
Targeted Next-Generation Sequencing of Liquid Biopsy Samples from Patients with NSCLC


Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Specimens
2.2. Specimen Collection, Nucleic Acid Extraction and Reverse Transcription
2.3. Library Preparation, Quantification and Pooling
2.4. Templating and Sequencing
2.5. Quality Control (QC) Metrics
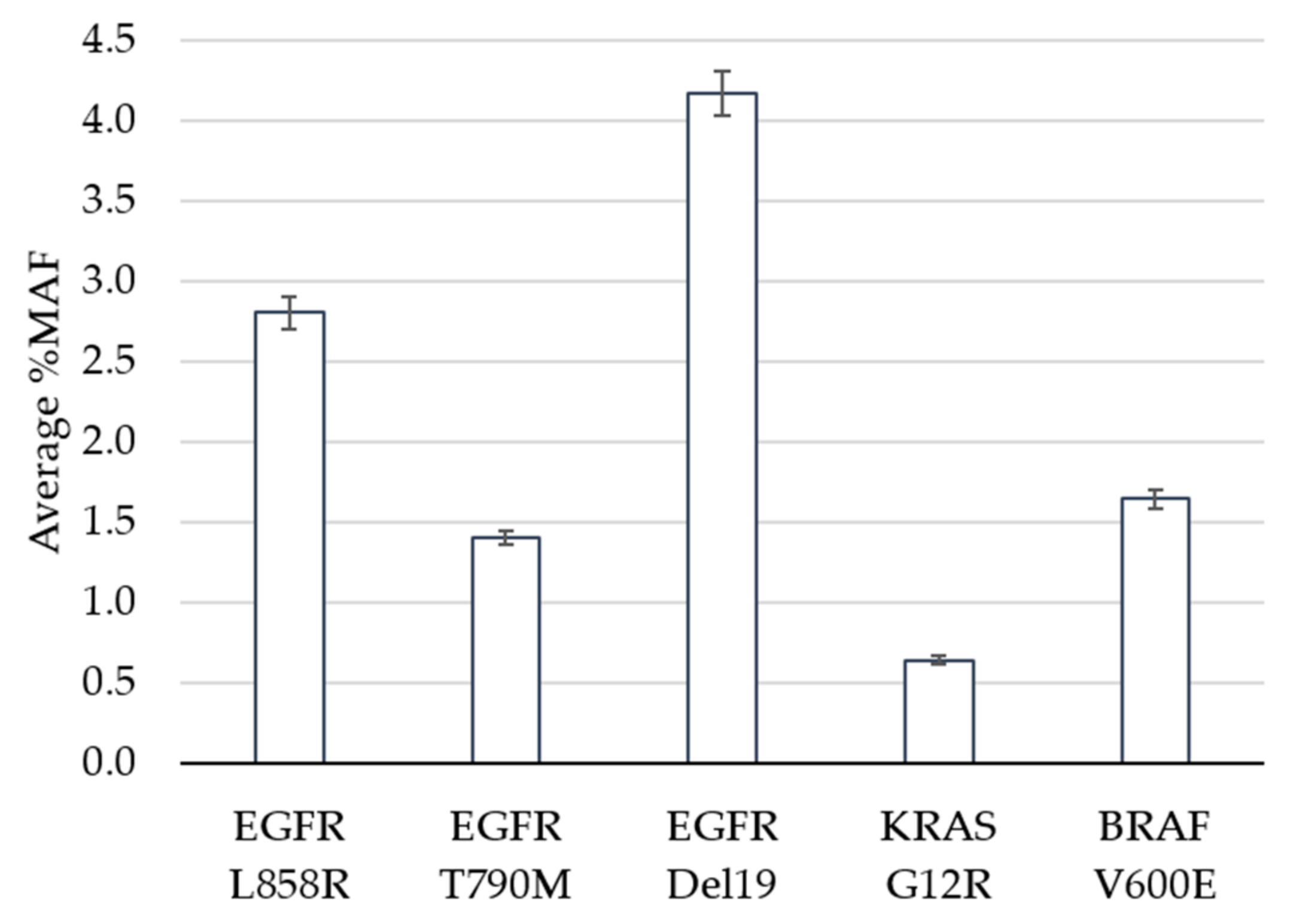
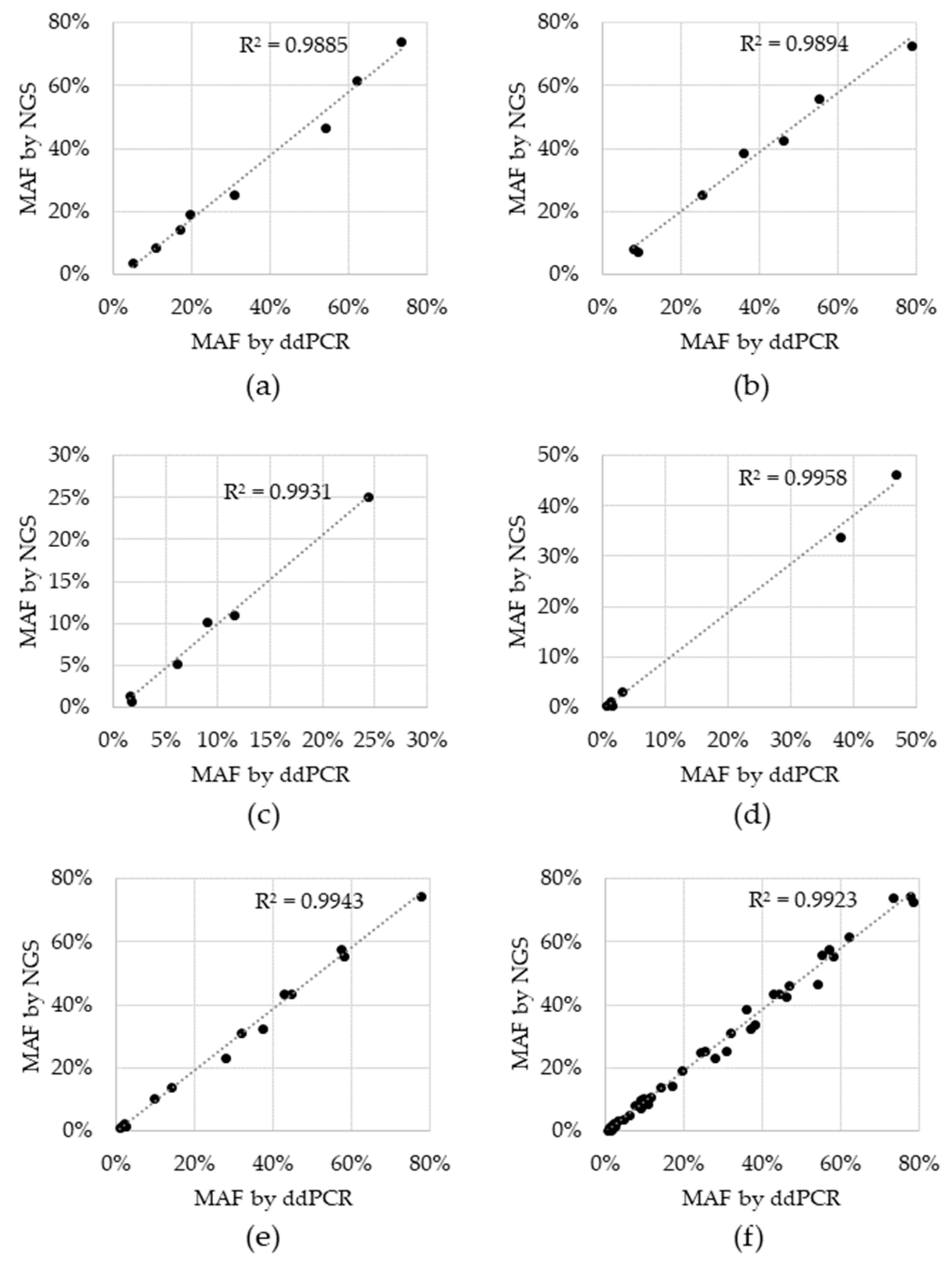
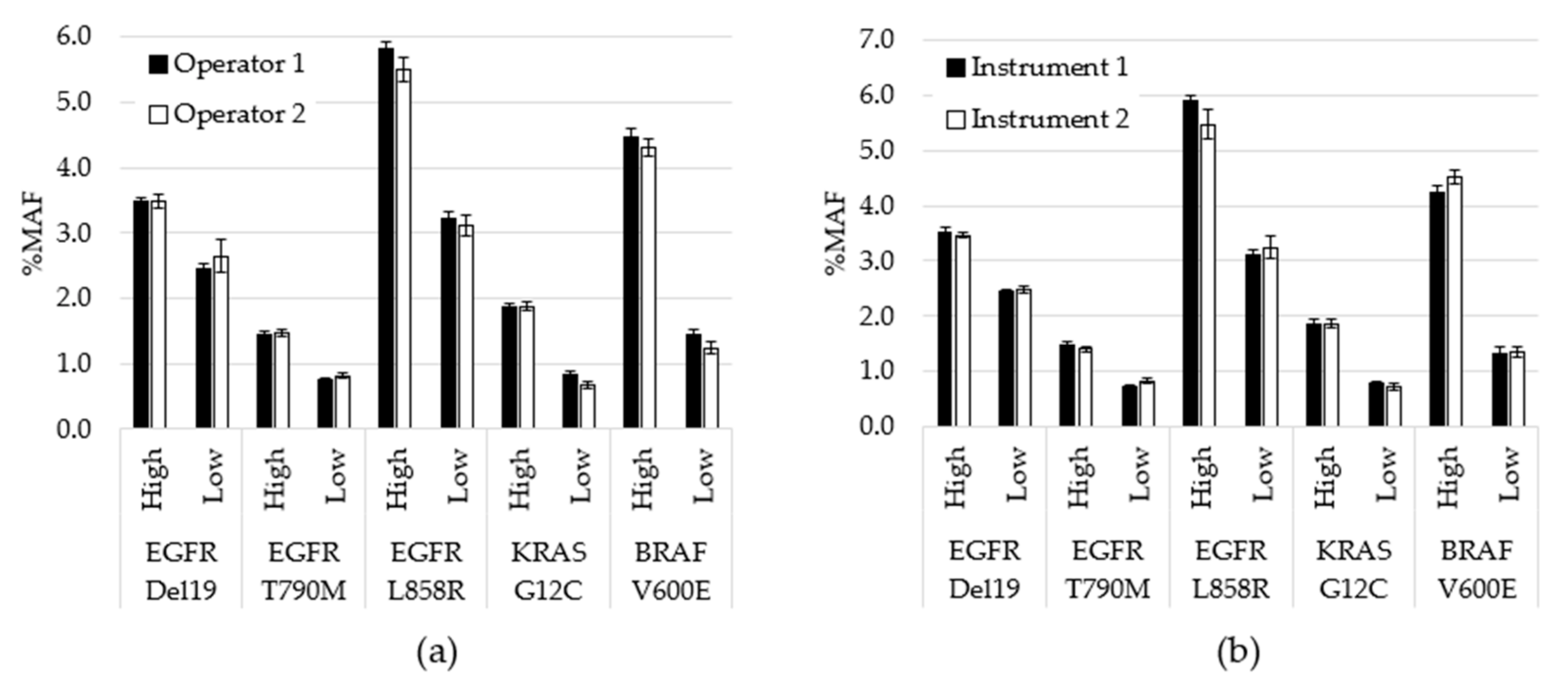
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swanton, C.; Govindan, R. Clinical Implications of Genomic Discoveries in Lung Cancer. N. Engl. J. Med. 2016, 374, 1864–1873. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Oellerich, M.; Schütz, E.; Beck, J.; Kanzow, P.; Plowman, P.N.; Weiss, G.J.; Walson, P.D. Using circulating cell-free DNA to monitor personalized cancer therapy. Crit. Rev. Clin. Lab. Sci. 2017, 54, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Novello, S.; Barlesi, F.; Califano, R.; Cufer, T.; Ekman, S.; Levra, M.G.; Kerr, K.; Popat, S.; Reck, M.; Senan, S.; et al. Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v1–v27. [Google Scholar] [CrossRef]
- Saarenheimo, J.; Eigeliene, N.; Andersen, H.; Tiirola, M.; Jekunen, A. The Value of Liquid Biopsies for Guiding Therapy Decisions in Non-small Cell Lung Cancer. Front. Oncol. 2019, 9, 129. [Google Scholar] [CrossRef]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O'Connell, A.; Feeney, N.; Mach, S.L.; Janne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.B.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef]
