


A Comprehensive Review of the Latest Approaches to Managing Hypercholesterolemia: A Comparative Analysis of Conventional and Novel Treatments: Part I

 ,
,  ,
,  ,
,  , and
, and 
Abstract
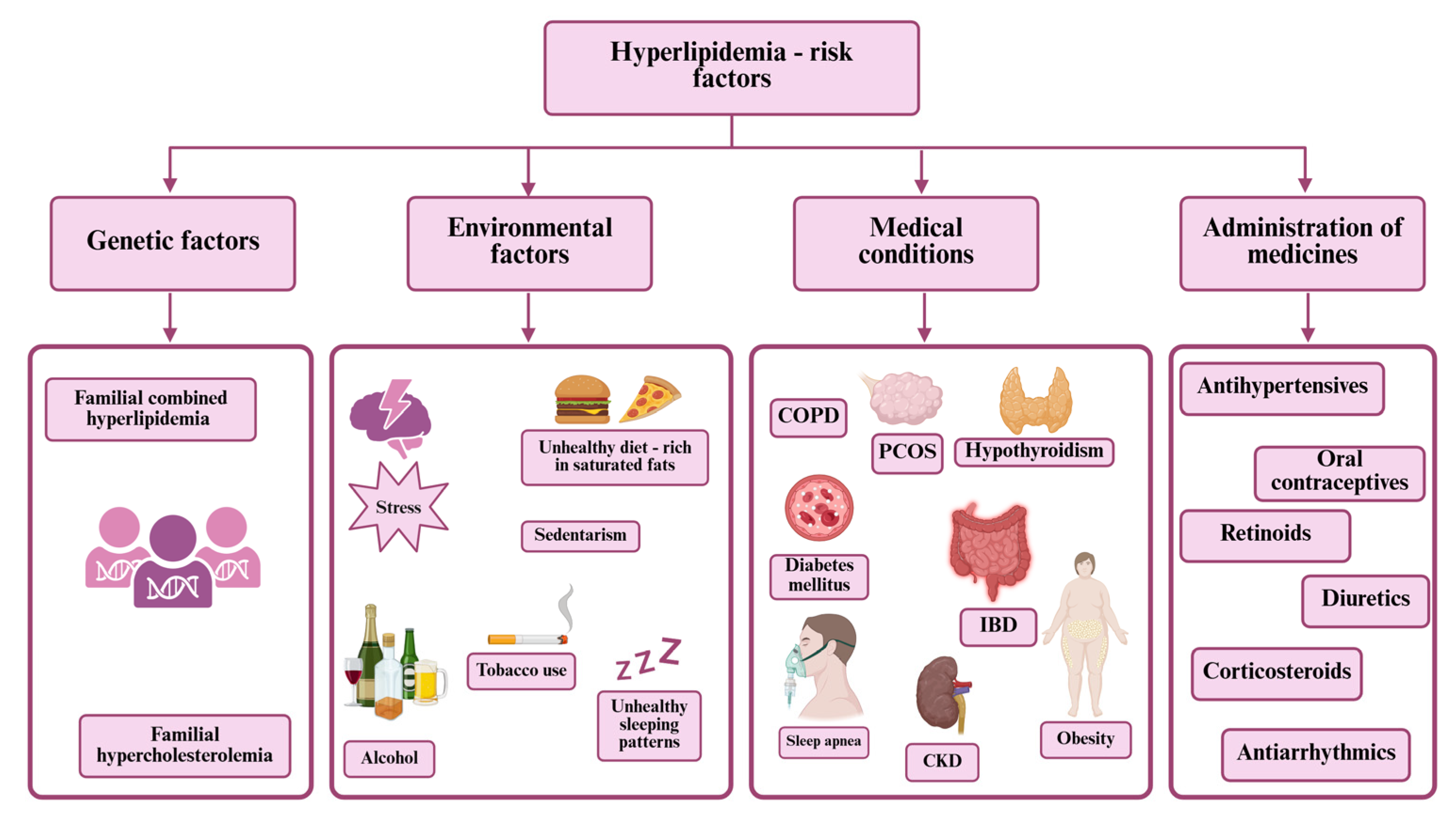
1. Introduction
2. Materials and Methods—Literature Search Strategy
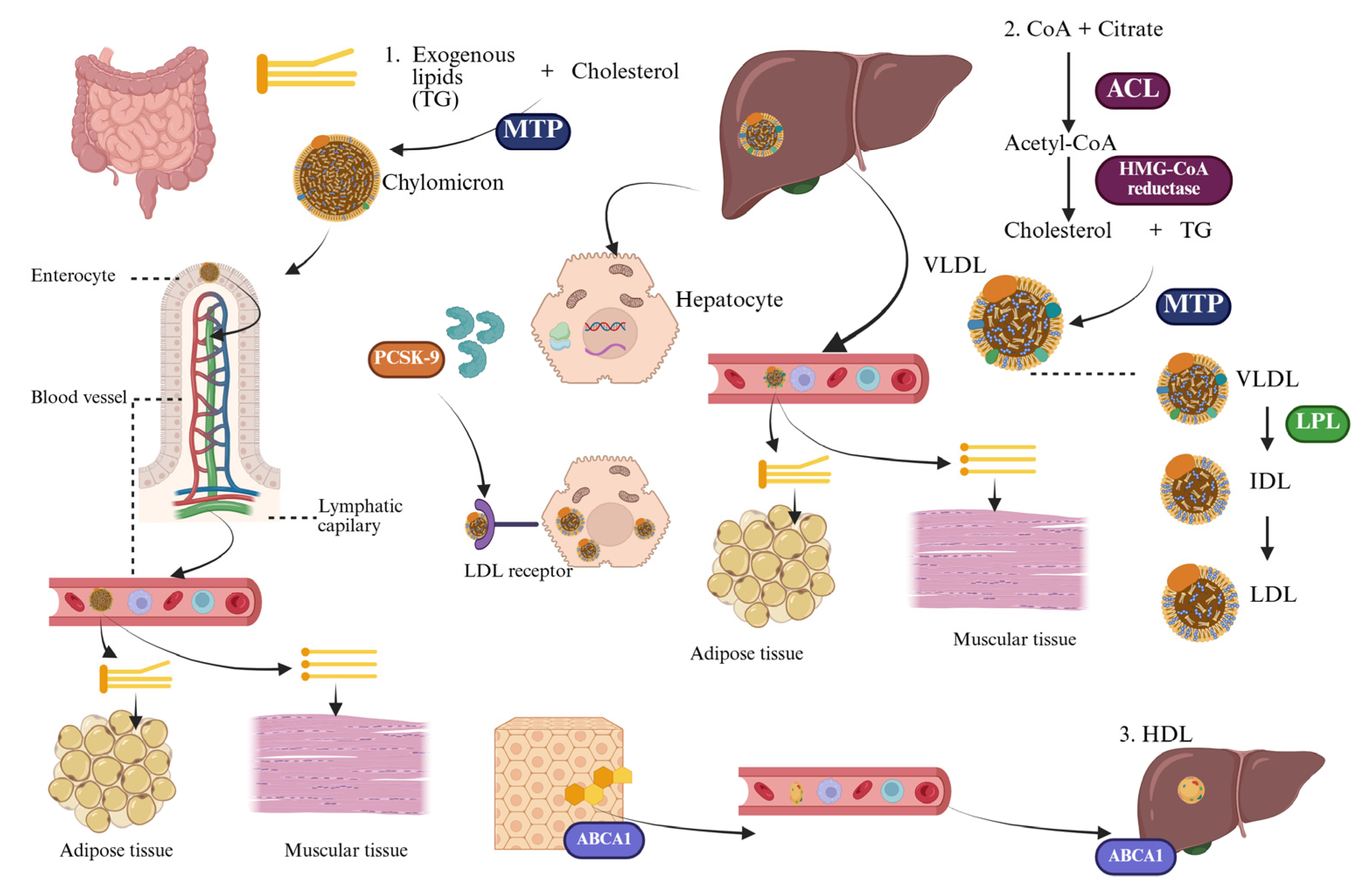
3. Lipid Metabolism and Atherosclerosis—An Integrated Pathophysiological Perspective
4. Complications of Hyperlipidemia
5. Cardiovascular Risk Assessment and Its Role in the Management of Dyslipidemia—Guidelines and Implications
6. Treatment
6.1. Non-Pharmaceutical Approach
6.2. Pharmaceutical Approach
6.2.1. Statins
6.2.2. Ezetimibe
7. Comparative Effectiveness and Safety of the Conventional Lipid-Lowering Therapies, and Pharmacoeconomic Considerations
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | Adenosine triphosphate-binding cassette |
| ABCA1 | ATP-binding cassette transporter A1 |
| ACC | American College of Cardiology |
| ACL | Adenosin triphosphate-citrate lyase |
| AHA | American Heart Association |
| ALT | Alanine Aminotransferase |
| ApoB | Apolipoprotein B |
| ASCVD | Atherosclerotic cardiovascular disease |
| AST | Aspartate Aminotransferase |
| AUC | Area under the curve |
| BCRP | Breast cancer-resistant protein |
| BMI | Body mass index |
| BP | Blood pressure |
| CAC | Coronary artery calcium |
| CAD | Coronary artery disease |
| CK | Creatine Kinase |
| CKD | Chronic kidney disease |
| COPD | Chronic Obstructive Pulmonary Disease |
| CPIC | Clinical Pharmacogenetics Implementation Consortium |
| CVD | Cardiovascular disease |
| CYP450 | Cytochrome P450 |
| DAST | Dietary Approaches to Stop Hypertension |
| DPWG | Dutch Pharmacogenetics Working Group |
| DRESS | Drug reaction with eosinophilia and systemic symptoms |
| ESC | European Society of Cardiology |
| FDA | Food and Drug Administration |
| FH | Familial hypercholesterolemia |
| FHS | Framingham Heart Study |
| HDL | High-density lipoprotein |
| HDL-C | High-density lipoprotein cholesterol |
| HeFH | Heterozygous familial hypercholesterolemia |
| HMG-CoA | β-hydroxy β-methylglutaryl-coenzyme A |
| HoFH | Homozygous familial hypercholesterolemia |
| IBD | Inflammatory Bowel Disease |
| IDL | Intermediate-density lipoproteins |
| IMPROVE-IT | Improved Reduction in Outcomes: Vytorin Efficacy International Trial |
| KDIGO | Kidney Disease: Improving Global Outcomes |
| LDL | Low-density lipoprotein |
| LDL-C | Low-density lipoprotein cholesterol |
| LDLR | Low-density lipoprotein receptor |
| LDLRAP | Low-density lipoprotein receptor adaptor protein |
| LFTs | Liver function tests |
| LPL | Lipoprotein lipase |
| MACE | Major Adverse Cardiovascular Events |
| MI | Myocardial infarction |
| MTP | Microsomal triglyceride transfer protein |
| NICE | National Institute for Health and Care Excellence |
| NPC1L1 | Niemann-Pick C1-like 1 |
| OATP | Organic anion-transporting polypeptide |
| oxLDL | Oxidized low-density lipoprotein |
| PAD | Peripheral arterial disease |
| PCE | Pooled Cohort Equations |
| PCOS | Polycystic ovary syndrome |
| PRE-CISE-IVUS | Plaque Regression with Cholesterol Absorption Inhibitor or Synthesis Inhibitor Evaluated by Intravascular Ultrasound |
| PUFAs | Polyunsaturated fats |
| SAMS | Statin-associated musculoskeletal symptoms |
| SHARPP | Study of Heart and Renal Protection |
| SR-BI | Scavenger receptor class B type I |
| TG | Triglycerides |
| UGT | Uridine diphosphate glucuronosyltransferases |
| ULN | Upper Limit of Normal |
| VLDL | Very-low-density lipoprotein |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.; Dubé, M.P.; Tardif, J.C. Precision Medicine to Change the Landscape of Cardiovascular Drug Development. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 337–343. [Google Scholar] [CrossRef]
- David, S.P. Two for the Heart: Dutch Pharmacogenetic Working Group Prescribing Guidance on Statins and Sulfonylureas to Reduce Cardiometabolic Risk. Eur. J. Hum. Genet. 2025, 33, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Młynarska, E.; Bojdo, K.; Frankenstein, H.; Kustosik, N.; Mstowska, W.; Przybylak, A.; Rysz, J.; Franczyk, B. Nanotechnology and Artificial Intelligence in Dyslipidemia Management—Cardiovascular Disease: Advances, Challenges, and Future Perspectives. J. Clin. Med. 2025, 14, 887. [Google Scholar] [CrossRef]
- Mormone, A.; Tortorella, G.; Esposito, F.; Caturano, A.; Marrone, A.; Cozzolino, D.; Galiero, R.; Marfella, R.; Sasso, F.C.; Rinaldi, L. Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments. Biomedicines 2024, 12, 432. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Alzibali, K.F.; Mahdi, A.M.M.; Alharbi, O.M.A.; Harbi, R.H.A.; Alkhaldi, H.S.M.; Alsayafi, Z.A.A.; Albisher, F.H.; Buqurayn, M.H.; Alharbi, M.M. Lipid-Lowering Medications for Managing Dyslipidemia: A Narrative Review. Cureus 2024, 16, e65202. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Nelson, A.J. Achieving More Optimal Lipid Control with Non-Statin Lipid Lowering Therapy. Curr. Atheroscler. Rep. 2025, 27, 32. [Google Scholar] [CrossRef]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef]
- Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; Saleheen, D.; et al. Lipid-Related Markers and Cardiovascular Disease Prediction. JAMA 2012, 307, 2499–2506. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Martin, S.S.; Aday, A.W.; Allen, N.B.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Bansal, N.; Beaton, A.Z.; et al. 2025 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2025, 151, 8. [Google Scholar] [CrossRef]
