Abstract
Cardiorenal syndrome (CRS) reflects the intricate and bidirectional interplay between cardiac and renal dysfunction, commonly resulting in diagnostic uncertainty, therapeutic dilemmas and poor outcomes. While traditional biomarkers like serum creatinine (Cr) and natriuretic peptides remain widely used, their limitations in specificity, timing and contextual interpretation often hinder optimal management. This narrative review synthesizes the current evidence on established and emerging biomarkers in CRS, with emphasis on their clinical relevance, integration into real-world practice, and potential to inform precision therapy. Markers of glomerular filtration rate beyond creatinine—such as cystatin C—offer more accurate assessment in frail or sarcopenic patients, while tubular injury markers such as NGAL, KIM-1, and urinary L-FABP (uL-FABP) provide early signals of structural renal damage. The FDA-approved NephroCheck® test—based on TIMP-2 and IGFBP7— enables risk stratification for imminent AKI up to 24 h before functional decline. Congestion-related markers such as CA125 and bio-adrenomedullin outperform natriuretic peptides in certain CRS phenotypes, particularly in right-sided heart failure or renally impaired patients. Fibrosis and inflammation markers (galectin-3, sST2, GDF-15) add prognostic insights, especially when combined with NT-proBNP or troponin. Rather than presenting biomarkers in isolation, this review proposes a framework that links them to specific clinical contexts—such as suspected decongestion-related renal worsening or persistent congestion despite therapy—to support actionable interpretation. A tailored, scenario-based, multi-marker strategy may enhance diagnostic precision and treatment safety in CRS. Future research should prioritize prospective biomarker-guided trials and standardized pathways for clinical integration.
1. Introduction
Heart failure (HF) and chronic kidney disease (CKD) are closely interconnected conditions that share a wide array of pathophysiological mechanisms, risk factors, and clinical features. Both are characterized by neurohormonal activation, endothelial dysfunction, inflammation, and fibrotic remodeling, and they often respond to the same pharmacological interventions [1,2]. Indeed, several therapeutic classes—including renin–angiotensin–aldosterone system inhibitors (RAASis), non-steroidal mineralocorticoid receptor antagonists (ns-MRAs), sodium-glucose cotransporter-2 inhibitors (SGLT2is), and glucagone-like-peptide-1 receptor agonists (GLP-1 RAs)—have demonstrated benefits in both HF and CKD, underscoring the overlap in their pathogenesis and treatment strategies [3,4,5,6].
From an epidemiological perspective, CKD affects approximately 844 million individuals globally, while HF affects around 64 million [7,8]. Notably, CKD coexists in nearly 50% of patients with HF, and the coexistence of both conditions confers a particularly poor prognosis [8,9]. It is now well established that the incidence of HF is significantly higher among individuals with CKD, while HF itself accelerates renal decline, particularly through hemodynamic alterations, venous congestion, and neurohormonal dysregulation [10].
An additional challenge is the asymptomatic nature of early-stage CKD, which often leads to delayed diagnosis at more advanced stages, when substantial nephron loss has already occurred. In a landmark study by Dalrymple et al. (2011), patients aged over 65 years with CKD were found to be six times more likely to die from cardiovascular causes than to progress to end-stage renal disease (ESRD), illustrating that most patients with progressive CKD die before reaching kidney failure [11,12].
These complex, bidirectional interactions between the heart and kidneys have led to the conceptualization of cardiorenal syndrome (CRS)—a term first officially defined by the National Heart, Lung, and Blood Institute in 2004 [13]. CRS encompasses acute or chronic dysfunction in the heart or kidneys that induces dysfunction in the other organ. It is currently classified into five subtypes based on the primary failing organ (heart vs. kidney), the chronicity (acute versus chronic), and whether the dysfunction is secondary to a systemic condition affecting both organs (Appendix A) [1,14,15]. Although this classification was intended to aid in diagnosis and therapeutic decision-making, in reality, it offers limited practical value. While it helps conceptually organize the different pathways of heart–kidney interaction, clinical presentations are often more complex, with overlapping features and dynamic transitions between acute and chronic states that do not fit neatly into predefined categories [13].
More recently, the concept of cardiovascular–kidney–metabolic (CKM) syndrome has emerged to capture the shared metabolic derangements and systemic drivers that link these conditions. This broader framework recognizes the interconnected nature of cardiovascular, renal, and metabolic health, and highlights the need for integrated diagnostic and therapeutic strategies [16].
In this evolving landscape, there is a growing need for tools that can more accurately reflect the complexity and heterogeneity of cardiorenal interactions. Traditional markers such as serum creatinine, estimated glomerular filtration rate (GFR), and natriuretic peptides (NPs) provide important information but have well-recognized limitations, particularly in the early detection, risk stratification, and monitoring of CRS. As our understanding of the pathophysiology deepens, a range of novel biomarkers has emerged, offering the potential to better characterize disease trajectories, identify sub-phenotypes, and guide individualized therapy. This narrative review will explore the most recent advances in biomarker research within the context of CRS, with a focus on their clinical relevance and future applications.
2. Literature Review Strategy
The literature review was conducted using a narrative approach focused on clinically relevant biomarkers in the context of CRS. Searches were performed in PubMed, Scopus, and Web of Science using combinations of keywords such as “cardiorenal syndrome,” “biomarkers,” “heart failure,” “acute kidney injury,” “chronic kidney disease,” and specific marker names (e.g., NGAL, KIM-1, cystatin C, CA125, bio-ADM, FGF-23, etc.).
Priority was given to:
- ○
- Original research, systematic reviews, and meta-analyses published in the past 10–15 years
- ○
- Landmark cohort studies, randomized controlled trials, and FDA-related regulatory approvals
- ○
- Publications in high-impact cardiology and nephrology journals
References were selected based on clinical applicability, study quality, and relevance to the CRS framework. Where available, we extracted and contextualized cut-off values, assay types, and prognostic data to support practical implementation. Special emphasis was placed on biomarker interpretation in dynamic clinical settings, including decongestion, worsening renal function (WRF) assessment, and acute kidney injury (AKI) risk stratification.
3. Beyond Creatinine and Natriuretic Peptides: Gaps in Cardiorenal Assessment
3.1. Limitations of Creatinine and Estimated Glomerular Filtration Rate
The gold standard of measuring GFR is through exogenous markers such as iothalamate or inulin, as these molecules are freely filtered and largely without renal secretion or/and absorption. However, this is cumbersome and impractical for routine use in clinical practice [17]. In routine clinical practice, renal function is almost exclusively assessed by serum creatinine (Cr) and Cr-based estimations of the GFR [18].
Creatinine is a product of muscle catabolism that is freely filtered by the glomerulus, with additional minor tubular secretion. It is cheap and easy to measure. While the use of creatinine-based GFR estimates has been instrumental in defining kidney disease as a global health problem, creatinine is not always the ideal surrogate marker for GFR [19]. This approach has well-known limitations such as Cr analytic assay variability, Cr secretion by the proximal renal tubules (rendering it an imperfect biomarker for glomerular filtration), as well as dependency of serum Cr levels on muscle mass, age, sex, diet, and physical activity [18]. Moreover, serum Cr is a relatively late and insensitive marker of AKI, often increasing only after substantial loss of kidney function has occurred, when early recognition of AKI is crucial to allow timely intervention [20]. Serum Cr levels may also transiently increase in HF, reflecting hemodynamic changes rather than structural injury [17,18]. Observational studies also have shown that patients discharged with unresolved congestion, despite modest Cr rises, have worse outcomes than those fully decongested, while the same principle applies in the initiation/uptitration of neurohormonal blockers. Therefore, Cr and eGFR must be interpreted in clinical context, alongside markers of congestion and tubular injury [19,21,22].
3.2. Nephron Dynamics and Renal Reserve
Mechanistically, GFR is the product of the number of functional nephrons and the single-nephron GFR (SNGFR). Each kidney is endowed with approximately one million nephrons at birth. This large nephron pool allows for substantial renal reserve capacity, enabling compensation for nephron loss through an increase in SNGFR, thereby preserving total GFR during the early phases of nephron reduction. Total GFR thus represents a dynamic equilibrium between nephron quantity and individual nephron function, modulated by systemic and intraglomerular hemodynamics as well as neurohormonal influences [17,18]. In contrast, the number of viable nephrons declines progressively with age—estimated at 5000–10,000 per year—resulting in a gradual depletion of renal reserve and, eventually a decline in overall GFR [18,23]. Although compensatory hyperfiltration at the single-nephron level may initially maintain GFR, this adaptive response is finite and, over time, contributes to glomerular damage, podocyte loss and the progression of chronic kidney disease [24].
In its purest sense, CKD is defined as a greater loss of functional nephrons than would be anticipated through healthy aging. A GFR < 60 mL/min/1.73 m2 is therefore used as a reasonable surrogate, with this cutoff corresponding to a >50% loss of functional nephrons at a normal single-nephron GFR. Importantly, an increased single-nephron GFR may compensate for a loss of functional nephrons, reducing the impact on the total GFR assessed by serum Cr, whereas albuminuria is a distinct measure reflecting glomerular and tubular damage and/or function [18,25]. The hypothesis is that in such patients, increased urinary albumin (≥30 mg/g Cr) reflects an increased single-nephron GFR (i.e., glomerular hyper-filtration) and, consequently, a lower number of functional nephrons than reflected by the total GFR, such as in diabetic kidney disease. Moreover, glomerular hyper-filtration is a key pathophysiological mechanism underlying further CKD progression, as it results in accelerated podocyte loss and a dysfunctional glomerular basement membrane. Albuminuria and its implications will be analyzed in a specific section [18,25].
3.3. Limitations of Natriuretic Peptides and Troponins
In parallel, natriuretic peptides, particularly NT-proBNP, and cardiac troponins, such as high-sensitivity cardiac troponin I (hs-cTnI), are commonly employed for risk stratification and diagnosis in HF, yet their utility in capturing the complexity of CRS is limited. NT-proBNP reflects myocardial wall stress and volume overload, but its levels are influenced by age, obesity, and renal function [15]. In patients with CKD or CRS, elevated NT-proBNP may not reliably differentiate between cardiac congestion and renal impairment, as impaired clearance contributes to persistently high levels regardless of actual hemodynamic status [26]. Similarly, hs-cTnI is a sensitive marker of myocardial injury and is often elevated in CRS due to subclinical myocardial stress, but it lacks specificity for reversible versus irreversible injury, and its interpretation can be challenging in the context of coexisting renal dysfunction. Thus, while both biomarkers offer prognostic information, they have limitations in guiding therapy in CRS [15].
4. A Primer on Novel Biomarker Classes in CRS
4.1. Glomerular Filtration Markers Beyond Creatinine
4.1.1. Cystatin-C
Cystatin C is a low-molecular-weight cysteine protease inhibitor produced by all nucleated cells at a relatively constant rate. It is freely filtered by the glomerulus and almost completely reabsorbed and metabolized by proximal tubular epithelial cells, making it an attractive endogenous marker of GFR [27]. Unlike serum creatinine, cystatin C is minimally influenced by muscle mass, age, or nutritional status, which enhances its reliability in patients with sarcopenia, cachexia, or other conditions where creatinine-based estimates may be misleading [15,17,28].
Because of its stability and reduced dependence on non-GFR factors, cystatin C has gained traction as a marker for cardiovascular (CV) and renal risk stratification. Elevated cystatin C levels have been associated with worse outcomes in a range of clinical scenarios, including coronary artery disease (CAD), acute and chronic HF, and chronic kidney disease [15,29]. In patients hospitalized for acute decompensated HF (ADHF), serum cystatin C levels at admission were found to be superior to both serum creatinine and B-type natriuretic peptide (BNP) in predicting all-cause mortality and rehospitalization [30].
Nevertheless, cystatin C is not without limitations. Its levels can be affected by several non-GFR determinants, including thyroid dysfunction, obesity, systemic inflammation, and corticosteroid use [15,17]. A recent study in 293 HF patients found that cystatin C levels, although less affected by muscle mass than creatinine, still showed some correlation with it—potentially reflecting underlying metabolic and inflammatory processes rather than direct muscle mass influence [31].
One of the most compelling insights into the utility of cystatin C in HF comes from population-based studies. In a large real-world cohort from Sweden, involving over 158.000 individuals, discordance between eGFRCr and eGFRCys was common and clinically meaningful. In 65% of cases, eGFRCys was lower than eGFRCr, with over a quarter of patients showing a difference exceeding 27%. This discrepancy was most pronounced in older individuals with multiple comorbidities—a profile commonly seen in HF populations—and was independently associated with higher risk of acute kidney injury (AKI), progression to kidney failure, HF events, and all-cause mortality [32]. These findings underscore the potential of cystatin-C to uncover hidden renal dysfunction in patients where creatinine may overestimate true GFR [33].
The importance of using cystatin C in conjunction with creatinine was further highlighted in a subanalysis of the PARADIGM-HF trial. When comparing the 2021 CKD-EPI combined creatinine–cystatin C equation to the creatinine-only equation, substantial individual reclassification occurred, particularly in sicker patients. A lower GFR estimated by the combined equation was associated with higher levels of natriuretic peptides and troponin, poorer quality of life as assessed by the Kansas City Cardiomyopathy Questionnaire (KCCQ), and a higher risk of mortality. These discrepancies were likely due to a less pronounced increase in creatinine compared to cystatin C in patients with greater neurohormonal activation—possibly as a result of decreased creatinine production or increased tubular secretion in advanced HF [34].
In summary, while cystatin C is not entirely exempt from confounding factors, it remains a more sensitive and less muscle-mass-dependent marker of GFR compared to serum Cr. It may enhance early detection of renal impairment, refine risk stratification, and guide clinical decisions in heart failure—particularly in frail, elderly, or cachectic individuals. Furthermore, the observed discordance between creatinine and cystatin C-based eGFR may serve as an indirect marker of frailty and worse prognosis, reinforcing the clinical value of incorporating cystatin C into routine assessment of patients with CRS [33] (Table 1).

Table 1.
Key points for the use of Cystatin-C in patients with heart failure [15,17,28,31,34,35].
4.1.2. Βeta-2-Microglobulin (β2-M)
Beta-2-microglobulin (β2M) is a low-molecular-weight protein produced by all nucleated cells, continuously released into the circulation, freely filtered by the glomerulus, and almost entirely reabsorbed and metabolized by proximal tubular cells, making it a useful marker of GFR [36].