- Sacher, A.G.; Komatsubara, K.M.; Oxnard, G.R. Application of Plasma Genotyping Technologies in Non–Small Cell Lung Cancer: A Practical Review. J. Thorac. Oncol. 2017, 12, 1344–1356. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Narayan, P.; Ghosh, S.; Philip, R.; Barrett, J.C.; McCormack, R.T.; Odegaard, J.I.; Oxnard, G.R.; Pracht, L.J.; Williams, P.M.; Kelloff, G.J.; et al. State of the Science and Future Directions for Liquid Biopsies in Drug Development. Oncologist 2020, 25, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Soda, N.; Rehm, B.H.; Sonar, P.; Nguyen, N.-T.; Shiddiky, M.J.A. Advanced liquid biopsy technologies for circulating biomarker detection. J. Mater. Chem. B 2019, 7, 6670–6704. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Gómez-Rueda, A.; Jantus-Lewintre, E.; Isla, D.; Camps, C.; Ramos, I.; Trigo, J.; Bernabé, R.; Juan-Vidal, O.; Sanchez-Torres, J.M.; et al. Clinical utility of plasma-based digital next-generation sequencing in oncogene-driven non-small-cell lung cancer patients with tyrosine kinase inhibitor resistance. Lung Cancer 2019, 134, 72–78. [Google Scholar] [CrossRef]
- Bowling, M.; Arastu, H.; Edwards, V.; Jackson, L.; Thurston, S.; Reese, J.; Mellert, H.; Pestano, G.A.; Walker, P. Longitudinal monitoring for the emergence of epidermal growth factor C797S resistance mutations in non-small cell lung cancer using blood-based droplet digital PCR. Cancer Drug Resist. 2019, 2, 912–916. [Google Scholar] [CrossRef]