- Prabhakar, A.P.; Vedamurthy, D.; Kalra, D.K. Impact of Lipid Lowering Therapies on the Primary Prevention of Atherosclerotic Cardiovascular Disease. Curr. Cardiovasc. Risk Rep. 2025, 19, 12. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Hindi, N.N.; Alenbawi, J.; Nemer, G. Pharmacogenomics Variability of Lipid-Lowering Therapies in Familial Hypercholesterolemia. J. Pers. Med. 2021, 11, 877. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Fazio, S. From Lipids to Inflammation: New Approaches to Reducing Atherosclerotic Risk. Circ. Res. 2016, 118, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, E1082–E1143. [Google Scholar] [CrossRef]
- Currie, G.; Delles, C. Precision Medicine and Personalized Medicine in Cardiovascular Disease. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1065, pp. 589–605. [Google Scholar] [CrossRef]
- Sethi, Y.; Patel, N.; Kaka, N.; Kaiwan, O.; Kar, J.; Moinuddin, A.; Goel, A.; Chopra, H.; Cavalu, S. Precision Medicine and the Future of Cardiovascular Diseases: A Clinically Oriented Comprehensive Review. J. Clin. Med. 2023, 12, 1799. [Google Scholar] [CrossRef]
- Karnati, S.A.; Wee, A.; Shirke, M.M.; Harky, A. Racial Disparities and Cardiovascular Disease: One Size Fits All Approach? J. Card. Surg. 2020, 35, 3530–3538. [Google Scholar] [CrossRef]
- Gaggini, M.; Gorini, F.; Vassalle, C. Lipids in Atherosclerosis: Pathophysiology and the Role of Calculated Lipid Indices in Assessing Cardiovascular Risk in Patients with Hyperlipidemia. Int. J. Mol. Sci. 2023, 24, 75. [Google Scholar] [CrossRef]
- Álvarez-López, H.; Ruiz-Gastélum, E.; Díaz-Aragón, A. Conociendo Los Mecanismos Básicos Del Metabolismo de Los Lípidos. Cardiovasc. Metab. Sci. 2021, 32, 147–152. [Google Scholar] [CrossRef]
- Chandel, N.S. Lipid Metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040576. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Pratico, D.; Lin, L.; Mantzoros, C.S.; Bahijri, S.; Tuomilehto, J.; Ren, J. Inflammation in Atherosclerosis: Pathophysiology and Mechanisms. Cell Death Dis. 2024, 15, 817. [Google Scholar] [CrossRef] [PubMed]
- Dakal, T.C.; Xiao, F.; Bhusal, C.K.; Sabapathy, P.C.; Segal, R.; Chen, J.; Bai, X. Lipids Dysregulation in Diseases: Core Concepts, Targets and Treatment Strategies. Lipids Health Dis. 2025, 24, 61. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, Q.; Zhou, J. Mechanisms of Abnormal Lipid Metabolism in the Pathogenesis of Disease. Int. J. Mol. Sci. 2024, 25, 8465. [Google Scholar] [CrossRef]
- Tasouli-Drakou, V.; Ogurek, I.; Shaikh, T.; Ringor, M.; DiCaro, M.V.; Lei, K.C. Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms. Int. J. Mol. Sci. 2025, 26, 1364. [Google Scholar] [CrossRef]
- Henriques, J.; Amaro, A.M.; Piedade, A.P. Understanding Atherosclerosis Pathophysiology: Can Additive Manufacturing Be Helpful? Polymers 2023, 15, 480. [Google Scholar] [CrossRef]
- Xiao, J.; Deng, Y.M.; Liu, X.R.; Cao, J.P.; Zhou, M.; Tang, Y.L.; Xiong, W.H.; Jiang, Z.S.; Tang, Z.H.; Liu, L.S. PCSK9: A New Participant in Lipophagy in Regulating Atherosclerosis? Clin. Chim. Acta 2019, 495, 358–364. [Google Scholar] [CrossRef]
- Patriki, D.; Saravi, S.S.S.; Camici, G.G.; Liberale, L.; Beer, J.H. PCSK 9: A Link Between Inflammation and Atherosclerosis. Curr. Med. Chem. 2021, 29, 251–267. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Sabouri-Rad, S.; Gotto, A.M.; Pirro, M.; Banach, M.; Awan, Z.; Barreto, G.E.; Sahebkar, A. PCSK9 and Inflammation: A Review of Experimental and Clinical Evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 237–245. [Google Scholar] [CrossRef]
- Stock, J. Familial Hypercholesterolemia: An Urgent Public Health Priority. Atherosclerosis 2020, 308, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, M.H.; Santos, R.D. Functional Testing of Familial Hypercholesterolemia-Related Variants: From Bench to Clinics. JACC Basic. Transl. Sci. 2025, 10, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Tichý, L.; Freiberger, T.; Blaha, V.; Satny, M.; Hubacek, J.A. Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 2020, 11, 574474. [Google Scholar] [CrossRef] [PubMed]
- Pećin, I.; Hartgers, M.L.; Hovingh, G.K.; Dent, R.; Reiner, E. Prevention of Cardiovascular Disease in Patients with Familial Hypercholesterolaemia: The Role of PCSK9 Inhibitors. Eur. J. Prev. Cardiol. 2017, 24, 1383–1401. [Google Scholar] [CrossRef]
- Marusic, T.; Sustar, U.; Sadiq, F.; Kotori, V.; Mlinaric, M.; Kovac, J.; Shafi, S.; Khan, I.; Cevc, M.; Trebusak Podkrajsek, K.; et al. Genetic and Clinical Characteristics of Patients with Homozygous and Compound Heterozygous Familial Hypercholesterolemia from Three Different Populations: Case Series. Front. Genet. 2020, 11, 572176. [Google Scholar] [CrossRef]
- McGowan, M.P.; Hosseini Dehkordi, S.H.; Moriarty, P.M.; Duell, P.B. Diagnosis and Treatment of Heterozygous Familial Hypercholesterolemia. J. Am. Heart Assoc. 2019, 8, e013225. [Google Scholar] [CrossRef]
- Brown, L.; Ruel, I.; Baass, A.; Bergeron, J.; Brunham, L.R.; Cermakova, L.; Couture, P.; Gaudet, D.; Francis, G.A.; Hegele, R.A.; et al. Homozygous Familial Hypercholesterolemia in Canada: An Observational Study. JACC Adv. 2023, 2, 100309. [Google Scholar] [CrossRef]
- Chaudhry, A.; Trinder, M.; Vesely, K.; Cermakova, L.; Jackson, L.; Wang, J.; Hegele, R.A.; Brunham, L.R. Genetic Identification of Homozygous Familial Hypercholesterolemia by Long-Read Sequencing among Patients with Clinically Diagnosed Heterozygous Familial Hypercholesterolemia. Circ. Genom. Precis. Med. 2023, 16, E003887. [Google Scholar] [CrossRef]