Serum β2M levels rise progressively with declining GFR, and its kinetics—similar to cystatin C—support its use as an alternative marker of glomerular filtration, particularly where creatinine-based estimates are less reliable. Unlike creatinine, β2M is not secreted by the renal tubules, minimizing potential biases in GFR estimation, and it does not appear to be influenced by sex [37]. However, despite these theoretical benefits, β2M concentrations can still be influenced by non-renal factors, including inflammation, malignancy and autoimmune diseases—all of which increase β2M production and turnover [36].
From a laboratory perspective, β2M is typically measured using immunoassays, though standardization is less advanced compared with cystatin C. The marker is generally stable in stored serum and plasma, but pre-analytical factors such as hemolysis or systemic inflammation may influence results. Furthermore, at higher GFR levels, β2M may also underestimate renal function, likely related to variability in extra-renal production and clearance pathways. These limitations mean that while β2M is a valid GFR marker, cystatin C often remains the preferred choice for routine estimation of kidney function beyond creatinine [36,38].
In addition to its filtration marker role, serum β2M has prognostic relevance. It is strongly associated with all-cause mortality, cardiovascular events, and progression to end-stage renal disease (ESRD) in both CKD populations and healthy individuals [36,39,40,41,42]. For example, in the CRIC Study, higher serum β2M independently predicted ESRD, mortality, and incident cardiovascular disease, occasionally outperforming creatinine-based eGFR [40]. In dialysis patients, β2M levels reflect residual renal function and correlate with cardiovascular outcomes, although they are also influenced by dialytic clearance efficiency, underlying inflammation, and the risk of dialysis-related β2M amyloidosis—a condition characterized by β2M fibril deposition with osteoarticular, dermatologic, gastrointestinal, and cardiovascular manifestations [36,43].
Urinary β2M, historically used to assess proximal tubular injury, is less commonly employed today due to its instability in acidic urine, limited specificity for tubular over glomerular injury and modest predictive performance [36].
In summary, β2M is a valuable alternative filtration marker and a potent prognostic biomarker in CKD and CRS. However, its clinical application is constrained by non-renal influences and greater familiarity with cystatin C in clinical settings. The combined assessment of β2M with other filtration markers may enhance risk stratification, particularly where creatinine or cystatin C measurements are ambiguous or less informative [38,41].
4.1.3. Proenkephalin (PENK)
Proenkephalin (PENK) is an endogenous opioid peptide and a stable surrogate of active enkephalins, synthesized primarily in the central nervous system. In the kidney, enkephalins modulate renal perfusion, diuresis, and natriuresis by activating delta-opioid receptors, especially in the proximal tubules, where receptor density is high [44,45]. PENK itself is freely filtered at the glomerulus and is not significantly influenced by age or sex in most cohorts, making it a promising marker of GFR. Studies have consistently shown a strong correlation between PENK levels and measured GFR across diverse populations, including those with CKD, HF, and post-cardiac surgery patients [45,46].
Beyond reflecting renal filtration, PENK levels may rise as a counterregulatory response to renal hypoperfusion or oxidative stress, conditions common in HF and CRS [45,47]. Its concentration has been associated with the risk of AKI in critically ill patients and may outperform traditional markers in early AKI detection [48,49]. Furthermore, elevated PENK levels have been linked to adverse outcomes in ADHF, including worsening renal function, rehospitalization, and all-cause mortality. Notably, PENK levels also correlate with other renal biomarkers such as NGAL, indicating its relevance in tubular stress contexts. Most studies quantify PENK using sandwich immunoassays, but no universally accepted clinical cut-off has been established, and reference ranges remain assay- and laboratory-specific [47,48,50,51].
While PENK tracks measured GFR across diverse cohorts, elevations in advanced HF may also reflect neurohormonal activation and systemic stress, limiting its specificity as a pure filtration surrogate. There is also variability in cutoff thresholds across studies, and its utility in guiding therapy remains unproven. While promising for risk stratification in CRS and acute HF, further research is needed to clarify its prognostic value and define standardized reference ranges [44,45,46,48].
4.2. Markers of Tubular Injury
4.2.1. Albuminuria
Albumin is a small, negatively charged plasma protein that plays a key role in maintaining oncotic pressure and transporting endogenous and exogenous molecules. Under normal conditions, the glomerular filtration barrier—composed of fenestrated endothelial cells, the glomerular basement membrane (GBM), and interdigitating podocytes connected by slit diaphragms—effectively prevents the passage of albumin into the urine. Disruption of this barrier leads to increased urinary albumin excretion, a phenomenon known as albuminuria [25,52]. Clinically, albuminuria is defined by a urinary albumin-to-creatinine ratio (UACR) ≥ 30 mg/g and is categorized into moderately increased-A2 (30–300 mg/g) and severely increased-A3 (>300 mg/g) levels [5]. Among patients with chronic HF, elevated UACR is observed in approximately 25% to 44% of cases, highlighting its high prevalence in this population [18,53,54].
Although albuminuria is traditionally considered a marker of glomerular barrier dysfunction, proximal tubular abnormalities may also contribute to increased urinary albumin excretion in specific clinical contexts. For example, in inherited Fanconi syndrome or drug-induced nephrotoxicity (e.g., Tenofovir, Aminoglycosides, Cisplatin), albuminuria may arise due to impaired tubular reabsorption [55]. Additionally, in diabetes mellitus, early dysfunction of megalin–cubilin receptor-mediated endocytosis in proximal tubules may reduce albumin reuptake, contributing to microalbuminuria even before overt glomerular damage. Thus, albuminuria may in some cases reflect early tubular dysfunction in addition to glomerular injury, especially in diseases characterized by complex nephron involvement [56].
Beyond its role as a marker of glomerular and proximal tubular damage, albuminuria reflects broader systemic processes involved in cardiovascular disease (CVD). The pathophysiological mechanisms linking albuminuria to HF are multifactorial, encompassing glomerular injury, systemic endothelial dysfunction, low-grade inflammation, neurohormonal activation—particularly of the renin–angiotensin–aldosterone system (RAAS) causing directly podocyte injury—and elevated central venous pressure leading to renal congestion [57,58].
Albuminuria is prevalent in both HF with reduced ejection fraction (HFrEF) and preserved ejection fraction (HFpEF), and is associated with adverse outcomes regardless of eGFR. Even mild elevations within the traditionally “normal” range (<30 mg/g) have been associated with an increased risk of heart failure and adverse cardiovascular outcomes, reinforcing the role of albuminuria as a sensitive marker of both early and progressive cardiorenal dysfunction [25,59]. Albuminuria thus serves not only as a marker of glomerular injury but also as an independent prognostic indicator in CRS [25,57]. Notably, albuminuria frequently occurs even in patients without diabetes or overt renal impairment, underscoring its utility as an early indicator of cardiorenal stress. Cohort data, including these from the Framingham Heart Study, CHARM and CHART-2 trials, have shown that even microalbuminuria (30–300 mg/g) is associated with increased risk of incident HF, hospitalization, and mortality [10,53,59,60,61]. In HFpEF populations specifically, such as those studied in TOPCAT, albuminuria was present in up to one-third of patients and independently predicted HF hospital readmission [62].
Recent evidence also suggests that albuminuria may reflect systemic congestion rather than intrinsic kidney damage alone. In HF cohorts, elevated UACR has been closely associated with markers of volume overload such as NT-proBNP, bio-adrenomedullin, peripheral edema, and echocardiographic signs of increased right-sided pressures. These associations persist independently of GFR and tubular injury markers, suggesting that albuminuria may also reflect venous congestion and systemic endothelial dysfunction in CRS, rather than intrinsic renal injury alone [58]. Levels often improve with decongestion [62], and this therapeutic responsiveness can help distinguish congestion-driven albuminuria from intrinsic nephropathy [63].
4.2.2. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
Neutrophil Gelatinase-Associated Lipocalin (NGAL), also known as lipocalin-2, is a 25-kDa glycoprotein primarily secreted by activated neutrophils and also expressed in a wide array of tissues, including renal tubular cells, cardiomyocytes, hepatocytes, and adipocytes. It is involved in iron transport, innate immunity, and cellular stress responses [64].
NGAL is one of the most extensively studied biomarkers for AKI, especially due to its rapid release following tubular injury [20]. Transcriptomic studies in animal models have shown that NGAL is among the earliest genes induced after kidney insult, particularly in distal nephron segments [65,66]. In both plasma and urine, NGAL levels rise within 2–6 h of renal injury, well before serum creatinine increases, thus enabling earlier detection of AKI [64]. Commercial assays for NGAL have been approved in several regions for AKI detection, and its clinical utility is being increasingly recognized in the context of CRS. Assay platforms include chemiluminescent microparticle immunoassay (CMIA), ELISA, and point-of-care devices, though availability and regulatory approval vary across regions [20,64].
In patients with HF, elevated NGAL levels—both plasma and urinary—have been consistently associated with impaired renal function, increased NT-proBNP, urinary albumin excretion, and higher mortality risk [67,68,69]. NGAL is particularly useful in identifying patients at risk of developing CRS type 1, in which acute HF precipitates AKI. Studies have shown that NGAL levels at admission can predict subsequent AKI in acute decompensated HF and correlate with worsening renal function and clinical outcomes [64,70,71,72,73]. In CRS type 2, NGAL may also contribute to pathogenesis through modulation of matrix metalloproteinase-9 (MMP-9) activity and extracellular matrix remodeling. This interaction promotes extracellular matrix degradation and remodeling, processes that are implicated in renal fibrosis and progression of CKD [64,74].
While NGAL is a highly sensitive biomarker, its expression is also upregulated in response to epithelial injury, inflammation, and ischemia. This broad responsiveness—although useful in signaling global pathophysiological stress—reduces specificity for renal tubular origin. Plasma NGAL may be particularly affected by systemic inflammation, anemia and sepsis, whereas urinary NGAL appears more specific for tubular injury in HF/CRS populations [15,20,75]. Therefore, clinical interpretation of NGAL levels requires careful consideration of the broader clinical context, and combining NGAL with complementary markers (e.g., NT-proBNP, C-reactive protein, etc.) may enhance diagnostic precision [15].
4.2.3. Kidney Injury Molecule-1 (KIM-1)
Kidney Injury Molecule-1 (KIM-1), also known as T-cell immunoglobulin mucin receptor 1 (TIM-1), is a 38.7-kDa type I transmembrane glycoprotein primarily expressed on the apical surface of proximal tubular epithelial cells. In healthy kidneys, KIM-1 is barely detectable, but its expression increases significantly after ischemic or toxic injury [76,77]. KIM-1 may also be present in other tissues such as the intestine and biliary system, but its expression in these organs is markedly lower compared to the kidney [76]. Following renal injury, KIM-1 is shed into the urine and blood, where it can be measured as a non-invasive biomarker [77,78]. Beyond its renal role, KIM-1 participates in immunoregulation through interactions with T-cell subsets and may exert anti-inflammatory and phagocytic functions during epithelial injury [76,79].
KIM-1 is one of the most promising markers of proximal tubular injury, particularly in the early stages of acute kidney injury AKI [80,81]. It is rapidly upregulated after ischemia–reperfusion injury and may precede histological changes [82]. Urinary KIM-1 has shown diagnostic and prognostic value in multiple settings, including post-cardiac surgery AKI, diabetic nephropathy, kidney transplant rejection, and nephrotoxic drug exposure (aminoglycosides, non-steroidal anti-inflammatory drugs, polymyxin etc.) [15,78,80]. In patients with HF, elevated urinary KIM-1 levels have been associated with WRF and increased mortality risk. Some studies demonstrated that urinary KIM-1 could identify HF patients at high risk of post-discharge mortality and CRS progression [69,83].
Despite its high specificity, its sensitivity for detecting AKI is moderate, and diagnostic accuracy may be influenced by comorbidities, proteinuria, and inflammatory conditions. Additionally, discrepancies between urinary and plasma KIM-1 levels complicate interpretation. In clinical practice, urinary KIM-1 is generally preferred for early tubular injury screening, whereas plasma KIM-1 should be interpreted with caution in acute decompensated HF (ADHF) until further validation is available [78,84,85,86].
4.2.4. Liver-Type Fatty Acid Binding Protein (L-FABP)
Liver-type fatty acid binding protein (L-FABP) is a 14-kDa cytoplasmic protein primarily expressed in the proximal tubular epithelial cells of the kidney. Under physiological conditions, L-FABP facilitates the intracellular transport and metabolism of long-chain fatty acids, protecting the tubular cells from lipotoxicity. Its expression is upregulated in response to hypoxia, oxidative stress, and ischemia—key mechanisms underlying both acute and chronic kidney injury [87,88].
In the setting of renal ischemia–reperfusion, tubular injury, or proteinuric kidney disease, L-FABP is released into the urine (uL-FABP), where it serves as a sensitive marker of early tubular stress. Unlike many other markers that reflect glomerular injury, uL-FABP is more closely associated with tubular dysfunction, offering unique insight into the pathophysiological processes of the renal tubulointerstitium [89,90]. In animal and human studies, urinary L-FABP levels have been shown to correlate with the severity of tubular damage and fibrotic changes, even in the absence of overt proteinuria [90].
From a clinical perspective, uL-FABP has demonstrated utility in both acute and chronic settings. It has been validated as an early marker of AKI, particularly in perioperative states, contrast-induced nephropathy, and critically ill populations. In patients with ADHF, elevated urinary L-FABP levels are predictive of AKI development and are associated with worse prognosis [90]. Several studies have shown that patients with higher L-FABP levels at admission were more likely to experience AKI, all-cause mortality, or adverse renal outcomes. In this context, L-FABP reflects underlying tubular stress that may not yet be apparent through traditional markers like serum Cr [89,91].
From a laboratory perspective, urinary L-FABP requires prompt sample processing or stabilization, as the protein is prone to degradation if stored at room temperature. In clinical studies, results are often reported by tertiles or quartiles of risk rather than universal cut-offs, since threshold values vary substantially depending on the assay platform [92,93].
In CKD, L-FABP has also shown promise as a predictor of disease progression, particularly in individuals with diabetic nephropathy or cardiovascular comorbidities. Importantly, uL-FABP may be especially helpful in stratifying risk among CKD patients with little or no albuminuria, where traditional markers may underestimate renal damage. Its role in reflecting ongoing oxidative stress and lipid-mediated tubular injury supports its use in early intervention strategies and long-term monitoring [88,90,94,95,96,97].
Moreover, the combination of L-FABP with established cardiac biomarkers such as NT-proBNP has been proposed as a powerful tool for identifying patients at risk of CRS. Studies in cardiac intensive care unit populations have shown that patients with elevations in both biomarkers were several times more likely to develop AKI than those with lower levels, suggesting additive predictive value and pathophysiological complementarity [98].