- Jackson, L.P.; Tjoa, B.A.; Mellert, H.; Pestano, G.A. Development of a TCR beta repertoire assay for profiling liquid biopsies from NSCLC donors. Cancer Drug Resist. 2020, 3, 563–571. [Google Scholar] [CrossRef]
- Das, K.; Fernando, M.R.; Basiaga, S.; Wigginton, S.M.; Williams, T.L. Effects of a novel cell stabilizing reagent on DNA amplification by PCR as compared to traditional stabilizing reagents. Acta Histochem. 2014, 116, 55–60. [Google Scholar] [CrossRef]
- Norton, S.; Lechner, J.; Williams, T.; Fernando, M.R. A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR. Clin. Biochem. 2013, 46, 1561–1565. [Google Scholar] [CrossRef]
- Norton, S.E.; Luna, K.K.; Lechner, J.M.; Qin, J.; Fernando, M.R. A New Blood Collection Device Minimizes Cellular DNA Release During Sample Storage and Shipping When Compared to a Standard Device. J. Clin. Lab. Anal. 2013, 27, 305–311. [Google Scholar] [CrossRef]
- Mellert, H.; Foreman, T.; Jackson, L.; Maar, D.; Thurston, S.; Koch, K.; Weaver, A.; Cooper, S.; Dupuis, N.; Sathyanarayana, U.G.; et al. Development and Clinical Utility of a Blood-Based Test Service for the Rapid Identification of Actionable Mutations in Non–Small Cell Lung Carcinoma. J. Mol. Diagn. 2017, 19, 404–416. [Google Scholar] [CrossRef]