- Borén, J.; John Chapman, M.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease: Pathophysiological, Genetic, and Therapeutic Insights: A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Abera, A.; Worede, A.; Hirigo, A.T.; Alemayehu, R.; Ambachew, S. Dyslipidemia and Associated Factors among Adult Cardiac Patients: A Hospital-Based Comparative Cross-Sectional Study. Eur. J. Med. Res. 2024, 29, 237. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Sanders, L.; Lawler, O.; Riley, T.; Maki, K. Hyperlipidemia. In Encyclopedia of Human Nutrition, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 1–4, pp. 361–379. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. Science 1979 2019, 111, 166–186. [Google Scholar] [PubMed]
- Yao, Y.S.; Li, T.D.; Zeng, Z.H. Mechanisms Underlying Direct Actions of Hyperlipidemia on Myocardium: An Updated Review. Lipids Health Dis. 2020, 19, 23. [Google Scholar] [CrossRef]
- Dampier, C.D.; Smith, W.R.; Wager, C.G.; Kim, H.Y.; Bell, M.C.; Miller, S.T.; Weiner, D.L.; Minniti, C.P.; Krishnamurti, L.; Ataga, K.I.; et al. IMPROVE Trial: A Randomized Controlled Trial of Patient-Controlled Analgesia for Sickle Cell Painful Episodes: Rationale, Design Challenges, Initial Experience, and Recommendations for Future Studies. Clin. Trials 2013, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Miceli, G.; Basso, M.G.; Pintus, C.; Pennacchio, A.R.; Cocciola, E.; Cuffaro, M.; Profita, M.; Rizzo, G.; Tuttolomondo, A. Molecular Pathways of Vulnerable Carotid Plaques at Risk of Ischemic Stroke: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 4351. [Google Scholar] [CrossRef]
- Wu, X.H.; Chen, X.Y.; Wang, L.J.; Wong, K.S. Intracranial Artery Calcification and Its Clinical Significance. J. Clin. Neurol. 2016, 12, 253–261. [Google Scholar] [CrossRef]
- He, Z.; Luo, J.; Lv, M.; Li, Q.; Ke, W.; Niu, X.; Zhang, Z. Characteristics and Evaluation of Atherosclerotic Plaques: An Overview of State-of-the-Art Techniques. Front. Neurol. 2023, 14, 1159288. [Google Scholar] [CrossRef]
- Yeramaneni, S.; Kleindorfer, D.; Sucharew, H.; Alwell, K.; Moomaw, C.; Flaherty, M.; Woo, D.; Adeoye, O.; Ferioli, S.; Rosa, F.D.L.R.L.; et al. Abstract T MP114: Hyperlipidemia and Statin Use: Is There an Effect on Mortality Following an Ischemic Stroke? Stroke 2015, 46, ATMP114. [Google Scholar] [CrossRef]
- Aday, A.W.; Everett, B.M. Dyslipidemia Profiles in Patients with Peripheral Artery Disease. Curr. Cardiol. Rep. 2019, 21, 42. [Google Scholar] [CrossRef]
- Balkanay, O.O.; Ömeroğlu, S.N. Approach to Peripheral Arterial Disease in the Elderly. Turk Kardiyol. Dern. Ars. 2017, 45, 96–101. [Google Scholar] [CrossRef]
- Dalal, J.; Padmanabhan, T.N.C.; Jain, P.; Patil, S.; Vasnawala, H.; Gulati, A. LIPITENSION: Interplay between Dyslipidemia and Hypertension. Indian J. Endocrinol. Metab. 2012, 16, 240. [Google Scholar] [CrossRef]
- Borghi, C.; Fogacci, F.; Agnoletti, D.; Cicero, A.F.G. Hypertension and Dyslipidemia Combined Therapeutic Approaches. High Blood Press. Cardiovasc. Prev. 2022, 29, 221–230. [Google Scholar] [CrossRef]
- Zubirán, R.; Cruz-Bautista, I.; Aguilar-Salinas, C.A. Interaction Between Primary Hyperlipidemias and Type 2 Diabetes: Therapeutic Implications. Diabetes Ther. 2024, 15, 1979–2000. [Google Scholar] [CrossRef]
- Rout, P.; Jialal, I. Diabetic Nephropathy. In StatPearls; StatPearls: St. Petersburg, FL, USA, 2025. [Google Scholar] [PubMed]
- Chait, A.; Eckel, R.H.; Vrablik, M.; Zambon, A. Lipid-Lowering in Diabetes: An Update. Atherosclerosis 2024, 394, 117313. [Google Scholar] [CrossRef]
- Rajab, H.A.; Al-Kuraishy, H.M.; Shokr, M.M.; Al-Gareeb, A.I.; Al-Harchan, N.A.; Alruwaili, M.; Papadakis, M.; Alexiou, A.; Batiha, G.E.S. Statins for Vascular Dementia: A Hype or Hope. Neuroscience 2025, 567, 45–55. [Google Scholar] [CrossRef]
- Westphal Filho, F.L.; Moss Lopes, P.R.; Menegaz de Almeida, A.; Sano, V.K.T.; Tamashiro, F.M.; Gonçalves, O.R.; de Moraes, F.C.A.; Kreuz, M.; Kelly, F.A.; Silveira Feitoza, P.V. Statin Use and Dementia Risk: A Systematic Review and Updated Meta-Analysis. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2025, 11, e70039. [Google Scholar] [CrossRef]
- Wang, W.; Li, X. Cognitive Function in Dyslipidemia Patients: Exploring the Impact of Statins. Front. Neurol. 2024, 15, 1436010. [Google Scholar] [CrossRef]
- Wee, J.; Sukudom, S.; Bhat, S.; Marklund, M.; Peiris, N.J.; Hoyos, C.M.; Patel, S.; Naismith, S.L.; Dwivedi, G.; Misra, A. The Relationship between Midlife Dyslipidemia and Lifetime Incidence of Dementia: A Systematic Review and Meta-Analysis of Cohort Studies. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2023, 15, e12395. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.G.; Patten, D.K.; Berlau, D.J. The Role of Statins in Both Cognitive Impairment and Protection against Dementia: A Tale of Two Mechanisms. Transl. Neurodegener. 2018, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Lecordier, S.; Manrique-Castano, D.; El Moghrabi, Y.; ElAli, A. Neurovascular Alterations in Vascular Dementia: Emphasis on Risk Factors. Front. Aging Neurosci. 2021, 13, 727590. [Google Scholar] [CrossRef] [PubMed]
- Ip, B.Y.M.; Ko, H.; Lam, B.Y.K.; Au, L.W.C.; Lau, A.Y.L.; Huang, J.; Kwok, A.J.; Leng, X.; Cai, Y.; Leung, T.W.H.; et al. Current and Future Treatments of Vascular Cognitive Impairment. Stroke 2024, 55, 822–839. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Y.; Zong, J.; Zhang, B.; Li, X.; Qi, H.; Yu, T.; Li, Y. Dendritic Cells Immunotargeted Therapy for Atherosclerosis. Acta Pharm. Sin. B 2025, 15, 792–808. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef]
- Li, B.; Li, W.; Li, X.; Zhou, H. Inflammation: A Novel Therapeutic Target/Direction in Atherosclerosis. Curr. Pharm. Des. 2017, 23, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 2 June 2025).