Despite its growing potential, certain limitations remain. While uL-FABP is sensitive and correlates well with tubular injury, its role as a monitoring tool during therapy remains to be fully elucidated. Furthermore, large-scale validation in diverse HF and CKD populations is still needed. Nevertheless, current evidence supports the integration of L-FABP into cardiorenal risk assessment, particularly in settings where subclinical kidney injury precedes overt declines in glomerular filtration [89,90,98].
4.2.5. Tissue Inhibitor of Metalloproteinases-2 (TIMP-2) and Insulin-like Growth Factor Binding Protein 7 (IGFBP7): Early Markers of Tubular Stress
Tissue inhibitor of metalloproteinases-2 (TIMP-2) and insulin-like growth factor-binding protein 7 (IGFBP7) are markers of early distal and proximal tubular stress respectively. Both are involved in G1 cell cycle arrest, a protective mechanism that prevents cell division during injury. Unlike traditional markers, they do not reflect established injury but rather cellular stress preceding overt tubular damage [99]. Combined as [TIMP-2] × [IGFBP7], these biomarkers form the basis of the NephroCheck® test, which has been Food and Drug Administration (FDA)-cleared for assessing the risk of moderate to severe AKI (KDIGO stage 2–3) within 12–24 h in ICU and post-cardiac surgery settings (Table 2). Their elevation prompts early preventive strategies, including optimization of hemodynamics, avoidance of nephrotoxins, and enhanced monitoring [100,101,102,103]. In clinical studies, this combination has shown strong predictive performance in high-risk populations, including patients with sepsis, shock, and those undergoing major surgery or kidney transplantation. Elevated [TIMP-2] × [IGFBP7] levels are also associated with prolonged delayed graft function and worse prognosis in transplant recipients [99].
While they are not substitutes for serum creatinine or urine output, they offer a valuable early warning signal—enabling proactive interventions before structural kidney damage becomes evident. Their role is particularly relevant in critically ill or hemodynamically unstable patients, and expanding use in pediatric and neonatal populations has also been reported [101,103,104,105].
4.3. Markers of Inflammation and Fibrosis
4.3.1. Interleukin-18
Interleukin-18 (IL-18) is a pro-inflammatory cytokine from the interleukin-1 (IL-1) family, structurally and functionally similar to IL-1β. It is produced in an inactive form by various cell types, including monocytes, macrophages, and renal tubular epithelial cells, and becomes biologically active after cleavage by caspase-1. IL-18 plays a central role in immune regulation by promoting interferon-gamma (IFN-γ) release and driving a Th1-type immune response through the myeloid-differentiation factor 88(MyD88)/nuclear factor kappa B (NF-κB) signaling pathway. Under normal conditions, its activity is modulated by IL-18 binding protein (IL-18BP), but in many inflammatory diseases, this balance is disrupted, leading to excessive IL-18 activity [106,107].
Beyond its role in immune regulation, IL-18 has been implicated in the pathogenesis of cardiovascular disease. Higher circulating IL-18 levels have been linked to an increased risk of coronary artery disease, acute myocardial infarction, and the development or progression of HF. Several large cohort studies and meta-analyses have confirmed its association with adverse cardiovascular outcomes and mortality, even in the general population [108].
In the kidney, IL-18 is predominantly released from proximal tubular cells in response to ischemic or inflammatory injury. Urinary IL-18 (uIL-18), released predominantly from proximal tubular cells, has been more extensively validated than plasma IL-18 for detecting renal inflammation. Measurement of uIL-18 offers a window into tubular injury and has been studied extensively as a biomarker for AKI. In patients with ADHF, elevated uIL-18 has been independently associated with a higher risk of AKI development [109]. One multicenter study found that patients with higher uIL-18 levels had a 3.6-fold increased risk of AKI compared to those with lower levels, after adjusting for clinical confounders [110]. Despite these associations, IL-18 lacks organ specificity, as levels may also rise in systemic inflammatory states, infections, and autoimmune disease. Its predictive performance improves when interpreted alongside complementary markers of tubular injury (e.g., NGAL, KIM-1), supporting its use within a biomarker panel rather than as a stand-alone test [109].
4.3.2. Soluble Suppression of Tumorigenicity-2 (sST2)
Soluble ST2 (sST2) is a circulating isoform of the ST2 glycoprotein, a member of the interleukin-1 receptor family. It functions as a decoy receptor for interleukin-33 (IL-33), a cytokine released by damaged or stressed cells that plays a protective role in the heart by reducing inflammation and fibrosis. By binding IL-33 and preventing its interaction with membrane-bound ST2 receptors, sST2 neutralizes its cardioprotective effects and contributes to adverse cardiac remodeling. As such, elevated sST2 levels are considered a marker of myocardial stress, inflammation, and fibrosis [111].
In HF, sST2 has been extensively validated as a prognostic biomarker [112,113]. Unlike natriuretic peptides, sST2 is not significantly affected by age, renal function, or body mass index, making it particularly attractive for risk stratification. Elevated sST2 levels at hospital admission have been associated with increased mortality and rehospitalization in both acute and chronic HF settings [114,115]. Studies suggest that serial monitoring of sST2 may provide additional prognostic information, as a reduction in sST2 after treatment is linked to improved outcomes, independent of changes in NT-proBNP [113]. In clinical practice, sST2 concentrations are most often measured using the Presage® ST2 assay, which is FDA-cleared and widely used to guide decision-making. Levels <35 ng/mL are associated with low likelihood of acute HF, while values >70 ng/mL reflect significant neurohormonal activation and fibrosis, supporting the need for hospitalization and intensified therapy. In addition, a relative reduction of >30% during hospitalization has been proposed as a treatment response signal. These thresholds are intended as prognostic and triage guides rather than diagnostic absolutes (Table 2) [112,116,117].
In patients with CRS, sST2 also holds promise. It has been associated with both cardiovascular events and the development of chronic kidney disease [15]. In individuals with renal dysfunction, elevated sST2 levels predict progression of HF and adverse outcomes, suggesting that it may serve as an integrative biomarker across the heart–kidney axis [118,119]. Moreover, in cardiac intensive care unit settings, combining sST2 with other markers such as NT-proBNP has improved the prediction of AKI and in-hospital events [111].
In summary, sST2 is a robust, dynamic biomarker that reflects myocardial fibrosis and inflammation and offers independent prognostic value in HF. Its utility extends to patients with renal dysfunction and CRS, where it may aid in risk stratification and guide therapeutic decisions [111,113].
4.3.3. Galectin-3 (Gal-3)
Galectin-3 (Gal-3) is a 30-kDa β-galactoside-binding lectin encoded by the LGALS3 gene and secreted mainly by activated macrophages. It plays key roles in inflammation, immune regulation, cell proliferation, and fibrosis [111]. Unlike many cardiac biomarkers, Gal-3 is not heart-specific and is broadly expressed across epithelial and immune cells, including in the kidney and vascular endothelium. Its ability to bind carbohydrate structures allows it to mediate both cell–cell and cell–matrix interactions, contributing to tissue remodeling and fibrotic responses [120].
In HF, Gal-3 has gained attention as a biomarker of myocardial fibrosis and adverse remodeling. Elevated Gal-3 levels have been associated with higher morbidity and mortality across a wide spectrum of cardiovascular conditions, including acute and chronic HF, myocardial infarction, and congenital heart disease [111,113]. Several studies have reported that higher Gal-3 levels correlate with HF severity and predict adverse outcomes independently of natriuretic peptides [111,113]. Unlike NT-proBNP, Gal-3 levels are less influenced by hemodynamic stress and may better reflect structural and inflammatory changes. However, emerging data suggest that its prognostic value may depend on renal function. In one study, Gal-3 levels lost their predictive utility in patients with preserved kidney function, while remaining informative in those with reduced eGFR. The proposed mortality risk thresholds were ~20 ng/mL for patients with eGFR ≥ 60 mL/min/1.73 m2 and ~30 ng/mL for those with eGFR < 60 mL/min/1.73 m2, underscoring the need to interpret Gal-3 in the context of renal status (Table 2) [121,122].
Gal-3 levels are also elevated in patients with both acute and chronic kidney disease (CKD), often increasing with declining renal function [113,121]. It is believed to contribute to tubulointerstitial fibrosis and inflammation, and its expression has been linked to immune cell infiltration and proteinuria in glomerular diseases. In translational studies, Gal-3 has shown strong associations with AKI severity, proteinuria burden, and progression of CKD, including in patients with diabetes, cardiovascular disease, and autoimmune nephropathies. Both serum and urinary Gal-3 concentrations have demonstrated predictive value for AKI in intensive care, cardiac surgery, and sepsis settings, with area under the curve (AUC) values exceeding 0.85 in some cohorts [120].
In HF patients, Gal-3 appears to have both diagnostic and prognostic utility, but its predictive accuracy may decline in the presence of renal dysfunction [121]. This overlap reflects its dual involvement in both cardiac and renal fibrosis and complicates its interpretation as a stand-alone marker. However, when used alongside other biomarkers such as NT-proBNP or NGAL, Gal-3 can enhance risk stratification, particularly in identifying patients at risk for long-term cardiorenal deterioration. By contrast, its short-term utility for guiding decongestion is limited compared with markers such as sST2 or bio-ADM [121,123].
Overall, Galectin-3 reflects underlying fibrotic and inflammatory processes common to both the heart and kidney. While it is not specific to one organ system, its broad involvement in cardiorenal pathophysiology makes it a valuable biomarker in identifying high-risk patients and potentially guiding antifibrotic or anti-inflammatory therapeutic strategies. Further studies are needed to validate its role in treatment monitoring and to clarify its utility across varying stages of cardiorenal disease [113,120].
4.3.4. Fibroblast Growth Factor-23 (FGF-23) and Klotho Axis
FGF-23 is a 32-kDa hormone primarily secreted by osteocytes and osteoblasts in the bone. Its primary physiological role is to maintain phosphate homeostasis and regulate vitamin D metabolism by acting on the kidneys. FGF-23 requires the presence of the co-receptor α-klotho to activate fibroblast growth factor receptors (FGFRs), most abundantly expressed in the renal tubules. In this context, FGF-23 reduces phosphate reabsorption in the proximal tubules and suppresses the production of 1,25-dihydroxyvitamin D. However, in chronic kidney disease (CKD), FGF-23 levels rise early, often before overt changes in phosphate or parathyroid hormone (PTH) levels, representing one of the first signs of disrupted mineral metabolism and bone mineral disease [124]. In advanced CKD, FGF-23 levels can increase up to 1000-fold and become maladaptive, contributing to systemic effects beyond mineral metabolism, particularly on the cardiovascular system [124,125]. Emerging data suggest that an imbalance between FGF-23 and Klotho may activate Wnt/β-catenin signaling, a pathway involved in cardiac hypertrophy, fibrosis, and endothelial dysfunction. These effects are thought to be mediated through Klotho-independent activation of FGFR4 in the myocardium, leading to cardiomyocyte hypertrophy, calcium dysregulation, and adverse remodeling. Klotho deficiency, common in CKD, appears to amplify these effects. Cross-talk with the renin–angiotensin system may further contribute to volume overload and blood pressure dysregulation [125,126].
Several studies have linked elevated FGF-23 levels to both HF with preserved ejection fraction (HFpEF) and HF with reduced ejection fraction (HFrEF), especially in individuals with CKD. In the CRIC cohort, FGF-23 independently predicted incident HF across all ejection fraction (EF) subtypes, reinforcing its value as a cardiorenal connector. Mechanistically, FGF-23 may promote adverse remodeling through LVH and inflammation, and may indirectly impair cardiac function by reducing Klotho expression [127].
From a diagnostic perspective, FGF-23 holds promise as a biomarker for early CKD-related cardiovascular risk, but its implementation in clinical practice is limited by assay variability (e.g., intact vs. C-terminal FGF-23 assays), lack of standardized cutoffs, and the complexity of its dual endocrine and paracrine actions. Furthermore, while observational data are strong, causal inference remains uncertain, as most evidence is derived from observational cohorts or post hoc analyses rather than randomized controlled trials [124,125,128,129]. Indirect evidence of benefit comes from studies in hemodialysis patients treated with phosphate binders and calcimimetics. In the EVOLVE post hoc analysis, a ≥30% reduction in FGF-23 with cinacalcet was associated with lower cardiovascular event rates and mortality [130]. Similarly, in randomized trials with etelcalcetide, potent FGF-23 suppression (~75%) was linked to reduced progression of left ventricular hypertrophy compared to alphacalcidol, despite equivalent parathyroid hormone control [131].
Additionally, fibroblast growth factor-21 (FGF-21), another member of the same family, has emerged as a stress-induced hormone with potential relevance in CRS. While it may exert counterregulatory effects by reducing oxidative stress and fibrosis, elevated FGF-21 levels likely reflect underlying metabolic stress and disease severity, serving more as a biomarker of poor prognosis rather than a direct mediator, based on current evidence [132,133,134].
In summary, FGF-23 is a pivotal marker and possible mediator in the interplay between kidney dysfunction and heart failure. Its early elevation in CKD and its strong association with adverse cardiovascular outcomes highlight its potential utility in risk stratification and possibly future therapeutic strategies in cardiorenal syndrome. Though assay heterogeneity and uncertain causality currently limit its clinical application [125,126,135].
4.3.5. Growth Differentiation Factor-15 (GDF-15)
GDF-15 is a stress-responsive cytokine belonging to the transforming growth factor-beta (TGF-β) superfamily. Under physiological conditions, it is weakly expressed across tissues, with higher expression in the placenta and low baseline levels in circulation. However, its expression rises markedly in response to cellular stress, tissue injury, inflammation, and oxidative stress [136,137]. Although initially characterized for its anti-inflammatory effects on macrophages, GDF-15 is now recognized as a pleiotropic molecule involved in diverse pathological states, including cardiovascular diseases, kidney disease, cancer, and metabolic disorders. Importantly, circulating GDF-15 levels are strongly influenced by non-disease factors such as age, smoking, cancer, and exposure to medications including metformin and chemotherapy. These confounders must be considered when interpreting elevated values in clinical practice [136,138].
In kidney pathophysiology, both plasma and urinary GDF-15 levels increase in acute and chronic kidney disease, reflecting ongoing inflammation, oxidative stress, and tissue injury [136]. Experimental studies suggest that GDF-15 may exert protective renal effects by limiting fibrosis, reducing oxidative stress, and preserving tubular integrity, partly through modulation of the PI3K/Akt and NF-κB pathways [136,139]. However, in clinical settings, elevated GDF-15 levels typically signal worse outcomes. Higher serum GDF-15 concentrations have been independently associated with CKD progression, the need for renal replacement therapy, and adverse cardiovascular events [140,141,142,143,144].