- Macías, M.; Cañada-Higueras, E.; Alegre, E.; Bielsa, A.; Gracia, J.; Patiño-García, A.; Ferrer-Costa, R.; Sendino, T.; Andueza, M.P.; Mateos, B.; et al. Performance comparison of two next-generation sequencing panels to detect actionable mutations in cell-free DNA in cancer patients. Clin. Chem. Lab. Med. 2020, 58, 1341–1348. [Google Scholar] [CrossRef]
- Chin, Y.M.; Takahashi, Y.; Chan, H.T.; Otaki, M.; Fujishima, M.; Shibayama, T.; Miki, Y.; Ueno, T.; Nakamura, Y.; Low, S. Ultradeep targeted sequencing of circulating tumor DNA in plasma of early and advanced breast cancer. Cancer Sci. 2021, 112, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Kojima, Y.; Okimoto, K.; Amemiya, K.; Mochizuki, H.; Omata, M. Comparison between two amplicon-based sequencing panels of different scales in the detection of somatic mutations associated with gastric cancer. BMC Genom. 2016, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of Capture and Amplicon-Based Approaches for the Development of a Targeted Next-Generation Sequencing Pipeline to Personalize Lymphoma Management. J. Mol. Diagn. 2018, 20, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Se-quencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn 2017, 19, 4–23. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variant | Expected | Detected | Hit Rate |
|---|---|---|---|
| EGFR L858R | 24 | 24 | 100% |
| EGFR T790M | 24 | 24 | 100% |
| EGFR E746-A750 | 23 | 23 | 100% |
| BRAF V600E | 24 | 24 | 100% |
| KRAS G12R | 24 | 24 | 100% |