- Thongtang, N.; Sukmawan, R.; Llanes, E.J.B.; Lee, Z.V. Dyslipidemia Management for Primary Prevention of Cardiovascular Events: Best in-Clinic Practices. Prev. Med. Rep. 2022, 27, 101819. [Google Scholar] [CrossRef]
- Visseren, F.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Komnianou, A.; Kyriakoulis, K.G.; Menti, A.; Dimakakos, E.; Stergiou, G.S.; Kollias, A. Cardiovascular Risk Assessment and Lipid-Lowering Therapy Recommendations in Primary Prevention. J. Clin. Med. 2025, 14, 2220. [Google Scholar] [CrossRef]
- Alloubani, A.; Nimer, R.; Samara, R. Relationship between Hyperlipidemia, Cardiovascular Disease and Stroke: A Systematic Review. Curr. Cardiol. Rev. 2020, 17, e051121189015. [Google Scholar] [CrossRef]
- Reiter-Brennan, C.; Osei, A.D.; Uddin, S.M.I.; Orimoloye, O.A.; Obisesan, O.H.; Mirbolouk, M.; Blaha, M.J.; Dzaye, O. ACC/AHA Lipid Guidelines: Personalized Care to Prevent Cardiovascular Disease. Cleve Clin. J. Med. 2020, 87, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Navar-Boggan, A.M.; Peterson, E.D.; D’Agostino, R.B.; Neely, B.; Sniderman, A.D.; Pencina, M.J. Hyperlipidemia in Early Adulthood Increases Long-Term Risk of Coronary Heart Disease. Circulation 2015, 131, 451–458. [Google Scholar] [CrossRef]
- Nelson, R.H. Hyperlipidemia as a Risk Factor for Cardiovascular Disease. Prim. Care Clin. Off. Pract. 2013, 40, 195–211. [Google Scholar] [CrossRef] [PubMed]
- NICE Guideline CG181. Cardiovascular Disease: Risk Assessment and Reduction, Including Lipid Modification; NICE: Hoboken, NJ, USA, 2023. [Google Scholar]
- Henry, J.P.; Gabriel, L.; Luchian, M.L.; Higny, J.; Benoit, M.; Xhaët, O.; Blommaert, D.; Telbis, A.M.; Robaye, B.; Guedes, A.; et al. Evaluating the Efficacy of a Pre-Established Lipid-Lowering Algorithm in Managing Hypercholesterolemia in Patients at Very High Cardiovascular Risk. J. Pers. Med. 2024, 14, 1044. [Google Scholar] [CrossRef] [PubMed]
- Dybiec, J.; Baran, W.; Dąbek, B.; Fularski, P.; Młynarska, E.; Radzioch, E.; Rysz, J.; Franczyk, B. Advances in Treatment of Dyslipidemia. Int. J. Mol. Sci. 2023, 24, 13288. [Google Scholar] [CrossRef]
- Yang, Y.J. An Overview of Current Physical Activity Recommendations in Primary Care. Korean J. Fam. Med. 2019, 40, 135–142. [Google Scholar] [CrossRef]
- Michos, E.D.; Lopez-Jimenez, F.; Gulati, M. Role of Glucagon-like Peptide-1 Receptor Agonists in Achieving Weight Loss and Improving Cardiovascular Outcomes in People with Overweight and Obesity. J. Am. Heart Assoc. 2023, 12, e029282. [Google Scholar] [CrossRef]
- Feier, C.; Vonica, R.; Faur, A.; Streinu, D.; Muntean, C. Assessment of Thyroid Carcinogenic Risk and Safety Profile of GLP1-RA Semaglutide (Ozempic) Therapy for Diabetes Mellitus and Obesity: A Systematic Literature Review. Int. J. Mol. Sci. 2024, 25, 4346. [Google Scholar] [CrossRef]
- Youk, K.M.; Kim, J.; Cho, Y.-S.; Park, D.J. Gastric Cancer After Bariatric Surgeries. J. Metab. Bariatr. Surg. 2022, 11, 20. [Google Scholar] [CrossRef]
- Prodan-Bărbulescu, C.; Faur, F.I.; Varga, N.-I.; Hajjar, R.; Pașca, P.; Ghenciu, L.-A.; Feier, C.I.V.; Dema, A.; Fărcuț, N.; Bolintineanu, S.; et al. A Histopathological and Surgical Analysis of Gastric Cancer: A Two-Year Experience in a Single Center. Cancers 2025, 17, 2219. [Google Scholar] [CrossRef]
- Frișan, A.-C.; Simonescu, M.; Lazăr, M.-A.; Crișan, S.; Mornoș, A.; Șoșdean, R.; Morar, A.-R.; Brie, D.-M.; Luca, C.-T.; Mornoș, C. Prognostic Significance of Left Ventricular Global Work Efficiency in Obese Patients with Acute ST-Segment Elevation Myocardial Infarction—A Pilot Study. Diagnostics 2025, 15, 1512. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Van Craenenbroeck, E.M.; Antoniades, C.; Blüher, M.; Gorter, T.M.; Hanssen, H.; Marx, N.; McDonagh, T.A.; Mingrone, G.; Rosengren, A.; et al. Obesity and Cardiovascular Disease: An ESC Clinical Consensus Statement. Eur. Heart J. 2024, 45, 4063–4098. [Google Scholar] [CrossRef] [PubMed]
- Chiavaroli, L.; Viguiliouk, E.; Nishi, S.K.; Mejia, S.B.; Rahelić, D.; Kahleová, H.; Salas-Salvadó, J.; Kendall, C.W.C.; Sievenpiper, J.L. DASH Dietary Pattern and Cardiometabolic Outcomes: An Umbrella Review of Systematic Reviews and Meta-Analyses. Nutrients 2019, 11, 338. [Google Scholar] [CrossRef]
- Dhaka, V.; Gulia, N.; Ahlawat, K.S.; Khatkar, B.S. Trans Fats-Sources, Health Risks and Alternative Approach—A Review. J. Food Sci. Technol. 2011, 48, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK570127/ (accessed on 20 July 2025).