In heart failure, GDF-15 is considered a robust prognostic marker. Elevated levels correlate with worse outcomes across both preserved and reduced ejection fraction phenotypes and are particularly predictive in patients with coexisting CKD [137,141,142,145]. GDF-15 levels have also been linked to right ventricular dysfunction, congestion, and cachexia in advanced heart failure, reflecting its association with systemic metabolic stress [143]. Notably, serial measurements of GDF-15, especially when combined with natriuretic peptides like NT-proBNP, improve risk stratification in acute heart failure, outperforming single time-point assessments. While dedicated studies on GDF-15 in CRS are limited, its dual association with cardiac and renal dysfunction, as well as inflammation and fibrosis, highlights its potential as a unifying biomarker in CRS and warrants further investigation [137,142].
Despite its predictive power, the interpretation of GDF-15 remains complex. As a systemic stress marker, it lacks organ specificity, and its exact role—whether adaptive or maladaptive—within the cardiovascular and renal systems is still under investigation [136]. Elevated GDF-15 may reflect both the severity of the underlying disease and a compensatory response to injury. Additionally, its levels increase with age, smoking, cancer, and certain treatments like metformin or chemotherapy, which can confound clinical interpretation [136,138].
In summary, GDF-15 is a promising biomarker in cardiorenal syndrome, capturing the systemic stress and inflammatory burden that accompany disease progression. While it enhances prognostic evaluation, particularly when combined with other biomarkers, its non-specific nature and susceptibility to confounding factors currently limit its use as a stand-alone tool, reinforcing its value as part of a multi-marker strategy [138].
4.4. Markers of Congestion
4.4.1. Carbohydrate Antigen 125 (CA125)
Carbohydrate antigen 125 (CA125), also known as MUC16, is a high-molecular-weight transmembrane glycoprotein originally recognized as a tumor marker in ovarian and colorectal cancer. Beyond oncology, CA125 is physiologically expressed by mesothelial cells lining the pericardium, pleura, and peritoneum, where it contributes to epithelial lubrication and defense against mechanical stress. In heart failure (HF), particularly in the context of right-sided dysfunction and systemic congestion, CA125 levels rise in response to mechanical stretch of mesothelial surfaces and inflammatory stimuli. Venous congestion leads to serosal effusions, bowel wall edema, and endothelial activation, which together with the release of pro-inflammatory cytokines (IL-6, IL-10, etc.) stimulate CA125 production [146,147].
Unlike natriuretic peptides, CA125 levels are less influenced by left ventricular ejection fraction (LVEF), renal function, age, or obesity, making it a distinct biomarker of tissue congestion rather than intravascular volume status [146,148]. In acute HF, elevated CA125 (>35 U/mL) correlates with congestion severity, right ventricular dysfunction, pleural effusions, and peripheral edema (Table 2). This marker has demonstrated prognostic utility, predicting mortality and rehospitalization independently of natriuretic peptides, particularly in patients with CRS or renal dysfunction where NT-proBNP loses specificity. Importantly, CA125 demonstrates on-treatment kinetics. Levels typically fall with effective decongestive therapy, and a lack of decline—or a rise post-discharge—signals persistent or recurrent congestion. In such cases, clinical reassessment, diuretic uptitration, or closer follow-up should be considered [146,148,149,150,151].
Serial measurement of CA125 provides insight into decongestion status and residual congestion after hospital discharge. Clinical studies, including the CHANCE-HF trial, have shown that CA125-guided therapy—primarily in adjusting diuretic strategies—reduces HF readmissions [152]. Persistently elevated or rising CA125 levels after discharge are associated with worse outcomes. In chronic HF and HF with preserved ejection fraction (HFpEF), higher CA125 correlates with atrial enlargement, impaired renal function, and reduced exercise capacity [148,149]. However, CA125 is not heart-specific and can be elevated in non-cardiac conditions such as malignancies, liver cirrhosis, peritoneal inflammation, and infections. Therefore, interpretation requires a comprehensive clinical assessment. Moreover, while it effectively reflects serosal and tissue congestion, CA125 is less sensitive to isolated intravascular congestion [146,149].
In clinical practice, CA125 serves as a complementary tool to natriuretic peptides and imaging, especially useful in elderly patients, those with renal dysfunction, or where right-sided HF predominates. A baseline and follow-up measurement post-decongestion can help stratify risk, optimize therapy, and potentially improve outcomes. While further research is needed to standardize its use, current evidence supports the clinical value of CA125 in assessing congestion and guiding management in HF and CRS [146,153].
4.4.2. Bioactive Adrenomedullin (Bio-ADM) and Mid-Regional Pro-Adrenomedullin (MR-ProADM)
Adrenomedullin (ADM) is a vasodilatory peptide secreted predominantly by vascular endothelial cells in response to volume overload, inflammation, and endothelial stress. Its biologically active form, bio-ADM, directly reflects vascular integrity and endothelial stress, but it is technically challenging to measure in routine practice. The stable, inactive fragment midregional pro-adrenomedullin (MR-proADM) is more widely used in clinical research due to its stability, yet both forms reflect vascular and tissue congestion in HF [154,155].
Elevated bio-ADM levels have been shown to correlate with clinical and hemodynamic markers of congestion, such as peripheral edema, orthopnea, and elevated right atrial pressures [156,157]. Bio-ADM also correlates with pulmonary capillary wedge pressure and right atrial mean pressure. A cut-off of 34 pg/mL has been proposed in research cohorts for identifying significant congestion, but it is not guideline-endorsed (Table 2) [158]. Similarly, MR-proADM is associated with pulmonary artery pressures and inversely with pulmonary artery compliance, particularly in HFpEF [158]. MR-proADM has demonstrated superior accuracy compared to natriuretic peptides in predicting 90-day and 1-year mortality in HF cohorts [155]. Importantly, bio-ADM levels remain elevated throughout hospitalization for acute HF and are modestly predictive of mortality and rehospitalization. Combining bio-ADM with NT-proBNP may enhance prognostic precision compared to either biomarker alone [15]. The STRONG-HF trial further emphasized that reductions in bio-ADM tracked with clinical decongestion over time [156]. Beyond its biomarker role, bio-ADM is under investigation as a therapeutic target. Adrecizumab, a non-neutralizing monoclonal antibody, binds circulating bio-ADM, shifting it from the interstitium to the vasculature, potentially enhancing vascular integrity [154]. Ongoing trials are assessing its role in conditions marked by severe interstitial congestion, such as acute HF and sepsis.
In clinical practice, while bio-ADM and MR-proADM are not yet part of standard care, MR-proADM is more feasible for clinical adoption due to its stability and availability, while bio-ADM offers stronger mechanistic insight but limited accessibility. Both show promise as adjuncts for evaluating residual congestion, particularly post-discharge, where they may guide diuretic strategies and risk stratification in CRS [155].
4.4.3. Copeptin (CPP)
Copeptin is the C-terminal part of the arginine vasopressin (AVP) precursor and is released in equimolar amounts with AVP, making it a reliable surrogate marker of vasopressin activity. AVP plays a crucial role in fluid homeostasis, urine osmolarity, vasoconstriction, and stress responses, but its direct measurement is limited by its instability and short half-life. In contrast, copeptin is highly stable, easily measurable in plasma or serum, and unaffected by factors such as platelet binding or circadian rhythm, rendering it clinically practical [159,160].
The release of copeptin is triggered by osmotic, hemodynamic, and stress stimuli. As such, copeptin reflects systemic stress, neurohormonal activation, and volume status—pathophysiological hallmarks of HF, kidney disease, and the broader cardiorenal-metabolic spectrum. In healthy individuals, copeptin inversely correlates with glomerular filtration rate (GFR), and elevated levels are consistently observed in CKD, cardiovascular diseases (CVD), and metabolic disorders such as diabetes and obesity [159,161].
In HF, copeptin levels increase with disease severity, correlate with NYHA class, and have been associated with worse cardiac function, particularly in HFrEF. Studies have shown that copeptin predicts mortality and rehospitalization in acute and chronic HF, often outperforming traditional markers like NPs in short-term prognostic settings particularly among patients presenting with acute dyspnea. Copeptin also shows potential as a prognostic marker in patients with CRS, where it may reflect the compounded stress and fluid dysregulation between heart and kidney dysfunction [160,162].
Nevertheless, the clinical utility of copeptin remains somewhat limited by several factors. While it consistently correlates with adverse outcomes, its incremental prognostic value beyond established markers such as NT-proBNP is modest and inconsistent across studies. Studies have yielded inconsistent results regarding its ability to guide therapy, such as beta-blocker titration or vasopressin antagonism. Moreover, copeptin levels can rise in various non-cardiac conditions (e.g., sepsis, stroke, AKI), which may confound its interpretation in HF populations [159,160].
In summary, copeptin is a stable, easily measurable biomarker reflecting AVP-driven neurohormonal activation and systemic stress, with consistent associations with HF severity and prognosis. Its role is most promising in risk stratification and research-based multi-marker panels, but its integration into routine clinical practice is constrained by modest add-on value, limited specificity and the absence of therapeutic cut-offs [159,160].
Table 2.
Proposed cut-off values for selected biomarkers and their suggested clinical applications. This table presents biomarker thresholds derived from key clinical studies, highlighting their potential utility in guiding diagnosis, prognosis, and treatment decisions in heart failure and cardiorenal syndrome. While these values are not yet universally validated or standardized, they offer a practical framework for interpretation in appropriate clinical contexts. Bio-ADM: Bioactive adrenomedullin; CA125: Carbohydrate antigen 125; eGFR: Estimated glomerular filtration rate; HF: Heart failure; IGFBP7: Insulin-like growth factor binding protein 7; sST2: Soluble Suppression of Tumorigenicity-2; TIMP-2: Tissue inhibitor of metalloproteinases-2.
5. Micro-RNAs as Emerging Biomarkers and Therapeutic Targets in Cardiorenal Syndrome
MicroRNAs (miRNAs) are small, non-coding RNAs involved in gene regulation and have emerged as promising biomarkers and therapeutic targets in HF, kidney disease, and CRS. Their tissue specificity, stability in circulation, and close association with key pathophysiological processes—such as hypertrophy, fibrosis, inflammation, and metabolic dysfunction—highlight their clinical potential [163,164].
Among them, miR-21 is the most studied, with strong evidence linking it to cardiac and renal fibrosis through pathways like TGF-β and extracellular signal-regulated kinase (ERK)- mitogen-activated protein kinase (MAPK). Its inhibition via antisense oligonucleotides (ASOs) has shown favorable effects in preclinical models of HF, CKD, and CRS. Similarly, other miRNAs, including miR-132 and miR-92a, have been implicated in myocardial remodeling and vascular repair. Circulating miRNAs are detectable in plasma and urine, offering non-invasive diagnostic potential. Distinct miRNA profiles have been associated with HFpEF, myocardial ischemia, and inflammatory cardiomyopathy, suggesting their role in risk stratification and precision medicine [164,165].
Therapeutically, miRNA mimics and inhibitors are under investigation, with clinical trials targeting miR-21 (e.g., in renal fibrosis) and miR-132 (in HF). However, challenges such as targeted delivery, off-target effects, and long-term safety need further research before widespread clinical application. In conclusion, miRNAs represent a promising approach in CRS, offering both diagnostic and therapeutic potential, though more research is needed before they can be used in clinical practice [164,166].
6. From Bench to Bedside: Clinical Application of Biomarkers in Cardiorenal Syndrome Management
The clinical utility of biomarkers in cardiorenal syndrome (CRS) lies not in their isolated measurement, but in their integrated interpretation within specific clinical contexts. This section moves beyond individual biomarker descriptions and provides a scenario-based framework for application, highlighting comparative strengths and guiding real-world decision-making [15,51].
6.1. Navigating Key Clinical Scenarios
Scenario 1: The Inpatient with “Worsening Renal Function”—The Decongestion Dilemma
A central challenge in managing hospitalized patients with CRS is differentiating true WRF—indicating structural kidney injury—from functional, hemodynamically mediated rises in serum creatinine during decongestive therapy. This distinction is especially important in critically ill or advanced heart failure patients, in whom inappropriate treatment withdrawal may compromise outcomes, while delayed recognition of true injury may allow irreversible damage to occur [17,21,22].
Upon presentation to the emergency department or admission unit, early biomarker assessment can help establish a risk baseline. Serum Cr and cystatin C should be interpreted together, particularly in patients with advanced HF. Cystatin C has been shown to be a more accurate estimator of GFR in frail or sarcopenic individuals, where creatinine-based estimates may be misleading. Moreover, a significant discrepancy between creatinine- and cystatin C–derived eGFR may serve as an early indicator of worse prognosis, reflecting heightened neurohormonal activation and impaired renal reserve. These patients typically require careful decongestion with close pharmacological monitoring, as they are more prone to intolerance or side effects of therapy [31,32,34,167].
At this early stage, measurement of renal tubular injury biomarkers—such as plasma or urinary NGAL, urinary KIM-1, and uL-FABP—is advised. Elevated baseline levels suggest ongoing subclinical tubular damage, potentially due to venous congestion, inflammation, or ischemia. Identifying these high-risk patients allows clinicians to initiate more intensive decongestive strategies and to consider earlier initiation of guideline-directed medical therapy (GDMT), aiming to improve outcomes through more timely volume offloading and neurohormonal modulation [20,64,90,91,168].
Once decongestion is initiated, fluctuations in serum creatinine are frequently observed. The interpretation of these fluctuations must be guided by repeated clinical and biomarker assessments. Tubular biomarkers are particularly helpful in this context. A stable or only mildly fluctuating NGAL, KIM-1, or uL-FABP trajectory typically reflects functional hemodynamic changes and supports the continued use of diuretics. In contrast, a marked rise in any of these markers may have different implications depending on timing. In the early phase, it often reflects persistent congestion, indicating the need for more aggressive decongestion and hemodynamic reassessment. In later stages, it is more likely due to overdiuresis, drug-induced nephrotoxicity, or true WRF from hypoperfusion, requiring therapy adjustment, withdrawal of nephrotoxins, and in select cases, consideration of renal replacement therapy [17,81,168,169,170].
Albuminuria represents another important marker with both diagnostic and prognostic significance in this setting. As mentioned before, in HF patients, albuminuria may result from multiple mechanisms, including glomerular damage, impaired tubular reabsorption, and most notably, venous congestion and systemic endothelial dysfunction. Importantly, albuminuria levels tend to improve following decongestive therapy, which can serve as a supportive marker of treatment efficacy. For consistency, UACR is best measured from morning spot urine samples collected at a consistent time after diuretic administration [52,57,58,171].