| Variant | TP 1 | TN 2 | FP 3 | FN 4 | Total | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
| EGFR Del19 | 8 | 45 | 0 | 0 | 53 | 100.0% | 100.0% | 100.0% |
| EGFR L858R | 7 | 45 | 0 | 0 | 52 | 100.0% | 100.0% | 100.0% |
| EGFR T790M | 6 | 46 | 0 | 0 | 52 | 100.0% | 100.0% | 100.0% |
| BRAF V600E | 4 | 41 | 0 | 2 | 47 | 95.7% | 66.7% | 100.0% |
| KRAS G12C | 14 | 33 | 0 | 0 | 47 | 100.0% | 100.0% | 100.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mellert, H.; Reese, J.; Jackson, L.; Maxwell, V.; Tschida, C.; Pestano, G.A. Targeted Next-Generation Sequencing of Liquid Biopsy Samples from Patients with NSCLC. Diagnostics 2021, 11, 155. https://doi.org/10.3390/diagnostics11020155
Mellert H, Reese J, Jackson L, Maxwell V, Tschida C, Pestano GA. Targeted Next-Generation Sequencing of Liquid Biopsy Samples from Patients with NSCLC. Diagnostics. 2021; 11(2):155. https://doi.org/10.3390/diagnostics11020155
Chicago/Turabian StyleMellert, Hestia, Jordan Reese, Leisa Jackson, Victoria Maxwell, Chérie Tschida, and Gary A. Pestano. 2021. "Targeted Next-Generation Sequencing of Liquid Biopsy Samples from Patients with NSCLC" Diagnostics 11, no. 2: 155. https://doi.org/10.3390/diagnostics11020155
APA StyleMellert, H., Reese, J., Jackson, L., Maxwell, V., Tschida, C., & Pestano, G. A. (2021). Targeted Next-Generation Sequencing of Liquid Biopsy Samples from Patients with NSCLC. Diagnostics, 11(2), 155. https://doi.org/10.3390/diagnostics11020155