- Trautwein, E.A.; McKay, S. The Role of Specific Components of a Plant-Based Diet in Management of Dyslipidemia and the Impact on Cardiovascular Risk. Nutrients 2020, 12, 2671. [Google Scholar] [CrossRef] [PubMed]
- Castro-Barquero, S.; Ruiz-León, A.M.; Sierra-Pérez, M.; Estruch, R.; Casas, R. Dietary Strategies for Metabolic Syndrome: A Comprehensive Review. Nutrients 2020, 12, 2983. [Google Scholar] [CrossRef]
- Sadowska, A.; Osiński, P.; Roztocka, A.; Kaczmarz-Chojnacka, K.; Zapora, E.; Sawicka, D.; Car, H. Statins—From Fungi to Pharmacy. Int. J. Mol. Sci. 2024, 25, 466. [Google Scholar] [CrossRef]
- Pop, G.; Farcaș, A.; Butucă, A.; Morgovan, C.; Arseniu, A.M.; Pumnea, M.; Teodoru, M.; Gligor, F.G. Post-Marketing Surveillance of Statins—A Descriptive Analysis of Psychiatric Adverse Reactions in EudraVigilance. Pharmaceuticals 2022, 15, 1536. [Google Scholar] [CrossRef]
- Dec, A.; Niemiec, A.; Wojciechowska, E.; Maligłówka, M.; Bułdak, Ł.; Bołdys, A.; Okopień, B. Inclisiran—A Revolutionary Addition to a Cholesterol-Lowering Therapy. Int. J. Mol. Sci. 2023, 24, 6858. [Google Scholar] [CrossRef]
- Choudhary, A.; Rawat, U.; Kumar, P.; Mittal, P. Pleotropic Effects of Statins: The Dilemma of Wider Utilization of Statin. Egypt. Heart J. 2023, 75, 1. [Google Scholar] [CrossRef] [PubMed]
- Svec, A.; Adameova, A. Facts and Ideas on Statins with Respect to Their Lipophilicity: A Focus on Skeletal Muscle Cells and Bone besides Known Cardioprotection. Mol. Cell Biochem. 2023, 478, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Talarico, M.; Coppi, F.; Boriani, G. Protective Role of Statins in COVID 19 Patients: Importance of Pharmacokinetic Characteristics Rather than Intensity of Action. Intern. Emerg. Med. 2020, 15, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Song, S.L.; Hays, S.B.; Panton, C.E.; Mylona, E.K.; Kalligeros, M.; Shehadeh, F.; Mylonakis, E. Statin Use Is Associated with Decreased Risk of Invasive Mechanical Ventilation in COVID-19 Patients: A Preliminary Study. Pathogens 2020, 9, 759. [Google Scholar] [CrossRef]
- Morris, R.; Bu, K.; Han, W.; Wood, S.; Hernandez Velez, P.M.; Ward, J.; Crescitelli, A.; Martin, M.; Cheng, F. The Association Between Statin Drugs and Rhabdomyolysis: An Analysis of FDA Adverse Event Reporting System (FAERS) Data and Transcriptomic Profiles. Genes 2025, 16, 248. [Google Scholar] [CrossRef]
- Merćep, I.; Vujević, A.; Strikić, D.; Radman, I.; Pećin, I.; Reiner, Ž. Present and Future of Dyslipidaemia Treatment—A Review. J. Clin. Med. 2023, 12, 5839. [Google Scholar] [CrossRef]
- Agnello, F.; Ingala, S.; Laterra, G.; Scalia, L.; Barbanti, M. Novel and Emerging LDL-C Lowering Strategies: A New Era of Dyslipidemia Management. J. Clin. Med. 2024, 13, 1251. [Google Scholar] [CrossRef]
- Chen, X.; Liu, H.; Li, L.; A, G.; Sun, P.; Tan, D.S.Y.; Chan, M.Y.Y.; Foo, R.S.Y.; Fonarow, G.C.; Yang, Q.; et al. Atorvastatin versus Rosuvastatin in Acute Myocardial Infarction with Elevated Liver Enzymes: A Target Trial Emulation Study. Clin. Res. Cardiol. 2025, 114, 796–808. [Google Scholar] [CrossRef]
- Vinci, P.; Panizon, E.; Tosoni, L.M.; Cerrato, C.; Pellicori, F.; Mearelli, F.; Biasinutto, C.; Fiotti, N.; Di Girolamo, F.G.; Biolo, G. Statin-Associated Myopathy: Emphasis on Mechanisms and Targeted Therapy. Int. J. Mol. Sci. 2021, 22, 11687. [Google Scholar] [CrossRef]
- Antonenko, A.; Leahy, A.; Babenko, M.; Lyons, D. Low Dose Hydrophilic Statins Are the Preferred Agents for Females at Risk of Osteoporosis. Bone Rep. 2022, 16, 101152. [Google Scholar] [CrossRef]
- Lee, Y.J.; Hong, S.J.; Kang, W.C.; Hong, B.K.; Lee, J.Y.; Lee, J.B.; Cho, H.J.; Yoon, J.; Lee, S.J.; Ahn, C.M.; et al. Rosuvastatin versus Atorvastatin Treatment in Adults with Coronary Artery Disease: Secondary Analysis of the Randomised LODESTAR Trial. BMJ 2023, 383, e075837. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD Practice Guidance on the Clinical Assessment and Management of Nonalcoholic Fatty Liver Disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J.; Liou, I.; Reuben, A.; Suzuki, A.; Fiel, M.I.; Lee, W.; Navarro, V. AASLD Practice Guidance on Drug, Herbal, and Dietary Supplement–Induced Liver Injury. Hepatology 2023, 77, 1036–1065. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. Statin-Associated Muscle Symptoms: Impact on Statin Therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Bosco, G.; Di Giacomo Barbagallo, F.; Spampinato, S.; Lanzafame, L.; Di Pino, A.; Piro, S.; Purrello, F.; Scicali, R. Management of Statin Intolerant Patients in the Era of Novel Lipid Lowering Therapies: A Critical Approach in Clinical Practice. J. Clin. Med. 2023, 12, 2444. [Google Scholar] [CrossRef]
- Lewek, J.; Bielecka-Dabrowa, A.; Toth, P.P.; Banach, M. Dyslipidaemia Management in Pregnant Patients: A 2024 Update. Eur. Heart J. Open 2024, 4, oeae032. [Google Scholar] [CrossRef]
- Mauricio, R.; Khera, A. Statin Use in Pregnancy: Is It Time for a Paradigm Shift? Circulation 2022, 145, 496–498. [Google Scholar] [CrossRef]
- Cobos-Palacios, L.; Sanz-Cánovas, J.; Muñoz-Ubeda, M.; Lopez-Carmona, M.D.; Perez-Belmonte, L.M.; Lopez-Sampalo, A.; Gomez-Huelgas, R.; Bernal-Lopez, M.R. Statin Therapy in Very Old Patients: Lights and Shadows. Front. Cardiovasc. Med. 2021, 8, 779044. [Google Scholar] [CrossRef]
- Bonnet, F.; Bénard, A.; Poulizac, P.; Afonso, M.; Maillard, A.; Salvo, F.; Berdaï, D.; Salles, N.; Rousselot, N.; Marchi, S.; et al. Discontinuing Statins or Not in the Elderly? Study Protocol for a Randomized Controlled Trial. Trials 2020, 21, 342. [Google Scholar] [CrossRef]
- Awad, K.; Banach, M. The Optimal Time of Day for Statin Administration: A Review of Current Evidence. Curr. Opin. Lipidol 2018, 29, 340–345. [Google Scholar] [CrossRef]
- Delabays, B.; Trajanoska, K.; Walonoski, J.; Mooser, V. Cardiovascular Pharmacogenetics: From Discovery of Genetic Association to Clinical Adoption of Derived Test. Pharmacol Rev. 2024, 76, 791–827. [Google Scholar] [CrossRef]
- Pirmohamed, M. Pharmacogenomics: Current Status and Future Perspectives. Nat. Rev. Genet. 2023, 24, 350–362. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Pirmohamed, M. Precision Medicine in Cardiovascular Therapeutics: Evaluating the Role of Pharmacogenetic Analysis Prior to Drug Treatment. J. Intern Med. 2024, 295, 583–598. [Google Scholar] [CrossRef]
- Mauriello, A.; Ascrizzi, A.; Molinari, R.; Falco, L.; Caturano, A.; D’Andrea, A.; Russo, V. Pharmacogenomics of Cardiovascular Drugs for Atherothrombotic, Thromboembolic and Atherosclerotic Risk. Genes 2023, 14, 2057. [Google Scholar] [CrossRef]
- Beunk, L.; Nijenhuis, M.; Soree, B.; de Boer-Veger, N.J.; Buunk, A.M.; Guchelaar, H.J.; Houwink, E.J.F.; Risselada, A.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) Guideline for the Gene-Drug Interaction between CYP2D6, CYP2C19 and Non-SSRI/Non-TCA Antidepressants. Eur. J. Hum. Genet. 2024, 32, 1371–1377. [Google Scholar] [CrossRef]
- Magavern, E.F.; Kaski, J.C.; Turner, R.M.; Drexel, H.; Janmohamed, A.; Scourfield, A.; Burrage, D.; Floyd, C.N.; Adeyeye, E.; Tamargo, J.; et al. The Role of Pharmacogenomics in Contemporary Cardiovascular Therapy: A Position Statement from the European Society of Cardiology Working Group on Cardiovascular Pharmacotherapy. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Cooper-DeHoff, R.M.; Niemi, M.; Ramsey, L.B.; Luzum, J.A.; Tarkiainen, E.K.; Straka, R.J.; Gong, L.; Tuteja, S.; Wilke, R.A.; Wadelius, M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 Genotypes and Statin-Associated Musculoskeletal Symptoms. Clin. Pharmacol. Ther. 2022, 111, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Precision Medicine | FDA. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/precision-medicine (accessed on 3 June 2025).
- Lehtisalo, M.; Taskinen, S.; Tarkiainen, E.K.; Neuvonen, M.; Viinamäki, J.; Paile-Hyvärinen, M.; Lilius, T.O.; Tapaninen, T.; Backman, J.T.; Tornio, A.; et al. A Comprehensive Pharmacogenomic Study Indicates Roles for SLCO1B1, ABCG2 and SLCO2B1 in Rosuvastatin Pharmacokinetics. Br. J. Clin. Pharmacol. 2023, 89, 242–252. [Google Scholar] [CrossRef]
- Deng, F.; Sjöstedt, N.; Santo, M.; Neuvonen, M.; Niemi, M.; Kidron, H. Novel Inhibitors of Breast Cancer Resistance Protein (BCRP, ABCG2) among Marketed Drugs. Eur. J. Pharm. Sci. 2023, 181, 106362. [Google Scholar] [CrossRef]
- Maslub, M.G.; Radwan, M.A.; Daud, N.A.A.; Sha’aban, A. Association between CYP3A4/CYP3A5 Genetic Polymorphisms and Treatment Outcomes of Atorvastatin Worldwide: Is There Enough Research on the Egyptian Population? Eur. J. Med. Res. 2023, 28, 381. [Google Scholar] [CrossRef] [PubMed]
- Rocca, B.; Bigagli, E.; Cerbai, E. Ticagrelor and Statins: Dangerous Liaisons? Cardiovasc. Drugs Ther. 2024, 38, 1103–1109. [Google Scholar] [CrossRef]
- Bellosta, S.; Paoletti, R.; Corsini, A. Safety of Statins: Focus on Clinical Pharmacokinetics and Drug Interactions. Circulation 2004, 109, III50–III57. [Google Scholar] [CrossRef]
- Karlgren, M.; Vildhede, A.; Norinder, U.; Wisniewski, J.R.; Kimoto, E.; Lai, Y.; Haglund, U.; Artursson, P. Classification of Inhibitors of Hepatic Organic Anion Transporting Polypeptides (OATPs): Influence of Protein Expression on Drug-Drug Interactions. J. Med. Chem. 2012, 55, 4740–4763. [Google Scholar] [CrossRef] [PubMed]
- Wells, I.; Ezzet, S.; Yamout, N.; Boutros, M.; Ray, S.D. Side Effects of Antilipid Medications, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2021; Volume 43, ISBN 9780128241196. [Google Scholar]
- Kallapur, A.; Sallam, T. Pharmacotherapy in Familial Hypercholesterolemia—Current State and Emerging Paradigms. Trends Cardiovasc. Med. 2023, 33, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Salinas, C.A.; Gómez-Díaz, R.A.; Corral, P. New Therapies for Primary Hyperlipidemia. J. Clin. Endocrinol. Metab. 2022, 107, 1216–1224. [Google Scholar] [CrossRef]
- Penson, P.E.; Bruckert, E.; Marais, D.; Reiner, Ž.; Pirro, M.; Sahebkar, A.; Bajraktari, G.; Mirrakhimov, E.; Rizzo, M.; Mikhailidis, D.P.; et al. Step-by-Step Diagnosis and Management of the Nocebo/Drucebo Effect in Statin-Associated Muscle Symptoms Patients: A Position Paper from the International Lipid Expert Panel (ILEP). J. Cachexia Sarcopenia Muscle 2022, 13, 1596–1622. [Google Scholar] [CrossRef]
- Ezetimibe-Related Liver Injury, SJS, TEN and DRESS. React. Wkly. 2024, 2002, 6. [CrossRef]
- Kusters, D.M.; Caceres, M.; Coll, M.; Cuffie, C.; Gagné, C.; Jacobson, M.S.; Kwiterovich, P.O.; Lee, R.; Lowe, R.S.; Massaad, R.; et al. Efficacy and Safety of Ezetimibe Monotherapy in Children with Heterozygous Familial or Nonfamilial Hypercholesterolemia. J. Pediatr. 2015, 166, 1377–1384.e3. [Google Scholar] [CrossRef]
- Lewek, J.; Banach, M. Dyslipidemia Management in Pregnancy: Why Is It Not Covered in the Guidelines? Curr. Atheroscler. Rep. 2022, 24, 547–556. [Google Scholar] [CrossRef]
- Ouchi, Y.; Sasaki, J.; Arai, H.; Yokote, K.; Harada, K.; Katayama, Y.; Urabe, T.; Uchida, Y.; Hayashi, M.; Yokota, N.; et al. Ezetimibe Lipid-Lowering Trial on Prevention of Atherosclerotic Cardiovascular Disease in 75 or Older (EWTOPIA 75): A Randomized, Controlled Trial. Circulation 2019, 140, 992–1003. [Google Scholar] [CrossRef]
- Kosoglou, T.; Statkevich, P.; Johnson-Levonas, A.O.; Paolini, J.F.; Bergman, A.J.; Alton, K.B. Ezetimibe. Clin. Pharmacokinet. 2005, 44, 467–494. [Google Scholar] [CrossRef]
- Tsujita, K.; Sugiyama, S.; Sumida, H.; Shimomura, H.; Yamashita, T.; Yamanaga, K.; Komura, N.; Sakamoto, K.; Oka, H.; Nakao, K.; et al. Impact of Dual Lipid-Lowering Strategy with Ezetimibe and Atorvastatin on Coronary Plaque Regression in Patients with Percutaneous Coronary Intervention: The Multicenter Randomized Controlled PRECISE-IVUS Trial. J. Am. Coll. Cardiol. 2015, 66, 495–507. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- González-Iglesias, E.; Ochoa, D.; Navares-Gómez, M.; Zubiaur, P.; Aldama, M.; Torre, T.d.l.; Ríos-Rodríguez, M.d.l.; Soria-Chacartegui, P.; Rodríguez-Lopez, A.; Abad-Santos, F.; et al. Evaluation of the Role of Metabolizing Enzymes and Transporter Variants in Ezetimibe Pharmacokinetics. Front. Pharmacol. 2024, 15, 1414059. [Google Scholar] [CrossRef] [PubMed]
- Knežević, S.; Filippi-Arriaga, F.; Belančić, A.; Božina, T.; Mršić-Pelčić, J.; Vitezić, D. Metabolic Syndrome Drug Therapy: The Potential Interplay of Pharmacogenetics and Pharmacokinetic Interactions in Clinical Practice: A Narrative Review. Diabetology 2024, 5, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.J.; Spencer, G.; Nugent, R.; Kostova, D.; Richter, P. The Cost-Effectiveness of Hyperlipidemia Medication in Low- and Middle-Income Countries: A Review. Glob. Heart 2022, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Michaeli, D.T.; Michaeli, J.C.; Boch, T.; Michaeli, T. Cost-Effectiveness of Lipid-Lowering Therapies for Cardiovascular Prevention in Germany. Cardiovasc. Drugs Ther. 2023, 37, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Kazi, D.S.; Moran, A.E.; Coxson, P.G.; Penko, J.; Ollendorf, D.A.; Pearson, S.D.; Tice, J.A.; Guzman, D.; Bibbins-Domingo, K. Cost-Effectiveness of PCSK9 Inhibitor Therapy in Patients with Heterozygous Familial Hypercholesterolemia or Atherosclerotic Cardiovascular Disease. JAMA 2016, 316, 743. [Google Scholar] [CrossRef]
- Farnier, M.; Hovingh, G.K.; Langslet, G.; Dufour, R.; Baccara-Dinet, M.T.; Din-Bell, C.; Manvelian, G.; Guyton, J.R. Long-Term Safety and Efficacy of Alirocumab in Patients with Heterozygous Familial Hypercholesterolemia: An Open-Label Extension of the ODYSSEY Program. Atherosclerosis 2018, 278, 307–314. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Abushanab, D.; Al-Badriyeh, D.; Marquina, C.; Bailey, C.; Jaam, M.; Liew, D.; Ademi, Z. A Systematic Review of Cost-Effectiveness of Non-Statin Lipid-Lowering Drugs for Primary and Secondary Prevention of Cardiovascular Disease in Patients with Type 2 Diabetes Mellitus. Curr. Probl. Cardiol. 2023, 48, 101211. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Ruscica, M.; Fazio, S.; Corsini, A. Correction: Ferri et al. Low-Density Lipoprotein Cholesterol-Lowering Drugs: A Narrative Review. J. Clin. Med. 2024, 13, 943, Erratum in J. Clin. Med. 2024, 13, 4582. [Google Scholar] [CrossRef]
- Ezhov, M.V.; Sergienko, I.V.; Kryzhanovskiy, S.M.; Manko, K.S.; Timoshina, E.V. Comparative Efficacy and Safety of Statin Monotherapy and Statin plus Ezetimibe Combination in a Real-World Setting. Diseases 2023, 11, 168. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration Efficacy and Safety of More Intensive Lowering of LDL Cholesterol: A Meta-Analysis of Data from 170,000 Participants in 26 Randomised Trials. Lancet 2010, 376, 1670–1681. [CrossRef]
- Tamaki, N.; Ueno, H.; Morinaga, Y.; Shiiya, T.; Nakazato, M. Ezetimibe Ameliorates Atherosclerotic and Inflammatory Markers, Atherogenic Lipid Profiles, Insulin Sensitivity, and Liver Dysfunction in Japanese Patients with Hypercholesterolemia. J. Atheroscler. Thromb. 2012, 19, 532–538. [Google Scholar] [CrossRef]
- Tecson, K.M.; Kluger, A.Y.; Cassidy-Bushrow, A.E.; Liu, B.; Coleman, C.M.; Jones, L.K.; Jefferson, C.R.; VanWormer, J.J.; McCullough, P.A. Usefulness of Statins as Secondary Prevention Against Recurrent and Terminal Major Adverse Cardiovascular Events. Am. J. Cardiol. 2022, 176, 37–42. [Google Scholar] [CrossRef]
- Ashraf, J.; Ali Khan, M.; Minhaj, S.; Khatti, S.; Aarij, K.M.; Shehzad, M.; Khan, T.M. Statins and Abnormal Liver Function Tests: Is There a Correlation? Cureus 2020, 12, e10145. [Google Scholar] [CrossRef]
- Bruckert, E.; Ferrières, J. Evidence Supporting Primary Prevention of Cardiovascular Diseases with Statins: Gaps between Updated Clinical Results and Actual Practice. Arch. Cardiovasc. Dis. 2014, 107, 188–200. [Google Scholar] [CrossRef]
- Banach, M.; Jaiswal, V.; Ang, S.P.; Sawhney, A.; Deb, N.; Amarenco, P.; Gaita, D.; Reiner, Z.; Pećin, I.; Lavie, C.J.; et al. Impact of Lipid-Lowering Combination Therapy with Statins and Ezetimibe vs Statin Monotherapy on the Reduction of Cardiovascular Outcomes: A Meta-Analysis. Mayo Clin. Proc. 2025. [Google Scholar] [CrossRef]
- Nowak, M.M.; Niemczyk, M.; Florczyk, M.; Kurzyna, M.; Pączek, L. Effect of Statins on All-Cause Mortality in Adults: A Systematic Review and Meta-Analysis of Propensity Score-Matched Studies. J. Clin. Med. 2022, 11, 5643. [Google Scholar] [CrossRef]
- Sydhom, P.; Al-Quraishi, B.; El-Shawaf, M.; Osman, M.T.; Naji, N.; Awwad, N.; Shehata, N.; Osama, M.; Sergany, H.; Maurice, K.F.; et al. The Clinical Effectiveness and Safety of Low/Moderate-Intensity Statins & Ezetimibe Combination Therapy vs. High-Intensity Statin Monotherapy: A Systematic Review and Meta-Analysis. BMC Cardiovasc. Disord. 2024, 24, 660. [Google Scholar] [CrossRef]
- Yasmin, F.; Moeed, A.; Umar, M.; Zaidi, F.; Khan, M.S.; Alraies, M.C. Efficacy and Safety of Moderate-Intensity Statin and Ezetimibe Combination Therapy Versus High-Intensity Statin Monotherapy in Patients with Cardiovascular Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Lipid Atheroscler. 2025, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, H.; Mousavi, A.; Shojaei, S.; Tavakoli, K.; Salabat, D.; Farahani Rad, F.; Askari, M.K.; Nelson, J.; Ruzieh, M.; Hosseini, K. Safety and Effectiveness of High-Intensity Statins Versus Low/Moderate-Intensity Statins Plus Ezetimibe in Patients with Atherosclerotic Cardiovascular Disease for Reaching LDL-C Goals: A Systematic Review and Meta-Analysis. Clin. Cardiol. 2024, 47, e24334. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; McGuire, D.K.; Cannon, C.P.; Giugliano, R.P.; Lokhnygina, Y.; Page, C.B.; Tershakovec, A.M.; Braunwald, E.; Blazing, M.A. Impact of Ezetimibe on New-Onset Diabetes: A Substudy of IMPROVE-IT. J. Am. Heart Assoc. 2023, 12, e029593. [Google Scholar] [CrossRef]
- Li, L.; Huang, C.; Liu, W.; Li, J.; A, G.; Chen, X.; Jiang, S.; Fang, Y.; Foo, R.S.-Y.; Chan, M.Y.-Y.; et al. Time to Benefit of Intensive Lipid Lowering Therapy in Individuals with Cardiovascular Disease. J. Clin. Lipidol 2025, 19, 51–59. [Google Scholar] [CrossRef]
- Jose, J. Statins and Its Hepatic Effects: Newer Data, Implications, and Changing Recommendations. J. Pharm. Bioallied Sci. 2016, 8, 23. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Characteristic | Heterozygous FH (HeFH) | Homozygous FH (HoFH) |
|---|---|---|
| Prevalence | ~1 in 250 individuals | ~1 in 300,000 individuals |
| Genetic basis | one pathogenic mutation (LDLR, ApoB, or PCSK9) | two pathogenic mutations (same gene or compound heterozygous) |
| LDL-C levels | typically 190–400 mg/dL | frequently > 500 mg/dL |
| Age of onset | early adulthood | childhood |
| Clinical features | tendon xanthomas, premature CAD | cutaneous xanthomas, arcus cornealis, aortic stenosis, early MI |
| Response to statins | generally good | minimal to poor |
| Response to PCSK9 inhibitors | effective in most cases | limited efficacy, especially in null LDLR mutations |
| Need for advance therapy | rarely required | often necessary (lipoprotein apheresis, lomitapide, evinacumab) |
| Cardiovascular risk | increased 10–20× vs. general population | extremely high; events often before age 20 if untreated |
| Prognosis without treatment | events typically after age 30–40 | death or major cardiovascular events in adolescence/young adulthood |
| Clinical Context/Risk Category | LDL-C Target | Non-HDL-C Target |
|---|---|---|
| Established ASCVD (CAD, stroke, PAD) | <55 mg/dL (1.4 mmol/L) | <85 mg/dL (2.2 mmol/L) |
| Diabetes mellitus with target organ damage (nephropathy, retinopathy, neuropathy) | <55 mg/dL | <85 mg/dL |
| Severe CKD (GFR < 30 mL/min/1.73 m2) | <55 mg/dL | <85 mg/dL |
| Diabetes without organ damage | <70 mg/dL (1.8 mmol/L) | <100 mg/dL (2.6 mmol/L) |
| Moderate CKD (GFR 30–59 mL/min/1.73 m2) | <70 mg/dL | <100 mg/dL |
| Heterozygous FH without ASCVD | <70 mg/dL | <100 mg/dL |
| Heterozygous FH with ASCVD or major risk factor | <55 mg/dL | <85 mg/dL |
| Severe hypertriglyceridemia (>500 mg/dL) | LDL-C per CV risk | Non-HDL-C per CV risk |
| Cerebrovascular atherosclerotic disease (ischemic stroke) | <55 mg/dL | <85 mg/dL |
| Hypertension without ASCVD or diabetes | LDL-C per global risk | Non-HDL-C per global risk |
| Moderate global cardiovascular risk | <100 mg/dL (2.6 mmol/L) | <130 mg/dL (3.4 mmol/L) |
| Low global cardiovascular risk | <116 mg/dL (3.0 mmol/L) | <145 mg/dL (3.8 mmol/L) |
| Criteria | SCORE2 (ESC) | PCE (ACC/AHA) |
|---|---|---|
| Geographic/Population focus | Europe (calibrated for low-, moderate-, high-, very-high risk countries) | United States population (including adjustments for Black vs. White individuals) |
| Age group | 40–69 years (SCORE2), extended to 70+ with SCORE2-OP | 40–79 years |
| Type of risk estimated | 10-year risk of first fatal and non-fatal CVD events (MI, stroke) | 10-year risk of first ASCVD event (MI, stroke, cardiovascular death) |
| Variables included | Age, sex, smoking status, systolic blood pressure, total cholesterol, or LDL-C | Age, sex, race, total cholesterol, HDL-C, systolic BP, diabetes, smoking, blood pressure treatment |
| Output format | Percentage (%) risk; categorized into 4 levels—low, moderate, high, very high | Percentage (%) risk; categorized into 4 levels -low, borderline, intermediate, high |
| Risk categories | <5%—Low | <5%—Low |
| 5–10%—Moderate | 5–7.4%—Borderline | |
| 10–15%—High | 7.5–19.9%—Intermediate | |
| ≥15% or clinical ASCVD—Very high | ≥20%—High | |
| Use of modifiers | Lifetime risk, family history, social determinants, CAC score, imaging (optional) | Risk-enhancing factors (e.g., hs-CRP, CKD, Lp(a), CAC score) for refined decision-making |
| Risk Category/Patient Group | ESC/EAS (LDL-C Target) | ACC/AHA (Treatment Threshold) | NICE (Approach and Targets) |
|---|---|---|---|
| Very high risk (e.g., ASCVD, diabetes + target organ damage, CKD) | <55 mg/dL and ≥50% reduction | High-intensity statin ± ezetimibe/PCSK9i if LDL-C ≥ 70 mg/dL | High-intensity statin; consider ezetimibe or PCSK9i if ≥40% non-HDL-C reduction not achieved on maximal statin |
| High risk | <70 mg/dL | ≥50% LDL-C reduction with high-intensity statin | High-intensity statin as first line; ezetimibe if needed |
| Moderate risk | <100 mg/dL | Moderate-intensity statin personalized based on risk enhancers | Offer statin if ≥10% 10-year risk; aim for ≥40% non-HDL-C reduction |
| Low risk | <116 mg/dL | Lifestyle modification statin optional based on discussion | Lifestyle modification; statin only if significant risk factors present |
| Severe hypercholesterolemia (LDL-C ≥ 190 mg/dL) | Treat as high or very high risk | High-intensity statin add ezetimibe/PCSK9i if needed | High-intensity statin; consider ezetimibe; PCSK9i reserved for familial hypercholesterolemia meeting strict criteria |
| Diabetes mellitus (age 40–75) | Risk-based LDL-C targets | At least moderate-intensity statin high-intensity if multiple risks | High-intensity statin if QRISK ≥ 10% or evidence of end-organ damage |
| Trans Fatty Acids | Polyunsaturated Fatty Acids | Phytosterols |
|---|---|---|
| margarine | corn oil | seeds |
| crackers | soybean oil | fruits |
| animal products | sunflower oil | legumes |
| cakes | walnuts | nuts |
| potato chips | sunflower seeds | vegetables |
| lard | tofu | vegetable oils |
| biscuits | soybeans | vegetable oil-based margarines |
| popcorn | cereal grains |
| Guideline | Parameter | Threshold for Discontinuation |
|---|---|---|
| ESC/EAS | Transaminases (ALT/AST) | >3 × Upper Limit of Normal (ULN), confirmed on repeat testing |
| Creatine Kinase (CK) | >10 × ULN (with or without symptoms) or >5 × ULN with significant muscle symptoms | |
| AHA/ACC | Transaminases (ALT/AST) | Persistent elevation >3 × ULN |
| Creatine Kinase (CK) | >10 × ULN or >5 × ULN with muscle symptoms | |
| NICE | Transaminases (ALT/AST) | ALT >3 × ULN on repeat testing |
| Creatine Kinase (CK) | >10 × ULN regardless of symptoms, or >5 × ULN with muscle pain | |
| KDIGO | Transaminases (ALT/AST) | >3 × ULN |
| Creatine Kinase (CK) | >10 × ULN or symptomatic elevations |
| Isoenzyme/Transportor | CYP3A4 | CYP2C9 | OATP1B1 | BCRP |
|---|---|---|---|---|
| Statins | Atorvastatin, lovastatin, simvastatin | Fluvastatin, rosuvastatin | Atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, simvastatin, | Rosuvastatin, fluvastatin, atorvastatin |
| Inhibitors | Amiodarone, amlodipine, clarithromycin, cyclosporine A, danazol, diltiazem, erythromycin, fluconazole, fluoxetine, fluvoxamine, gemfibrozil, grapefruit juice, isoniazid, itraconazole, ketoconazole, mibefradil, midazolam, nefazodone, posaconazole, protease inhibitors, ranolazine, sertraline, tacrolimus, telithromycin, ticagrelor, tricyclic antidepressants, verapamil | Amiodarone, capecitabine, fluconazole, fluvoxamine, ketoconazole, metronidazole, miconazole, sulfamethoxazole + trimethoprim, voriconazole, zafirlukast | Clarithromicine, coumestrol, cyclosporine, diazepam, diethylstilbestrol, erythromycin, erlotinib, estrone-3-sulfate, gemfibrozil, genistein, ivermectin, ketoconazole, novobiocin, rifampicin, ritonavir, roxithromycin, sacubitril, telithromycin, tipranavir, verapamil, vemurafenib | Cyclosporine, dabigatran etexilate, elacridar, everolimus, ketoconazole, meloxicam, miconazole, rifampicin, tyrosine-kinase inhibitors (sunitinib, gefitinib, imatinib), vemurafenib, ziprasidone, |
| Inducers | Aprepitant, carbamazepine, cyclophosphamide, corticosteroids, efavirenz, nevirapine, phenytoin, pioglitazone, phenobarbital, St. John’s wort | Carbamazepine, phenobarbital, phenytoin, rifampin |
| Parameter | Statins | Ezetimibe |
|---|---|---|
| Average LDL-C Reduction | 30–60% | ~20% in monotherapy; additional 21–27% when added to statin (vs. placebo) |
| HDL-C Increase | 10–30% | 3–5% |
| MACE Reduction (vs. placebo) | 18% overall risk reduction (secondary prevention) >20% (primary prevention) | Decrease MACE risk when added to statins (e.g., IMPROVE-IT) |
| Effect on All-Cause Mortality | Significant reduction, especially in high-risk patients 65% reduction | No significant reduction alone; some benefit when combined |
| Myopathy Risk | 10–25% | Not significantly increased over placebo |
| Liver Enzyme Elevation | Borderline elevation of liver function tests (LFTs), more commonly with potent statins or high doses | Relatively rare Improves serum ALT levels |
| New-Onset Diabetes Risk | Slightly increased, especially with high-intensity statins | Improves insulin resistance |
| Other Common Side Effects | GI system disorders, | GI system disorders, arthralgia, headache |
| Use in Statin Intolerance | Limited (lower dose or alternate statin often tried) | Preferred non-statin option |
| Parameter | Statin Monotherapy | Statin + Ezetimibe Combination |
|---|---|---|
| Average LDL-C Reduction | 30–60% | An additional 23–24%, on average |
| HDL-C Increase | 10–30% | No significant differences |
| Additional LDL-C drop vs. doubling statin dose | - | Combination produces greater LDL-C drop than doubling statin dose |
| MACE Reduction (vs. placebo) | 18% overall risk reduction (secondary prevention) >20% (primary prevention) | 18% reduction (odd ratio [OR], 0.82) vs. statin alone in meta-analysis of 108,373 patients |
| All-cause mortality | Significantly reduced risk | Superior to statin monotherapy; 19% reduction (OR, 0.81) vs. statin monotherapy |
| Stroke reduction | Included in MACE outcomes | 17% reduction (OR, 0.83) |
| Time to prevent 1 MACE/100 patients | 19.6 months of intensive statin therapy | ~ 36 months |
| Adverse events/discontinuation | Mild muscle pain; elevated LFTs in ~1–3% of patients (<2 × the upper limit of normal); new-onset diabetes risk increased | Comparable AE profile to statin alone in meta-analysis; combination had fewer discontinuations in elderly/diabetics Lower risks of discontinuation |
| Myalgia/myopathy | 10–25% | Significant reduction in muscle-related AE for Low/Moderate-intensity Statins and Ezetimibe |
| Liver enzymes (ALT/AST) | Usually transient elevation of transaminase | Favorable to statin monotherapy No significant difference in ALT levels, greater increase in AST levels vs. high-intensity statin monotherapy |
| New-onset diabetes | Slightly increased risk with high-intensity statins | Reduced events of new-onset diabetes for low/moderate-intensity statins and ezetimibe No increase in the risk of new-onset diabetes when adding ezetimibe to statin therapy |
| GI, headache, others | Statin: GI symptoms, headache, fatigue; Ezetimibe: mild GI, URTI, arthralgia | Combined profile acceptable; no increase in overall adverse events |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nițu, E.-T.; Jianu, N.; Merlan, C.; Foica, D.; Sbârcea, L.; Buda, V.; Suciu, M.; Lombrea, A.; Movilă, D.E. A Comprehensive Review of the Latest Approaches to Managing Hypercholesterolemia: A Comparative Analysis of Conventional and Novel Treatments: Part I. Life 2025, 15, 1185. https://doi.org/10.3390/life15081185
Nițu E-T, Jianu N, Merlan C, Foica D, Sbârcea L, Buda V, Suciu M, Lombrea A, Movilă DE. A Comprehensive Review of the Latest Approaches to Managing Hypercholesterolemia: A Comparative Analysis of Conventional and Novel Treatments: Part I. Life. 2025; 15(8):1185. https://doi.org/10.3390/life15081185
Chicago/Turabian StyleNițu, Ema-Teodora, Narcisa Jianu, Cristina Merlan, Darius Foica, Laura Sbârcea, Valentina Buda, Maria Suciu, Adelina Lombrea, and Dana Emilia Movilă. 2025. "A Comprehensive Review of the Latest Approaches to Managing Hypercholesterolemia: A Comparative Analysis of Conventional and Novel Treatments: Part I" Life 15, no. 8: 1185. https://doi.org/10.3390/life15081185
APA StyleNițu, E.-T., Jianu, N., Merlan, C., Foica, D., Sbârcea, L., Buda, V., Suciu, M., Lombrea, A., & Movilă, D. E. (2025). A Comprehensive Review of the Latest Approaches to Managing Hypercholesterolemia: A Comparative Analysis of Conventional and Novel Treatments: Part I. Life, 15(8), 1185. https://doi.org/10.3390/life15081185