An emerging tool for early AKI risk prediction is the NephroCheck® test, which combines TIMP-2 and IGFBP-7. These markers reflect early cellular stress and G1 cell-cycle arrest in renal tubular cells—changes that precede overt tubular damage. The test result, expressed as [TIMP-2] × [IGFBP-7] in (ng/mL)2/1000, is interpreted along a validated risk continuum. A value below 0.3 indicates a low risk of developing moderate-to-severe AKI (KDIGO stage 2–3) within the following 12–24 h. Values above 2.0 signify a high risk of imminent AKI and should prompt urgent implementation of nephroprotective strategies (hemodynamic optimization, minimization or avoidance of nephrotoxic agents, etc.). While NephroCheck is primarily validated in high-risk populations—such as ICU patients, those undergoing major cardiac surgery, or individuals with septic shock—its clinical application in advanced HF is increasingly recognized. In this context, elevated NephroCheck values may indicate the need for more aggressive decongestion and timely GDMT initiation [101,102,105].
In summary, a patient-centered approach that integrates traditional renal markers with tubular injury biomarkers (NGAL, KIM-1, uL-FABP), filtration markers (cystatin C), albuminuria, and early stress markers (NephroCheck) throughout the course of hospitalization can improve diagnostic accuracy, allow safer titration of therapies, and enhance clinical decision-making in patients with CRS and suspected WRF [64,91,99,101,168] (Table 3, Figure 1).
Table 3.
Summary of selected biomarkers in CRS: clinical utility, diagnostic strengths, limitations, and practical considerations. AKI: Acute kidney injury; bio-ADM: Bioactive adrenomedullin; CA125: Carbohydrate antigen 125; CKD: Chronic kidney disease; Gal-3: Galectin-3; GDF-15: Growth differentiation factor-15; GFR: Glomerular filtration rate; ICU: Intensive Care Unit; IGFBP7: Insulin-like growth factor binding protein 7; IL-18: Interleukin-18; KIM-1: Kidney injury molecule-1; L-FABP: Liver-type Fatty Acid Binding Protein; MR-proADM: Mid-regional pro-adrenomedullin; NT-proBNP: PENK: Proenkephalin; sST2: Soluble Suppression of Tumorigenicity-2, TIMP-2: Tissue inhibitor of metalloproteinases-2.
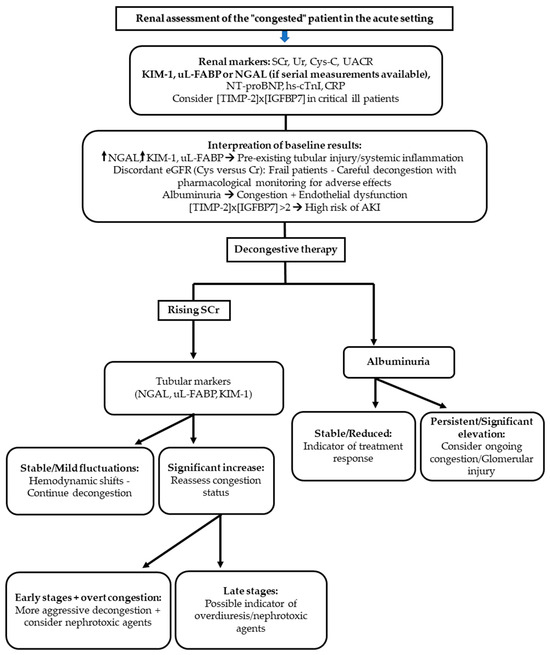
Figure 1.
Algorithm for renal assessment of the “congested” patient in the acute setting. Biomarkers complement conventional renal indices to guide diagnosis and management. Initial evaluation includes serum creatinine (SCr), urea (Ur), cystatin C (Cys-C), and urinary albumin-to-creatinine ratio (UACR), with tubular injury markers (NGAL, KIM-1, uL-FABP) and [TIMP-2] × [IGFBP7] considered in selected patients. Interpretation of baseline results enables recognition of pre-existing tubular injury, endothelial dysfunction, and risk of acute kidney injury (AKI). During decongestive therapy, trends in SCr, tubular markers, and albuminuria help differentiate hemodynamic shifts from true injury, distinguish early versus late stages of renal dysfunction, and tailor the intensity of therapy while minimizing nephrotoxic risk. AKI: acute kidney injury; CRP: C-reactive protein; Cys-C: cystatin C; hs-cTnI: high-sensitivity cardiac troponin I; KIM-1: kidney injury molecule-1; NGAL: neutrophil gelatinase-associated lipocalin; NT-proBNP: N-terminal pro-B-type natriuretic peptide; SCr: serum creatinine; TIMP-2 × IGFBP7: tissue inhibitor of metalloproteinases-2 × insulin-like growth factor binding protein-7; UACR: urinary albumin-to-creatinine ratio; Ur: urea; uL-FABP: urinary liver-type fatty acid–binding protein.
6.2. Scenario 2: The Acutely Dyspneic Patient: Diagnosing and Stratifying Congestion
In the emergency setting, a patient presenting with acute dyspnea requires rapid differentiation between cardiac and non-cardiac causes. Clinical examination remains the first step, with key findings such as orthopnea, rales, jugular venous distention, and peripheral edema pointing toward heart failure. These signs can be quickly corroborated by point-of-care ultrasound, using IVC size and collapsibility, lung B-lines, or renal venous Doppler to provide an immediate picture of intravascular and extravascular fluid overload [169,172].
At this stage, biomarkers refine the diagnostic certainty and stratify congestion. Soluble ST2 (sST2) is particularly useful to confirm that dyspnea is cardiac in origin. Low levels (<35 ng/mL) make acute HF unlikely, while values >70 ng/mL indicate marked myocardial stress and fibrosis, supporting the need for hospital admission and intensive therapy. Beyond diagnosis, a reduction of >30% in sST2 during hospitalization has been associated with improved outcomes, offering a practical therapeutic target [117].
Once acute HF is established, the focus shifts to characterizing the congestion status. Carbohydrate Antigen 125 (CA125) is a robust marker of tissue-level and serosal fluid overload, especially in right-sided HF or when natriuretic peptides are less informative due to renal dysfunction or preserved ejection fraction. Elevated levels (>35 U/mL) identify high-risk patients and, importantly, serial measurements track decongestion and recovery, guiding both inpatient therapy and follow-up intensity [146,152].
To further define systemic and vascular congestion, bio-ADM and its stable fragment MR-proADM provide insight into endothelial dysfunction and hemodynamic burden. High levels (>34 pg/mL) correlate with elevated filling pressures, persistent fluid overload, and increased mortality risk, particularly in HF with preserved EF. In cases where natriuretic peptides are inconclusive, bio-ADM adds complementary information, and when combined with NT-proBNP it strengthens both diagnostic and prognostic confidence [155,156,157].
In summary, the acutely dyspneic patient benefits from a layered, clinically integrated approach. Bedside examination and ultrasound guide the initial impression, while biomarkers provide precision: sST2 confirms the likelihood of acute HF, CA125 quantifies tissue and serosal congestion, and bio-ADM highlights vascular dysfunction. Used together, they help clinicians not only establish the diagnosis but also individualize therapy and monitoring, ensuring that treatment intensity matches the true burden of congestion (Figure 2, Table 3) [152,156].
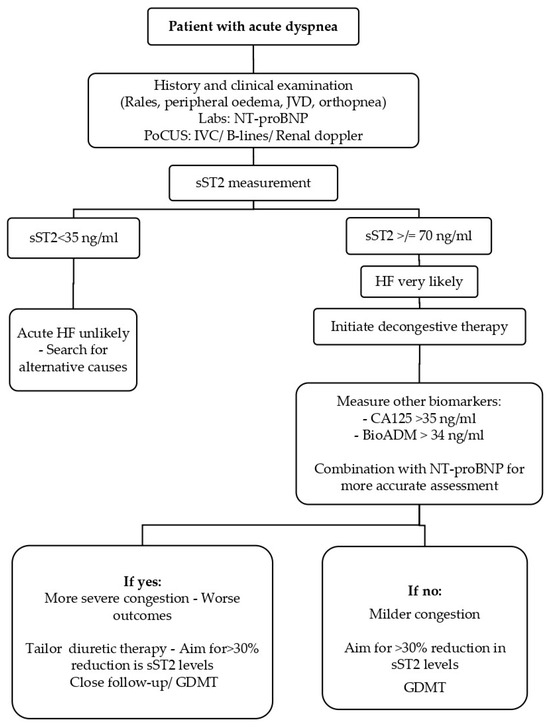
Figure 2.
Biomarker-guided algorithm for the patient with acute dyspnea. Clinical evaluation—including history, physical examination (rales, peripheral edema, jugular venous distension, orthopnea), laboratory testing (NT-proBNP), and point-of-care ultrasound (IVC, B-lines, renal Doppler)—is the first step in assessing a patient presenting with acute dyspnea. Measurement of soluble ST2 (sST2) refines the diagnostic likelihood of acute heart failure (HF): values < 35 ng/mL make HF unlikely, whereas levels ≥ 70 ng/mL indicate high probability and justify initiation of decongestive therapy. Additional biomarkers such as CA125 (>35 U/mL) and bioactive adrenomedullin (BioADM > 34 ng/mL), particularly in combination with NT-proBNP, help stratify congestion severity and prognosis. Tailoring therapy toward >30% reduction in sST2 levels, alongside guideline-directed medical therapy (GDMT), improves risk stratification and guides follow-up intensity. BioADM: bioactive adrenomedullin; CA125: carbohydrate antigen 125; GDMT: guideline-directed medical therapy; HF: heart failure; IVC: inferior vena cava; JVD: jugular venous distension; NT-proBNP: N-terminal pro-B-type natriuretic peptide; PoCUS: point-of-care ultrasound; sST2: soluble suppression of tumorigenicity-2.
6.3. Scenario 3: The Chronic Patient—Long Term Risk Stratification and Management
In stable outpatients, biomarkers help clinicians move beyond short-term decongestion and toward long-term risk assessment and tailored management. For prognostic enrichment, biomarkers reflecting inflammation such as IL-18 and GDF-15 highlight systemic processes that may drive progression of both heart and kidney failure. Their elevation should prompt not only optimization of GDMT for HF but also strict control of comorbidities, autoimmune activity, and lifestyle factors (salt, alcohol, smoking, and physical inactivity) [106,108,136,142].
Markers of fibrosis such as Galectin-3 and FGF-23 provide additional layers of information. Galectin-3 reflects cardiac and renal fibrosis, while FGF-23 has been associated with adverse ventricular remodeling and neurohormonal activation. Serial measurement of FGF-23, in particular, may offer insight into response to chronic HF therapies or device-based interventions such as cardiac resynchronization therapy [121,129,135].
Finally, proenkephalin (PENK) is emerging as a marker of neurohormonal activation and sympathetic drive. Elevated levels may support prioritizing therapies targeting the renin–angiotensin–aldosterone system, including ARNIs, ACE inhibitors/ARBs, and MRAs [47,50].
In summary, in the chronic HF outpatient, biomarkers complement clinical assessment by refining CKD classification, identifying inflammatory and fibrotic risk, and signaling neurohormonal activation. Together, they allow for a more individualized approach to long-term therapy and follow-up, ensuring that treatment intensity and monitoring are aligned with each patient’s evolving biological risk profile (Table 3).
6.4. Practical Considerations: Cost, Turnaround Time and Availability
The use of novel biomarkers in cardiorenal syndrome faces not only interpretative challenges but also practical and economic barriers. Formal cost-effectiveness analyses are almost entirely lacking, leaving a major evidence gap that hinders reimbursement and routine use [173,174].
6.4.1. Availability
CA-125 is universally accessible, and Cystatin C is increasingly routine. NephroCheck® and sST2 are FDA-cleared but remain limited to specialized centers. Most other markers (NGAL, KIM-1, L-FABP, bio-ADM, PENK) are largely confined to research with limited clinical utility [15,174].
6.4.2. Turnaround Time
NephroCheck® is rapid (<1 h), enabling point-of-care decisions [175,176]. Cystatin C and sST2 generally return results within a day, while research-based assays often take >24 h and are unsuitable for acute management [33,64,111].
6.4.3. Cost
Standard assays (CA-125, Cystatin C) are comparatively less expensive, whereas patented tests such as NephroCheck® and sST2 are considerably more costly, and their use should be justified by a demonstrable impact on patient management and outcomes (Table 4) [174,176].
Table 4.
Availability, turnaround time, and approximate cost of selected biomarkers relevant to cardiorenal syndrome. CA-125 and Cystatin C are widely available and relatively inexpensive, whereas soluble ST2 (sST2) and NephroCheck® remain more specialized, higher-cost assays with limited accessibility. Turnaround time varies from <1 h (NephroCheck®) to >24 h for many research-based assays. The table underscores the resource-dependent nature of biomarker testing, emphasizing that selection must be tailored to the clinical setting and cost justification CA-125: Carbohydrate antigen 125; ELISA: Enzyme-linked immunosorbent assay; IGFBP7: Insulin-like growth factor binding protein-7; POC: Point-of-care; sST2: Soluble suppression of tumorigenicity-2; TIMP-2: Tissue inhibitor of metalloproteinases-2; USD: United States dollars.
In summary, novel biomarkers currently fit best in tertiary care or clinical trial settings. Limited availability, variable turnaround time, and higher costs demand careful selection of context, while future research must address not only clinical validity but also economic value to enable broader adoption [15,174].
7. Conclusions and Future Perspectives
Cardiorenal syndrome (CRS) remains a major clinical challenge due to the complex and bidirectional interplay between cardiac and renal dysfunction, which conventional biomarkers like creatinine and natriuretic peptides fail to fully capture. This review has synthesized the evidence for a new generation of biomarkers that illuminate specific pathophysiological pathways—including glomerular filtration, tubular injury, congestion, inflammation, and fibrosis—providing a more nuanced understanding of CRS heterogeneity.
The clinical utility of these markers is maximized not in isolation, but when integrated into a scenario-based framework that addresses common dilemmas: differentiating structural renal injury from hemodynamically mediated creatinine changes during decongestion, unmasking tissue-level congestion in cases of diagnostic uncertainty, and refining long-term prognostic stratification. The combined application of biomarkers such as NGAL, KIM-1, uL-FABP, CA125, and sST2 offers a powerful strategy to move beyond the limitations of traditional tests, enabling more precise and personalized patient management.
However, significant barriers to routine implementation remain. These include the non-specificity of certain markers, a critical lack of robust health-economic data on cost-effectiveness, limited assay availability outside specialized centers, and an absence of standardized, context-specific cut-off values. To overcome these hurdles and translate promise into practice, future efforts must prioritize three key areas: (1) the design of prospective, biomarker-guided randomized controlled trials that test specific management algorithms against standard care; (2) the development and validation of accessible, rapid-turnaround assays suitable for point-of-care decision-making; and (3) the exploration of novel tools, such as microRNAs, for their dual diagnostic and therapeutic potential.
In conclusion, the journey from biomarker discovery to bedside application in CRS is ongoing. By embracing a multi-marker, phenotype-informed approach and addressing the extant practical and evidence-based gaps, the future management of CRS can evolve from reactive to proactive, ultimately improving outcomes for this high-risk population. While this review has focused primarily on biomarkers in acute and chronic cardiorenal syndromes, future research should also extend to renocardial processes, such as left ventricular hypertrophy in CKD and cardiac complications of dialysis, which remain underexplored.
Author Contributions
Conceptualization, G.A., M.H., E.F. and K.S.; methodology, G.A., M.B., E.L. and K.S.; investigation, G.A., M.S., M.B., I.P. and K.S.; writing—original draft preparation, G.A., M.B., M.H., I.P. and K.S.; writing—review and editing, Y.P., M.S., M.H. and K.S.; visualization, G.A., M.B. and M.S.; supervision, Y.P., E.F., E.L., M.H. and K.S.; project administration, G.A. and K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADHF | Acute decompensated heart failure |
| AF | Atrial Fibrillation |
| AKI | Acute kidney injury |
| ASOs | Antisense oligonucleotides |
| AVP | Arginine vasopressin |
| bio-ADM | Bioactive adrenomedullin |
| BNP | B-type natriuretic peptide |
| CA125 | Carbohydrate antigen 125 |
| CAD | Coronary artery disease |
| CHARM | Candesartan in Heart Failure—Assessment of Reduction in Mortality and Morbidity |
| CHART-2 | Chronic Heart Failure Analysis and Registry in the Tohoku District 2 |
| CKD | Chronic kidney disease |
| CMIA | Chemiluminescent microparticle immunoassay |
| CPP | Copeptin |
| Cr | Creatinine |
| CRS | Cardiorenal syndrome |
| CV | Cardiovascular |
| EF | Ejection fraction |
| eGFRCr | Creatinine-based estimated glomerular filtration rate |
| eGFRCys | Cystatin-based estimated glomerular filtration rate |
| ELISA | Enzyme-linked immunosorbent assay |
| ERK | Extracellular signal-regulated kinase |
| ESRD | End-Stage Renal disease |
| FDA | Food and Drug Administration |
| FGF-21 | Fibroblast Growth Factor-21 |
| FGF-23 | Fibroblast Growth Factor-23 |
| FGFRs | Fibroblast Growth Factor receptors |
| Gal-3 | Galectin-3 |
| GBM | Glomerular basement membrane |
| GDF-15 | Growth differentiation factor-15 |
| GDMT | Guideline directed medical therapy |
| GFR | Glomerular filtration rate |
| GLP-1 RA | Glucagone-like-peptide-1 receptor agonists |
| HF | Heart failure |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| Hc-cTnI | High-sensitive cardiac troponin I |
| ICU | Intensive Care Unit |
| IFN-γ | Interferon-γ |
| IGFBP7 | Insulin-like growth factor binding protein 7 |
| IL-1 | Interleukin-1 |
| IL-18 | Interleukin-18 |
| IL-18BP | Interleukin-18 binding protein |
| IL-33 | Interleukin-33 |
| IVC | Inferior vena cava |
| KCCQ | Kansas City Cardiomyopathy Questionnaire |
| KIM-1 | Kidney Injury Molecule-1 |
| L-FABP | Liver-type Fatty Acid Binding Protein |
| LVEF | Left ventricular ejection fraction |
| LVH | Left ventricular hypertrophy |
| MAPK | Mitogen-activated protein kinase |
| miRNAs | MicroRNAs |
| miR-21 | MicroRNA-21 |
| MMP-9 | Metalloproteinase-9 |
| MR-ProADM | Mid-regional pro-adrenomedullin |
| MyD88 | Myeloid differentiation factor 88 |
| NF-kB | Nuclear factor kappa B |
| NGAL | Neutrophil gelatinase associated lipocalin |
| Ns-MRAs | Non-steroidal mineralocorticoid receptors antagonists |
| NPs | Natriuretic peptides |
| NT-proBNP | N-terminal pro-B-type natriuretic peptide |
| PARADIGM-HF | Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure |
| PENK | Proenkephalin |
| PoCUS | Point of care ultrasound |
| PTH | Parathyroid hormone |
| RAAS | Renin–Angiotensin–Aldosterone System |
| SGLT2i | Sodium-glucose cotransporter-2 inhibitors |
| SNGFR | Single-nephron glomerular filtration rate |
| sST2 | Soluble Suppression of Tumorigenicity-2 |
| ΤAΤ | Turnaround time |
| TGF-β | Transforming growth factor-beta |
| TIM-1 | T-cell immunoglobulin mucin receptor 1 |
| TIMP-2 | Tissue inhibitor of metalloproteinases-2 |
| TOPCAT | Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist Trial |
| UACR | Urinary albumin to creatinine ratio |
| uL-FABP | Urinary Liver-type Fatty Acid Binding Protein |
| USD | United states dollars |
| WRF | Worsening renal function |
| β2-M | Beta-2-microglobulin |
Appendix A. Classification of Cardiorenal Syndrome
Cardiorenal syndrome (CRS) encompasses a spectrum of disorders where acute or chronic dysfunction of the heart and kidneys is closely interconnected. To facilitate clinical understanding, CRS is commonly divided into five phenotypes based on the primary affected organ, the time course, and whether a systemic process is involved. The Table A1 below summarizes these subtypes with their key features and representative clinical examples [13,14].
Table A1.
Classification and mechanisms of cardiorenal syndrome [1,2]. ACRS: Acute cardiorenal syndrome; AHF: Acute heart failure; AKI: Acute kidney injury; CKD: Chronic kidney disease, HF: Heart failure.
References
- McCullough, P.A.; Amin, A.; Pantalone, K.M.; Ronco, C. Cardiorenal Nexus: A Review with Focus on Combined Chronic Heart and Kidney Failure, and Insights From Recent Clinical Trials. J. Am. Heart Assoc. 2022, 11, e024139. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Ahmad, R.; Tantry, I.Q.; Ahmad, W.; Siddiqui, S.; Alam, M.; Abbas, K.; Moinuddin; Hassan, M.I.; Habib, S.; et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells 2024, 13, 1838. [Google Scholar] [CrossRef]
- Filippatos, G.; Anker, S.D.; Agarwal, R.; Ruilope, L.M.; Rossing, P.; Bakris, G.L.; Tasto, C.; Joseph, A.; Kolkhof, P.; Lage, A.; et al. Finerenone Reduces Risk of Incident Heart Failure in Patients with Chronic Kidney Disease and Type 2 Diabetes: Analyses From the FIGARO-DKD Trial. Circulation 2022, 145, 437–447. [Google Scholar] [CrossRef]
- Filippatos, G.; Anker, S.D.; Pitt, B.; Rossing, P.; Joseph, A.; Kolkhof, P.; Lambelet, M.; Lawatscheck, R.; Bakris, G.L.; Ruilope, L.M.; et al. Finerenone and Heart Failure Outcomes by Kidney Function/Albuminuria in Chronic Kidney Disease and Diabetes. JACC Heart Fail. 2022, 10, 860–870. [Google Scholar] [CrossRef]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication—Worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Vijay, K.; Neuen, B.L.; Lerma, E.V. Heart Failure in Patients with Diabetes and Chronic Kidney Disease: Challenges and Opportunities. Cardiorenal Med. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Lawson, C.A.; Seidu, S.; Zaccardi, F.; McCann, G.; Kadam, U.T.; Davies, M.J.; Lam, C.S.; Heerspink, H.L.; Khunti, K. Outcome trends in people with heart failure, type 2 diabetes mellitus and chronic kidney disease in the UK over twenty years. eClinicalMedicine 2021, 32, 100739. [Google Scholar] [CrossRef] [PubMed]
- Nayor, M.; Larson, M.G.; Wang, N.; Santhanakrishnan, R.; Lee, D.S.; Tsao, C.W.; Cheng, S.; Benjamin, E.J.; Vasan, R.S.; Levy, D.; et al. The association of chronic kidney disease and microalbuminuria with heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, L.S.; Katz, R.; Kestenbaum, B.; Shlipak, M.G.; Sarnak, M.J.; Stehman-Breen, C.; Seliger, S.; Siscovick, D.; Newman, A.B.; Fried, L. Chronic Kidney Disease and the Risk of End-Stage Renal Disease versus Death. J. Gen. Intern. Med. 2011, 26, 379–385. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F.; Adamczak, M.; de Oliveira, R.B.; Massy, Z.A.; Sarafidis, P.; Agarwal, R.; Mark, P.B.; Kotanko, P.; Ferro, C.J.; et al. Cardiovascular complications in chronic kidney disease: A review from the European Renal and Cardiovascular Medicine Working Group of the European Renal Association. Cardiovasc. Res. 2023, 119, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef] [PubMed]
- Mitsas, A.C.; Elzawawi, M.; Mavrogeni, S.; Boekels, M.; Khan, A.; Eldawy, M.; Stamatakis, I.; Kouris, D.; Daboul, B.; Gunkel, O.; et al. Heart Failure and Cardiorenal Syndrome: A Narrative Review on Pathophysiology, Diagnostic and Therapeutic Regimens—From a Cardiologist’s View. J. Clin. Med. 2022, 11, 7041. [Google Scholar] [CrossRef]
- Gallo, G.; Lanza, O.; Savoia, C. New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy. Int. J. Mol. Sci. 2023, 24, 5089. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. From cardiorenal to cardiovascular–kidney–metabolic syndromes. Eur. Heart J. 2025, 46, 682–684. [Google Scholar] [CrossRef]
- Mullens, W.; Damman, K.; Testani, J.M.; Martens, P.; Mueller, C.; Lassus, J.; Tang, W.H.W.; Skouri, H.; Verbrugge, F.H.; Orso, F.; et al. Evaluation of kidney function throughout the heart failure trajectory—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 584–603. [Google Scholar] [CrossRef]
- Verbrugge, F.H. Utility of Urine Biomarkers and Electrolytes for the Management of Heart Failure. Curr. Heart Fail. Rep. 2019, 16, 240–249. [Google Scholar] [CrossRef]
- Roehm, B.; McAdams, M.; Hedayati, S.S. Novel Biomarkers of Kidney Disease in Advanced Heart Failure: Beyond GFR and Proteinuria. Curr. Heart Fail. Rep. 2022, 19, 223–235. [Google Scholar] [CrossRef]
- Soni, S.S.; Cruz, D.; Bobek, I.; Chionh, C.Y.; Nalesso, F.; Lentini, P.; de Cal, M.; Corradi, V.; Virzi, G.; Ronco, C. NGAL: A biomarker of acute kidney injury and other systemic conditions. Int. Urol. Nephrol. 2010, 42, 141–150. [Google Scholar] [CrossRef]
- Metra, M.; Cotter, G.; Senger, S.; Edwards, C.; Cleland, J.G.; Ponikowski, P.; Cursack, G.C.; Milo, O.; Teerlink, J.R.; Givertz, M.M.; et al. Prognostic Significance of Creatinine Increases During an Acute Heart Failure Admission in Patients with and Without Residual Congestion: A Post Hoc Analysis of the PROTECT Data. Circ. Heart Fail. 2018, 11, e004644. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; Davison, B.; Bettari, L.; Sun, H.; Edwards, C.; Lazzarini, V.; Piovanelli, B.; Carubelli, V.; Bugatti, S.; Lombardi, C.; et al. Is worsening renal function an ominous prognostic sign in patients with acute heart failure? The role of congestion and its interaction with renal function. Circ. Heart Fail. 2012, 5, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Denic, A.; Mathew, J.; Lerman, L.O.; Lieske, J.C.; Larson, J.J.; Alexander, M.P.; Poggio, E.; Glassock, R.J.; Rule, A.D. Single-Nephron Glomerular Filtration Rate in Healthy Adults. N. Engl. J. Med. 2017, 376, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S. Redefining glomerular hyperfiltration: Pathophysiology, clinical implications, and novel perspectives. Hypertens. Res. 2025, 48, 1176–1178. [Google Scholar] [CrossRef]
- Min, K.-D.; Matsumoto, Y.; Asakura, M.; Ishihara, M. Rediscovery of the implication of albuminuria in heart failure: Emerging classic index for cardiorenal interaction. ESC Heart Fail. 2024, 11, 3470–3487. [Google Scholar] [CrossRef]
- Yoo, B.-S. Clinical Significance of B-type Natriuretic Peptide in Heart Failure. J. Lifestyle Med. 2014, 4, 34–38. [Google Scholar] [CrossRef][Green Version]
- Acute Kidney Injury: Novel Biomarkers and Potential Utility for Patient Care in Urology. Urology 2011, 77, 5–11. [CrossRef]
- Kane-Gill, S.L.; Amatullah, N.; Tran, T.; Karimzadeh, I. Contemporary considerations for nephrotoxin stewardship: Estimating kidney function and use of novel biomarkers. JACCP J. Am. Coll. Clin. Pharm. 2024, 7, 832–844. [Google Scholar] [CrossRef]
- Wu, X.; Xu, G.; Zhang, S. Association Between Cystatin C and Cardiac Function and Long-Term Prognosis in Patients with Chronic Heart Failure. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e919422-1–e919422-10. [Google Scholar] [CrossRef]
- Lassus, J.P.E.; Nieminen, M.S.; Peuhkurinen, K.; Pulkki, K.; Siirilä-Waris, K.; Sund, R.; Harjola, V.-P.; For the FINN-AKVA Study Group. Markers of renal function and acute kidney injury in acute heart failure: Definitions and impact on outcomes of the cardiorenal syndrome. Eur. Heart J. 2010, 31, 2791–2798. [Google Scholar] [CrossRef]
- Ivey-Miranda, J.B.; Inker, L.A.; Griffin, M.; Rao, V.; Maulion, C.; Turner, J.M.; Wilson, F.P.; Tang, W.H.W.; Levey, A.S.; Testani, J.M. Cystatin C and Muscle Mass in Patients with Heart Failure. J. Card. Fail. 2021, 27, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.-J.; Fu, E.L.; Sang, Y.; Ballew, S.; Evans, M.; Elinder, C.-G.; Barany, P.; Inker, L.A.; Levey, A.S.; Coresh, J.; et al. Discordances Between Creatinine- and Cystatin C–Based Estimated GFR and Adverse Clinical Outcomes in Routine Clinical Practice. Am. J. Kidney Dis. 2023, 82, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Pinsino, A.; Fabbri, M.; Braghieri, L.; Bohn, B.; Gaudig, A.J.; Kim, A.; Takeda, K.; Naka, Y.; Sayer, G.T.; Uriel, N.; et al. The difference between cystatin C- and creatinine-based assessment of kidney function in acute heart failure. ESC Heart Fail. 2022, 9, 3139–3148. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, P.; Butt, J.H.; Kondo, T.; Campo, G.; Desai, A.S.; Jhund, P.S.; Køber, L.; Lefkowitz, M.P.; Rouleau, J.L.; Solomon, S.D.; et al. Importance of cystatin C in estimating glomerular filtration rate: The PARADIGM-HF trial. Eur. Heart J. 2023, 44, 2202–2212. [Google Scholar] [CrossRef]
- Levey, A.S.; Inker, L.A. GFR as the “Gold Standard”: Estimated, Measured, and True. Am. J. Kidney Dis. 2016, 67, 9–12. [Google Scholar] [CrossRef]
- Argyropoulos, C.P.; Chen, S.S.; Ng, Y.-H.; Roumelioti, M.-E.; Shaffi, K.; Singh, P.P.; Tzamaloukas, A.H. Rediscovering Beta-2 Microglobulin As a Biomarker across the Spectrum of Kidney Diseases. Front. Med. 2017, 4, 73. [Google Scholar] [CrossRef]
- Juraschek, S.P.; Coresh, J.; Inker, L.A.; Levey, A.S.; Köttgen, A.; Foster, M.C.; Astor, B.C.; Eckfeldt, J.H.; Selvin, E. Comparison of Serum Concentrations of β-Trace Protein, β2-Microglobulin, Cystatin C, and Creatinine in the US Population. Clin. J. Am. Soc. Nephrol. 2013, 8, 584. [Google Scholar] [CrossRef]
- Inker, L.A.; Tighiouart, H.; Coresh, J.; Foster, M.C.; Anderson, A.H.; Beck, G.J.; Contreras, G.; Greene, T.; Karger, A.B.; Kusek, J.W.; et al. GFR Estimation Using β-Trace Protein and β2-Microglobulin in CKD. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2016, 67, 40–48. [Google Scholar] [CrossRef]
- Koh, E.S.; Lee, K.; Kim, S.H.; Kim, Y.O.; Jin, D.C.; Song, H.C.; Choi, E.J.; Kim, Y.L.; Kim, Y.S.; Kang, S.W.; et al. Serum β2-Microglobulin Predicts Mortality in Peritoneal Dialysis Patients: A Prospective Cohort Study. Am. J. Nephrol. 2015, 42, 91–98. [Google Scholar] [CrossRef]
- Foster, M.C.; Coresh, J.; Hsu, C.-Y.; Xie, D.; Levey, A.S.; Nelson, R.G.; Eckfeldt, J.H.; Vasan, R.S.; Kimmel, P.L.; Schelling, J.; et al. Serum β-Trace Protein and β2-Microglobulin as Predictors of ESRD, Mortality, and Cardiovascular Disease in Adults with CKD in the Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2016, 68, 68–76. [Google Scholar] [CrossRef]
- Vianello, A.; Caponi, L.; Galetta, F.; Franzoni, F.; Taddei, M.; Rossi, M.; Pietrini, P.; Santoro, G. β2-Microglobulin and TIMP1 Are Linked Together in Cardiorenal Remodeling and Failure. Cardiorenal Med. 2015, 5, 369260. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Lenglet, A.; Desjardins, L.; Neirynck, N.; Glorieux, G.; Lemke, H.-D.; Vanholder, R.; Diouf, M.; Choukroun, G.; Massy, Z.A.; et al. Plasma beta-2 microglobulin is associated with cardiovascular disease in uremic patients. Kidney Int. 2012, 82, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Portales-Castillo, I.; Yee, J.; Tanaka, H.; Fenves, A.Z. Beta-2 Microglobulin Amyloidosis: Past, Present, and Future. Kidney360 2020, 1, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Emmens, J.E.; ter Maaten, J.M.; Damman, K.; van Veldhuisen, D.J.; de Boer, R.A.; Struck, J.; Bergmann, A.; Sama, I.E.; Streng, K.W.; Anker, S.D.; et al. Proenkephalin, an Opioid System Surrogate, as a Novel Comprehensive Renal Marker in Heart Failure. Circ. Heart Fail. 2019, 12, e005544. [Google Scholar] [CrossRef] [PubMed]
- Siranart, N.; Laohasurayotin, K.; Phanthong, T.; Sowalertrat, W.; Ariyachaipanich, A.; Chokesuwattanaskul, R. Proenkephalin as a Novel Prognostic Marker in Heart Failure Patients: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2023, 24, 4887. [Google Scholar] [CrossRef]
- Khorashadi, M.; Beunders, R.; Pickkers, P.; Legrand, M. Proenkephalin: A New Biomarker for Glomerular Filtration Rate and Acute Kidney Injury. Nephron 2020, 144, 655–661. [Google Scholar] [CrossRef]
- Ng, L.L.; Squire, I.B.; Jones, D.J.L.; Cao, T.H.; Chan, D.C.S.; Sandhu, J.K.; Quinn, P.A.; Davies, J.E.; Struck, J.; Hartmann, O.; et al. Proenkephalin, Renal Dysfunction, and Prognosis in Patients with Acute Heart Failure. JACC 2017, 69, 56–69. [Google Scholar] [CrossRef]
- Lin, L.-C.; Chuan, M.-H.; Liu, J.-H.; Liao, H.-W.; Ng, L.L.; Magnusson, M.; Jujic, A.; Pan, H.-C.; Wu, V.-C.; Forni, L.G. Proenkephalin as a biomarker correlates with acute kidney injury: A systematic review with meta-analysis and trial sequential analysis. Crit. Care 2023, 27, 481. [Google Scholar] [CrossRef]
- Wenzl, F.A.; Wang, P.; Arrigo, M.; Parenica, J.; Jones, D.J.L.; Bruno, F.; Tarnowski, D.; Hartmann, O.; Boucek, L.; Lang, F.; et al. Proenkephalin improves cardio-renal risk prediction in acute coronary syndromes: The KID-ACS score. Eur. Heart J. 2025, 46, 38–54. [Google Scholar] [CrossRef]
- Matsiras, D.; Ventoulis, I.; Verras, C.; Bistola, V.; Bezati, S.; Fyntanidou, B.; Polyzogopoulou, E.; Parissis, J.T. Proenkephalin 119–159 in Heart Failure: From Pathophysiology to Clinical Implications. J. Clin. Med. 2025, 14, 2657. [Google Scholar] [CrossRef]
- Chung, E.Y.M.; Trinh, K.; Li, J.; Hahn, S.H.; Endre, Z.H.; Rogers, N.M.; Alexander, S.I. Biomarkers in Cardiorenal Syndrome and Potential Insights Into Novel Therapeutics. Front. Cardiovasc. Med. 2022, 9, 868658. [Google Scholar] [CrossRef]
- Barzilay, J.I.; Farag, Y.M.K.; Durthaler, J. Albuminuria: An Underappreciated Risk Factor for Cardiovascular Disease. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2024, 13, e030131. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.E.; Solomon, S.D.; Gerstein, H.C.; Zetterstrand, S.; Olofsson, B.; Michelson, E.L.; Granger, C.B.; Swedberg, K.; Pfeffer, M.A.; Yusuf, S.; et al. Albuminuria in chronic heart failure: Prevalence and prognostic importance. Lancet Lond. Engl. 2009, 374, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Masson, S.; Latini, R.; Milani, V.; Moretti, L.; Rossi, M.G.; Carbonieri, E.; Frisinghelli, A.; Minneci, C.; Valisi, M.; Maggioni, A.P.; et al. Prevalence and prognostic value of elevated urinary albumin excretion in patients with chronic heart failure: Data from the GISSI-Heart Failure trial. Circ. Heart Fail. 2010, 3, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Solano, A.; Lew, S.Q.; Ing, T.S. Dent–Wrong disease and other rare causes of the Fanconi syndrome. Clin. Kidney J. 2014, 7, 344–347. [Google Scholar] [CrossRef][Green Version]
- Raja, P.; Maxwell, A.P.; Brazil, D.P. The Potential of Albuminuria as a Biomarker of Diabetic Complications. Cardiovasc. Drugs Ther. 2021, 35, 455–466. [Google Scholar] [CrossRef]
- Khan, M.S.; Shahid, I.; Anker, S.D.; Fonarow, G.C.; Fudim, M.; Hall, M.E.; Hernandez, A.; Morris, A.A.; Shafi, T.; Weir, M.R.; et al. Albuminuria and Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2023, 81, 270–282. [Google Scholar] [CrossRef]
- Boorsma, E.M.; ter Maaten, J.M.; Damman, K.; van Essen, B.J.; Zannad, F.; van Veldhuisen, D.J.; Samani, N.J.; Dickstein, K.; Metra, M.; Filippatos, G.; et al. Albuminuria as a marker of systemic congestion in patients with heart failure. Eur. Heart J. 2023, 44, 368–380. [Google Scholar] [CrossRef]
- Blecker, S.; Matsushita, K.; Köttgen, A.; Loehr, L.R.; Bertoni, A.G.; Boulware, L.E.; Coresh, J. High-Normal Albuminuria and Risk of Heart Failure in the Community. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2011, 58, 47–55. [Google Scholar] [CrossRef]
- Tsuji, K.; Sakata, Y.; Nochioka, K.; Miura, M.; Yamauchi, T.; Onose, T.; Abe, R.; Oikawa, T.; Kasahara, S.; Sato, M.; et al. Characterization of heart failure patients with mid-range left ventricular ejection fraction-a report from the CHART-2 Study. Eur. J. Heart Fail. 2017, 19, 1258–1269. [Google Scholar] [CrossRef]
- Ingelsson, E.; Sundström, J.; Lind, L.; Risérus, U.; Larsson, A.; Basu, S.; Arnlöv, J. Low-grade albuminuria and the incidence of heart failure in a community-based cohort of elderly men. Eur. Heart J. 2007, 28, 1739–1745. [Google Scholar] [CrossRef]
- Selvaraj, S.; Claggett, B.; Shah, S.J.; Anand, I.; Rouleau, J.L.; O’Meara, E.; Desai, A.S.; Lewis, E.F.; Pitt, B.; Sweitzer, N.K.; et al. Prognostic Value of Albuminuria and Influence of Spironolactone in Heart Failure with Preserved Ejection Fraction. Circ. Heart Fail. 2018, 11, e005288. [Google Scholar] [CrossRef]
- Koyama, S.; Sato, Y.; Tanada, Y.; Fujiwara, H.; Takatsu, Y. Early evolution and correlates of urine albumin excretion in patients presenting with acutely decompensated heart failure. Circ. Heart Fail. 2013, 6, 227–232. [Google Scholar] [CrossRef]
- Romejko, K.; Markowska, M.; Niemczyk, S. The Review of Current Knowledge on Neutrophil Gelatinase-Associated Lipocalin (NGAL). Int. J. Mol. Sci. 2023, 24, 10470. [Google Scholar] [CrossRef]
- Filiopoulos, V.; Biblaki, D.; Vlassopoulos, D. Neutrophil gelatinase-associated lipocalin (NGAL): A promising biomarker of contrast-induced nephropathy after computed tomography. Ren. Fail. 2014, 36, 979–986. [Google Scholar] [CrossRef]
- Yuen, P.S.T.; Jo, S.-K.; Holly, M.K.; Hu, X.; Star, R.A. Ischemic and Nephrotoxic Acute Renal Failure are Distinguished by their Broad Transcriptomic Responses (102/160 char). Physiol. Genomics 2006, 25, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, B.; Malyszko, J.; Bachorzewska-Gajewska, H.; Malyszko, J.S.; Dobrzycki, S. Serum neutrophil gelatinase-associated lipocalin as a marker of renal function in patients with chronic heart failure and coronary artery disease. Kidney Blood Press. Res. 2009, 32, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; van Veldhuisen, D.J.; Navis, G.; Voors, A.A.; Hillege, H.L. Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur. J. Heart Fail. 2008, 10, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; Masson, S.; Hillege, H.L.; Maggioni, A.P.; Voors, A.A.; Opasich, C.; van Veldhuisen, D.J.; Montagna, L.; Cosmi, F.; Tognoni, G.; et al. Clinical outcome of renal tubular damage in chronic heart failure†. Eur. Heart J. 2011, 32, 2705–2712. [Google Scholar] [CrossRef]
- Shrestha, K.; Shao, Z.; Singh, D.; Dupont, M.; Tang, W.H.W. Relation of systemic and urinary neutrophil gelatinase-associated lipocalin levels to different aspects of impaired renal function in patients with acute decompensated heart failure. Am. J. Cardiol. 2012, 110, 1329–1335. [Google Scholar] [CrossRef]
- Zhang, Y.; He, D.; Zhang, W.; Xing, Y.; Guo, Y.; Wang, F.; Jia, J.; Yan, T.; Liu, Y.; Lin, S. ACE Inhibitor Benefit to Kidney and Cardiovascular Outcomes for Patients with Non-Dialysis Chronic Kidney Disease Stages 3–5: A Network Meta-Analysis of Randomised Clinical Trials. Drugs 2020, 80, 797–811. [Google Scholar] [CrossRef]
- Testani, J.M.; Tang, W.H.W. Biomarkers of Acute Kidney Injury in Chronic Heart Failure. JACC Heart Fail. 2013, 1, 425–426. [Google Scholar] [CrossRef]
- Nasonova, S.N.; Zhirov, I.V.; Ledyakhova, M.V.; Sharf, T.V.; Bosykh, E.G.; Masenko, V.P.; Tereshchenko, S.N. Early diagnosis of acute renal injury in patients with acute decompensation of chronic heart failure. Ter. Arkh. 2019, 91, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Buonafine, M.; Martinez-Martinez, E.; Jaisser, F. More than a simple biomarker: The role of NGAL in cardiovascular and renal diseases. Clin. Sci. 2018, 132, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin: A promising biomarker for human acute kidney injury. Biomark. Med. 2010, 4, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Brozat, J.F.; Harbalioğlu, N.; Hohlstein, P.; Abu Jhaisha, S.; Pollmanns, M.R.; Adams, J.K.; Wirtz, T.H.; Hamesch, K.; Yagmur, E.; Weiskirchen, R.; et al. Elevated Serum KIM-1 in Sepsis Correlates with Kidney Dysfunction and the Severity of Multi-Organ Critical Illness. Int. J. Mol. Sci. 2024, 25, 5819. [Google Scholar] [CrossRef]
- Goffredo, G.; Barone, R.; Di Terlizzi, V.; Correale, M.; Brunetti, N.D.; Iacoviello, M. Biomarkers in Cardiorenal Syndrome. J. Clin. Med. 2021, 10, 3433. [Google Scholar] [CrossRef]
- Fu, K.; Hu, Y.; Zhang, H.; Wang, C.; Lin, Z.; Lu, H.; Ji, X. Insights of Worsening Renal Function in Type 1 Cardiorenal Syndrome: From the Pathogenesis, Biomarkers to Treatment. Front. Cardiovasc. Med. 2021, 8, 760152. [Google Scholar] [CrossRef]
- Yeung, M.Y.; Ding, Q.; Brooks, C.R.; Xiao, S.; Workman, C.J.; Vignali, D.A.A.; Ueno, T.; Padera, R.F.; Kuchroo, V.K.; Najafian, N.; et al. TIM-1 signaling is required for Maintenance and Induction of regulatory B cells. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2015, 15, 942–953. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Radu, S.; Costea, C.F.; Ciocoiu, M.; Carauleanu, A.; Lacatusu, C.M.; Maranduca, M.A.; Floria, M.; Rezus, C. The Predictive Role of the Biomarker Kidney Molecule-1 (KIM-1) in Acute Kidney Injury (AKI) Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 2019, 20, 5238. [Google Scholar] [CrossRef]
- Coca, S.G.; Yalavarthy, R.; Concato, J.; Parikh, C.R. Biomarkers for the diagnosis and risk stratification of acute kidney injury: A systematic review. Kidney Int. 2008, 73, 1008–1016. [Google Scholar] [CrossRef]
- Lim, A.I.; Tang, S.C.W.; Lai, K.N.; Leung, J.C.K. Kidney injury molecule-1: More than just an injury marker of tubular epithelial cells? J. Cell. Physiol. 2013, 228, 917–924. [Google Scholar] [CrossRef]
- Sokolski, M.; Zymliński, R.; Biegus, J.; Siwołowski, P.; Nawrocka-Millward, S.; Todd, J.; Yerramilli, M.R.; Estis, J.; Jankowska, E.A.; Banasiak, W.; et al. Urinary levels of novel kidney biomarkers and risk of true worsening renal function and mortality in patients with acute heart failure. Eur. J. Heart Fail. 2017, 19, 760–767. [Google Scholar] [CrossRef]
- Ahmad, T.; Jackson, K.; Rao, V.S.; Tang, W.H.W.; Brisco-Bacik, M.A.; Chen, H.H.; Felker, G.M.; Hernandez, A.F.; O’Connor, C.M.; Sabbisetti, V.S.; et al. Worsening Renal Function in Patients with Acute Heart Failure Undergoing Aggressive Diuresis Is Not Associated with Tubular Injury. Circulation 2018, 137, 2016–2028. [Google Scholar] [CrossRef]
- Legrand, M.; Berardinis, B.D.; Gaggin, H.K.; Magrini, L.; Belcher, A.; Zancla, B.; Femia, A.; Simon, M.; Motiwala, S.; Sambhare, R.; et al. Evidence of Uncoupling between Renal Dysfunction and Injury in Cardiorenal Syndrome: Insights from the BIONICS Study. PLoS ONE 2014, 9, e112313. [Google Scholar] [CrossRef]
- Grodin, J.L.; Perez, A.L.; Wu, Y.; Hernandez, A.F.; Butler, J.; Metra, M.; Felker, G.M.; Voors, A.A.; McMurray, J.J.; Armstrong, P.W.; et al. Circulating Kidney Injury Molecule-1 Levels in Acute Heart Failure: Insights From the ASCEND-HF Trial (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure). JACC Heart Fail. 2015, 3, 777–785. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Siribamrungwong, M.; Doi, K.; Noiri, E.; Terrin, N.; Jaber, B.L. Performance of urinary liver-type fatty acid-binding protein in acute kidney injury: A meta-analysis. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2013, 61, 430–439. [Google Scholar] [CrossRef]
- Mitsides, N.; Mitra, V.; Saha, A.; Harris, S.; Kalra, P.A.; Mitra, S. Urinary Liver-Type Fatty Acid Binding Protein, a Biomarker for Disease Progression, Dialysis and Overall Mortality in Chronic Kidney Disease. J. Pers. Med. 2023, 13, 1481. [Google Scholar] [CrossRef]
- Sunayama, T.; Yatsu, S.; Matsue, Y.; Dotare, T.; Maeda, D.; Ishiwata, S.; Nakamura, Y.; Suda, S.; Kato, T.; Hiki, M.; et al. Urinary liver-type fatty acid-binding protein as a prognostic marker in patients with acute heart failure. ESC Heart Fail. 2022, 9, 442–449. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, Y.; Shao, X.; Ni, Z.; Mou, S. L-FABP: A novel biomarker of kidney disease. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 445, 85–90. [Google Scholar] [CrossRef]
- Hishikari, K.; Hikita, H.; Nakamura, S.; Nakagama, S.; Mizusawa, M.; Yamamoto, T.; Doi, J.; Hayashi, Y.; Utsugi, Y.; Araki, M.; et al. Urinary Liver-Type Fatty Acid-Binding Protein Level as a Predictive Biomarker of Acute Kidney Injury in Patients with Acute Decompensated Heart Failure. Cardiorenal Med. 2017, 7, 267–275. [Google Scholar] [CrossRef]
- Kamijo-Ikemori, A.; Sugaya, T.; Yasuda, T.; Kawata, T.; Ota, A.; Tatsunami, S.; Kaise, R.; Ishimitsu, T.; Tanaka, Y.; Kimura, K. Clinical Significance of Urinary Liver-Type Fatty Acid–Binding Protein in Diabetic Nephropathy of Type 2 Diabetic Patients. Diabetes Care 2011, 34, 691–696. [Google Scholar] [CrossRef]
- Kubota, Y.; Higashiyama, A.; Marumo, M.; Konishi, M.; Yamashita, Y.; Okamura, T.; Miyamoto, Y.; Wakabayashi, I. Relationship of urinary liver-type fatty acid-binding protein with cardiovascular risk factors in the Japanese population without chronic kidney disease: Sasayama study. BMC Nephrol. 2021, 22, 189. [Google Scholar] [CrossRef]
- Thi, T.N.D.; Gia, B.N.; Thi, H.L.L.; Thi, T.N.C.; Thanh, H.P. Evaluation of urinary L-FABP as an early marker for diabetic nephropathy in type 2 diabetic patients. J. Med. Biochem. 2020, 39, 224–230. [Google Scholar] [CrossRef]
- Kamijo-Ikemori, A.; Sugaya, T.; Ichikawa, D.; Hoshino, S.; Matsui, K.; Yokoyama, T.; Yasuda, T.; Hirata, K.; Kimura, K. Urinary liver type fatty acid binding protein in diabetic nephropathy. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 424, 104–108. [Google Scholar] [CrossRef]
- Kamijo, A.; Sugaya, T.; Hikawa, A.; Yamanouchi, M.; Hirata, Y.; Ishimitsu, T.; Numabe, A.; Takagi, M.; Hayakawa, H.; Tabei, F.; et al. Urinary liver-type fatty acid binding protein as a useful biomarker in chronic kidney disease. Mol. Cell. Biochem. 2006, 284, 175–182. [Google Scholar] [CrossRef]
- Tanaka, T.; Doi, K.; Maeda-Mamiya, R.; Negishi, K.; Portilla, D.; Sugaya, T.; Fujita, T.; Noiri, E. Urinary L-type fatty acid-binding protein can reflect renal tubulointerstitial injury. Am. J. Pathol. 2009, 174, 1203–1211. [Google Scholar] [CrossRef]
- Naruse, H.; Ishii, J.; Takahashi, H.; Kitagawa, F.; Nishimura, H.; Kawai, H.; Muramatsu, T.; Harada, M.; Yamada, A.; Motoyama, S.; et al. Predicting acute kidney injury using urinary liver-type fatty-acid binding protein and serum N-terminal pro-B-type natriuretic peptide levels in patients treated at medical cardiac intensive care units. Crit. Care 2018, 22, 197. [Google Scholar] [CrossRef]
- Delrue, C.; Speeckaert, M.M. Tissue Inhibitor of Metalloproteinases-2 (TIMP-2) as a Prognostic Biomarker in Acute Kidney Injury: A Narrative Review. Diagnostics 2024, 14, 1350. [Google Scholar] [CrossRef]
- Daubin, D.; Cristol, J.P.; Dupuy, A.M.; Kuster, N.; Besnard, N.; Platon, L.; Buzançais, A.; Brunot, V.; Garnier, F.; Jonquet, O.; et al. Urinary Biomarkers IGFBP7 and TIMP-2 for the Diagnostic Assessment of Transient and Persistent Acute Kidney Injury in Critically Ill Patients. PLoS ONE 2017, 12, e0169674. [Google Scholar] [CrossRef]
- Guzzi, L.M.; Bergler, T.; Binnall, B.; Engelman, D.T.; Forni, L.; Germain, M.J.; Gluck, E.; Göcze, I.; Joannidis, M.; Koyner, J.L.; et al. Clinical use of [TIMP-2]•[IGFBP7] biomarker testing to assess risk of acute kidney injury in critical care: Guidance from an expert panel. Crit. Care 2019, 23, 225. [Google Scholar] [CrossRef]
- Doukas, P.; Frese, J.P.; Eierhoff, T.; Hellfritsch, G.; Raude, B.; Jacobs, M.J.; Greiner, A.; Oberhuber, A.; Gombert, A. The NephroCheck bedside system for detecting stage 3 acute kidney injury after open thoracoabdominal aortic repair. Sci. Rep. 2023, 13, 11096. [Google Scholar] [CrossRef]
- Clinical adoption of Nephrocheck® in the early detection of acute kidney injury—Godi Ilaria, Kashani Kianoush, Boteanu Ruxandra, Martino Francesca, Carta Mariarosa, Giavarina Davide, Ronco Claudio. 2021. Available online: https://journals.sagepub.com/doi/full/10.1177/0004563220970032 (accessed on 18 August 2025).
- Atici, A.; Emet, S.; Cakmak, R.; Yuruyen, G.; Alibeyoglu, A.; Akarsu, M.; Arman, Y.; Kose, M.; Ozgun, E.; Ozcan, M.; et al. Type I cardiorenal syndrome in patients with acutely decompensated heart failure: The importance of new renal biomarkers. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3534–3543. [Google Scholar] [CrossRef]
- Schanz, M.; Shi, J.; Wasser, C.; Alscher, M.D.; Kimmel, M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin. Cardiol. 2017, 40, 485–491. [Google Scholar] [CrossRef]
- Hirooka, Y.; Nozaki, Y. Interleukin-18 in Inflammatory Kidney Disease. Front. Med. 2021, 8, 639103. [Google Scholar] [CrossRef]
- Lin, X.; Yuan, J.; Zhao, Y.; Zha, Y. Urine interleukin-18 in prediction of acute kidney injury: A systemic review and meta-analysis. J. Nephrol. 2015, 28, 7–16. [Google Scholar] [CrossRef]
- O’Brien, L.C.; Mezzaroma, E.; Van Tassell, B.W.; Marchetti, C.; Carbone, S.; Abbate, A.; Toldo, S. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol. Med. Camb. Mass 2014, 20, 221–229. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, W.; Zhang, J.; Xu, C.; Yu, S.; Mao, Z.; Wu, J.; Ye, C.; Mei, C.; Dai, B. Urinary interleukin 18 for detection of acute kidney injury: A meta-analysis. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2013, 62, 1058–1067. [Google Scholar] [CrossRef]
- Łagosz, P.; Biegus, J.; Urban, S.; Zymliński, R. Renal Assessment in Acute Cardiorenal Syndrome. Biomolecules 2023, 13, 239. [Google Scholar] [CrossRef]
- Bi, J.; Garg, V.; Yates, A.R. Galectin-3 and sST2 as Prognosticators for Heart Failure Requiring Extracorporeal Life Support: Jack n’ Jill. Biomolecules 2021, 11, 166. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wu, Y.; Grodin, J.L.; Hsu, A.P.; Hernandez, A.F.; Butler, J.; Metra, M.; Voors, A.A.; Felker, G.M.; Troughton, R.W.; et al. Prognostic Value of Baseline and Changes in Circulating Soluble ST2 Levels and the Effects of Nesiritide in Acute Decompensated Heart Failure. JACC Heart Fail. 2016, 4, 68–77. [Google Scholar] [CrossRef]
- Merino-Merino, A.; Gonzalez-Bernal, J.; Fernandez-Zoppino, D.; Saez-Maleta, R.; Perez-Rivera, J.-A. The Role of Galectin-3 and ST2 in Cardiology: A Short Review. Biomolecules 2021, 11, 1167. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; McCloskey, K.; Rame, J.E.; McIntosh, E.; Shahi, P.; Dries, D.L.; Tang, W.H.W.; Wu, A.H.B.; Fang, J.C.; et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ. Heart Fail. 2011, 4, 180–187. [Google Scholar] [CrossRef]
- Shah, R.V.; Chen-Tournoux, A.A.; Picard, M.H.; van Kimmenade, R.R.J.; Januzzi, J.L. Serum levels of the interleukin-1 receptor family member ST2, cardiac structure and function, and long-term mortality in patients with acute dyspnea. Circ. Heart Fail. 2009, 2, 311–319. [Google Scholar] [CrossRef]
- Aleksova, A.; Paldino, A.; Beltrami, A.P.; Padoan, L.; Iacoviello, M.; Sinagra, G.; Emdin, M.; Maisel, A.S. Cardiac Biomarkers in the Emergency Department: The Role of Soluble ST2 (sST2) in Acute Heart Failure and Acute Coronary Syndrome—There is Meat on the Bone. J. Clin. Med. 2019, 8, 270. [Google Scholar] [CrossRef]
- Januzzi, J.L.; Mebazaa, A.; Di Somma, S. ST2 and Prognosis in Acutely Decompensated Heart Failure: The International ST2 Consensus Panel. Am. J. Cardiol. 2015, 115, 26B–31B. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Zamora, E.; de Antonio, M.; Galán, A.; