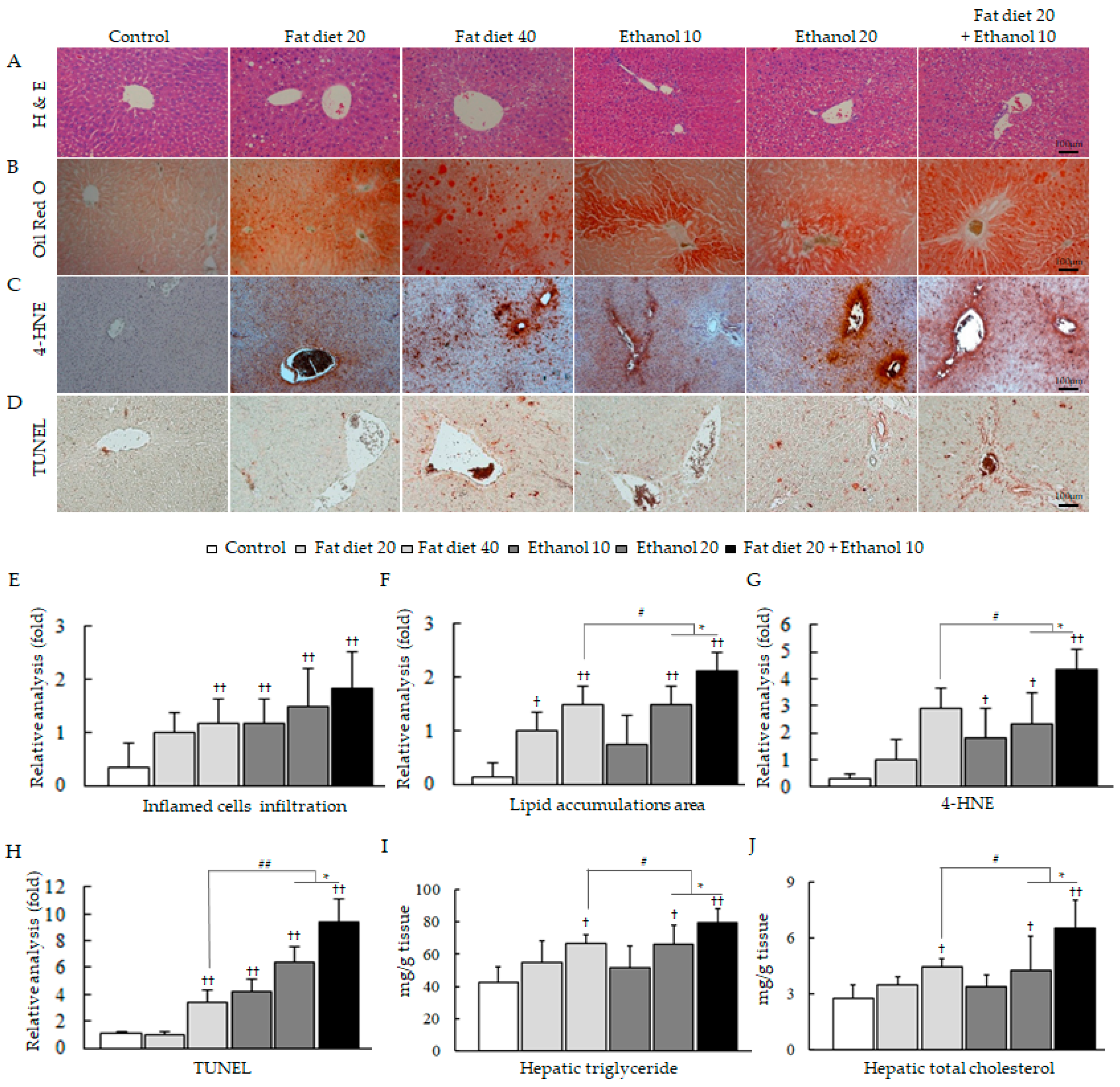
3.1. Synergistic Induction of Hepatosteatosis and Liver Injury
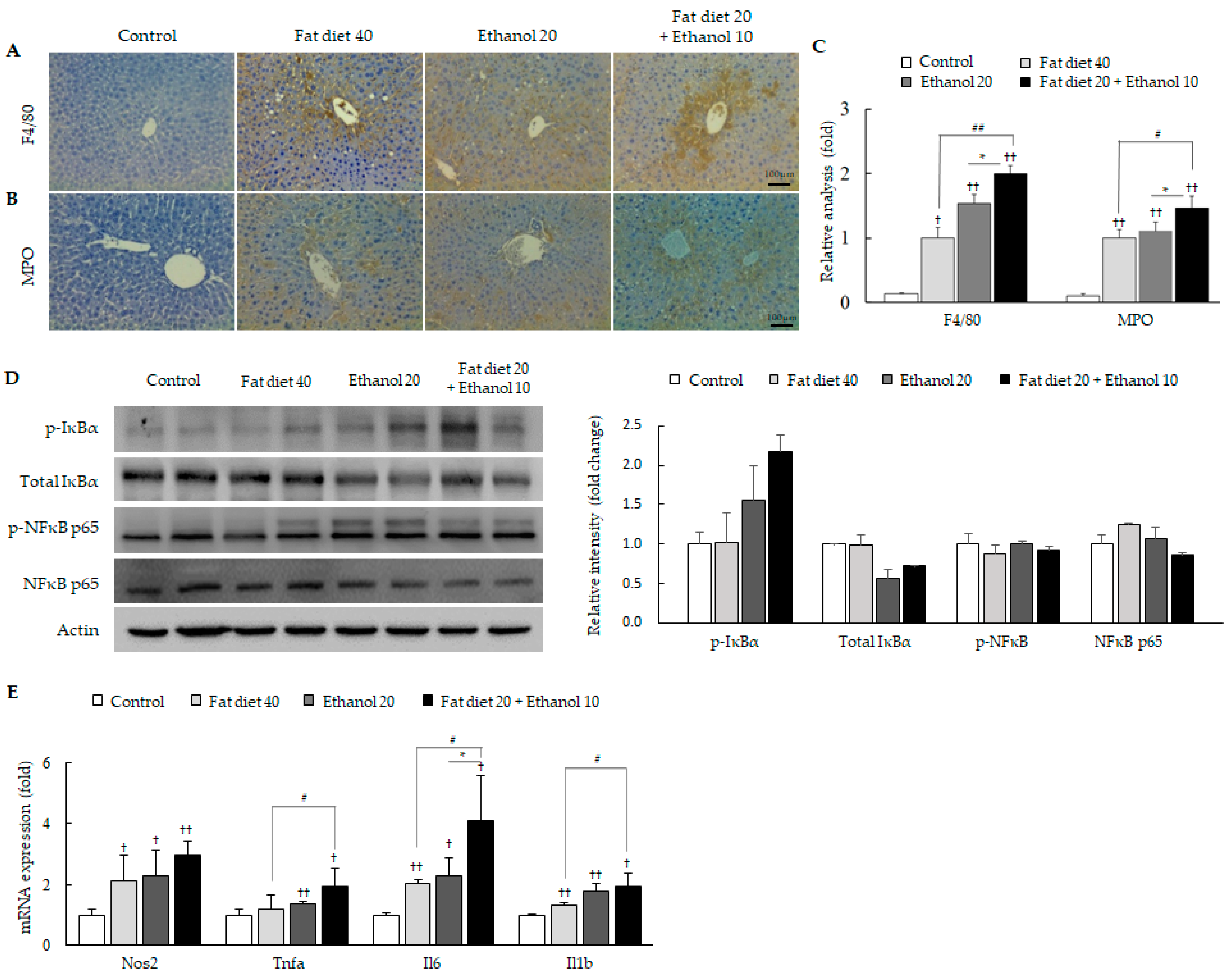
3.2. Exacerbation of Oxidative Stress and Hepatic Inflammation
3.3. Synergistic Induction of Endoplasmic Reticulum (ER) Stress
3.4. Induction of Mitochondria-Independent Apoptosis
3.5. Synergistic Alterations of Lipid Homeostasis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Footnotes
References
- Chief Medical Officer: First annual report on the NHS. Community Pract. 2013, 86, 6.
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol. Res. 2017, 38, 147–161. [Google Scholar]
- Luo, Z.; Li, L.; Ruan, B. Impact of the implementation of a vaccination strategy on hepatitis B virus infections in China over a 20-year period. Int. J. Infect. Dis. 2012, 16, e82–e88. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.H.; Jensen, D.M.; Manns, M.P.; Jacobson, I.; Kumada, H.; Toyota, J.; Heo, J.; Yoffe, B.; Sievert, W.; Bessone, F.; et al. Daclatasvir plus asunaprevir for HCV genotype 1b infection in patients with or without compensated cirrhosis: A pooled analysis. Liver Int. 2016, 36, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Blachier, M.; Leleu, H.; Peck-Radosavljevic, M.; Valla, D.C.; Roudot-Thoraval, F. The burden of liver disease in Europe: A review of available epidemiological data. J. Hepatol. 2013, 58, 593–608. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Dunn, W.; Angulo, P.; Sanderson, S.; Jamil, L.H.; Stadheim, L.; Rosen, C.; Malinchoc, M.; Kamath, P.S.; Shah, V.H. Utility of a new model to diagnose an alcohol basis for steatohepatitis. Gastroenterology 2006, 131, 1057–1063. [Google Scholar] [CrossRef]
- Cortez-Pinto, H.; Baptista, A.; Camilo, M.E.; De Moura, M.C. Nonalcoholic steatohepatitis—A long-term follow-up study: Comparison with alcoholic hepatitis in ambulatory and hospitalized patients. Dig. Dis. Sci. 2003, 48, 1909–1913. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Tremblay, A.; Wouters, E.; Wenker, M.; St-Pierre, S.; Bouchard, C.; Despres, J.P. Alcohol and a high-fat diet: A combination favoring overfeeding. Am. J. Clin. Nutr. 1995, 62, 639–644. [Google Scholar] [CrossRef]
- Wai-Sun Wong, V.; Lai-Hung Wong, G.; Woo, J.; Abrigo, J.M.; Ka-Man Chan, C.; She-Ting Shu, S.; Ka-Yu Leung, J.; Mei-Ling Chim, A.; Pik-Shan Kong, A.; Chung-Yan Lui, G.; et al. Impact of the new definition of metabolic associated fatty liver disease on the epidemiology of the disease. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Singh, S.P.; Anirvan, P.; Reddy, K.R.; Conjeevaram, H.S.; Marchesini, G.; Rinella, M.E.; Madan, K.; Petroni, M.L.; Al-Mahtab, M.; Caldwell, S.H.; et al. Non-alcoholic Fatty Liver Disease: Not Time for an Obituary Just Yet! J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.el. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, C.E.; Everhart, J.E. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin. Gastroenterol. Hepatol. 2005, 3, 1260–1268. [Google Scholar] [CrossRef]
- Alatalo, P.I.; Koivisto, H.M.; Hietala, J.P.; Puukka, K.S.; Bloigu, R.; Niemela, O.J. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am. J. Clin. Nutr. 2008, 88, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Sakurai, S.; Uemura, M.; Takekawa, T.; Morimoto, H.; Tamagawa, Y.; Fukui, H. Difference and similarity between non-alcoholic steatohepatitis and alcoholic liver disease. Alcohol. Clin. Exp. Res. 2005, 29, 259S–263S. [Google Scholar] [CrossRef]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef]
- Duly, A.M.; Alani, B.; Huang, E.Y.; Yee, C.; Haber, P.S.; McLennan, S.V.; Seth, D. Effect of multiple binge alcohol on diet-induced liver injury in a mouse model of obesity. Nutr. Diabetes 2015, 5, e154. [Google Scholar] [CrossRef]
- Gabele, E.; Dostert, K.; Dorn, C.; Patsenker, E.; Stickel, F.; Hellerbrand, C. A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol. Clin. Exp. Res. 2011, 35, 1361–1367. [Google Scholar] [CrossRef]
- Lu, S.C.; Huang, Z.Z.; Yang, J.M.; Tsukamoto, H. Effect of ethanol and high-fat feeding on hepatic gamma-glutamylcysteine synthetase subunit expression in the rat. Hepatology 1999, 30, 209–214. [Google Scholar] [CrossRef]
- Xu, J.; Lai, K.K.Y.; Verlinsky, A.; Lugea, A.; French, S.W.; Cooper, M.P.; Ji, C.; Tsukamoto, H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 2011, 55, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Xu, M.J.; Zhou, Z.; Cai, Y.; Li, M.; Wang, W.; Feng, D.; Bertola, A.; Wang, H.; Kunos, G.; et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology 2015, 62, 1070–1085. [Google Scholar] [CrossRef]
- Hozumi, Y.; Kawano, M.; Jordan, V.C. In vitro study of the effect of raloxifene on lipid metabolism compared with tamoxifen. Eur. J. Endocrinol. 2000, 143, 427–430. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, J.S.; Zheng, Z.; Mendez, R.; Ha, S.W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M. Nonalcoholic steatohepatitis: Definition and pathology. Semin. Liver Dis. 2001, 21, 3–16. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Scaglioni, F.; Ciccia, S.; Marino, M.; Bedogni, G.; Bellentani, S. ASH and NASH. Dig. Dis. 2011, 29, 202–210. [Google Scholar] [CrossRef]
- Toshikuni, N.; Tsutsumi, M.; Arisawa, T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8393–8406. [Google Scholar] [CrossRef] [PubMed]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sanchez-Campos, S.; Garcia-Mediavilla, M.V.; Fernandez-Bermejo, M.; Lozano-Rodriguez, T.; Vargas-Castrillon, J.; Buque, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef]
- Woods, S.C.; D’Alessio, D.A.; Tso, P.; Rushing, P.A.; Clegg, D.J.; Benoit, S.C.; Gotoh, K.; Liu, M.; Seeley, R.J. Consumption of a high-fat diet alters the homeostatic regulation of energy balance. Physiol. Behav. 2004, 83, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Giannini, E.G.; Testa, R.; Savarino, V. Liver enzyme alteration: A guide for clinicians. CMAJ 2005, 172, 367–379. [Google Scholar] [CrossRef]
- Lasker, S.; Rahman, M.M.; Parvez, F.; Zamila, M.; Miah, P.; Nahar, K.; Kabir, F.; Sharmin, S.B.; Subhan, N.; Ahsan, G.U.; et al. High-fat diet-induced metabolic syndrome and oxidative stress in obese rats are ameliorated by yogurt supplementation. Sci. Rep. 2019, 9, 20026. [Google Scholar] [CrossRef]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef]
- Wang, Z.; Dou, X.; Li, S.; Zhang, X.; Sun, X.; Zhou, Z.; Song, Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology 2014, 59, 1381–1392. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.L.; Xiao, M.; Yang, R.; Xie, K.Q. Critical Roles of Kupffer Cells in the Pathogenesis of Alcoholic Liver Disease: From Basic Science to Clinical Trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef]
- Ji, C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S1), S16–S24. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Gutterman, D.D. Mitochondria and reactive oxygen species: An evolution in function. Circ. Res. 2005, 97, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, P.S.; Cortez-Pinto, H.; Sola, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Asp. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Tsai, J.; Ford, E.S.; Zhao, G.; Li, C.; Greenlund, K.J.; Croft, J.B. Co-occurrence of obesity and patterns of alcohol use associated with elevated serum hepatic enzymes in US adults. J. Behav. Med. 2012, 35, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.; St-Pierre, S. The hyperphagic effect of a high-fat diet and alcohol intake persists after control for energy density. Am. J. Clin. Nutr. 1996, 63, 479–482. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contents | Control | Fat Diet 20% | Fat Diet 40% | Ethanol 10% | Ethanol 20% | Fat Diet 20% + Ethanol 10% |
|---|---|---|---|---|---|---|
| Final body weight (g) | 462.2 ± 14.8 | 484.3 ± 9.9 | 513.2 ± 21.7 †† | 439.5 ± 21.0 | 429.0 ± 11.9 †† | 473.3 ± 26.4 #,** |
| Liver weights (g) | 11.9 ± 0.5 | 12.8 ± 0.5 † | 13.9 ± 0.8 †† | 12.9 ± 0.7 † | 13.8 ± 0.4 †† | 14.6 ± 0.8 ††,#,** |
| Relative liver weight (%) | 2.58 ± 0.01 | 2.66 ± 0.01 | 2.69 ± 0.01 | 2.94 ± 0.01 †† | 3.21 ± 0.01 †† | 3.10 ± 0.01 ††,## |
| Visceral fat weights (g) | 18.8 ± 4.3 | 27.7 ± 5.0 †† | 38.2 ± 6.3 †† | 18.6 ± 3.3 | 20.2 ± 1.6 | 26.1 ± 3.9 †,##,* |
| AST (IU/L) | 166.5 ± 15.6 | 162.8 ± 14.3 | 169.5 ± 15.8 | 166.3 ± 24.6 | 189.0 ± 29.1 | 199.1 ± 47.5 † |
| ALT (IU/L) | 22.8 ± 2.6 | 26.8 ± 6.0 | 34.0 ± 11.4 | 25.8 ± 2.8 | 33.0 ± 4.0 | 47.2 ± 26.9 †† |
| ALP (IU/L) | 384.8 ±36.4 | 335.3 ± 41.3 | 357.8 ± 61.7 | 363.0 ± 71.2 | 345.3 ± 23.1 | 522.0 ± 71.9 ††,##,** |
| TC (mg/dL) | 58.7 ± 8.0 | 72.7 ± 7.9 † | 73.0 ± 8.4 † | 53.0 ± 8.2 | 64.2 ± 29.8 | 97.7 ± 16.1 ††,##,** |
| LDL (mg/mL) | 10.0 ± 0.9 | 9.5 ± 2.3 | 9.5 ± 1.2 | 11.5 ± 2.9 | 14.0 ± 2.6 † | 19.8 ± 3.1 ††,##,** |
| TG (mg/dL) | 47.3 ± 10.4 | 70.0 ± 19.4 † | 74.7 ± 16.9 †† | 66.5 ± 18.2 | 84.7 ± 26.7 †† | 96.7 ± 13.5 †,# |
| Glucose (mg/dL) | 95.6 ± 11.3 | 103.7 ± 9.1 | 104.0 ± 8.0 | 101.7 ± 11.1 | 120.3 ± 10.8 | 184.5 ± 18.4 ††,##,** |
| FFA (mEq/L) | 0.79 ± 0.18 | 0.82 ± 0.11 | 0.86 ± 0.14 | 1.13 ± 0.12 †† | 1.45 ± 0.14 †† | 1.63 ± 0.30 ††,##,** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-G.; Wang, J.-H.; Kim, H.-S.; Lee, J.-S.; Im, H.-J.; Lee, S.-B.; Lee, D.-s.; Hur, G.-M.; Son, C.-G. Animal Evidence for Synergistic Induction of Hepatic Injury by Dietary Fat and Alcohol Consumption and Its Potential Mechanisms. J. Pers. Med. 2021, 11, 287. https://doi.org/10.3390/jpm11040287
Kim H-G, Wang J-H, Kim H-S, Lee J-S, Im H-J, Lee S-B, Lee D-s, Hur G-M, Son C-G. Animal Evidence for Synergistic Induction of Hepatic Injury by Dietary Fat and Alcohol Consumption and Its Potential Mechanisms. Journal of Personalized Medicine. 2021; 11(4):287. https://doi.org/10.3390/jpm11040287
Chicago/Turabian StyleKim, Hyeong-Geug, Jing-Hua Wang, Hyo-Seon Kim, Jin-Seok Lee, Hwi-Jin Im, Sung-Bae Lee, Dong-soo Lee, Gang-Min Hur, and Chang-Gue Son. 2021. "Animal Evidence for Synergistic Induction of Hepatic Injury by Dietary Fat and Alcohol Consumption and Its Potential Mechanisms" Journal of Personalized Medicine 11, no. 4: 287. https://doi.org/10.3390/jpm11040287
APA StyleKim, H.-G., Wang, J.-H., Kim, H.-S., Lee, J.-S., Im, H.-J., Lee, S.-B., Lee, D.-s., Hur, G.-M., & Son, C.-G. (2021). Animal Evidence for Synergistic Induction of Hepatic Injury by Dietary Fat and Alcohol Consumption and Its Potential Mechanisms. Journal of Personalized Medicine, 11(4), 287. https://doi.org/10.3390/jpm11040287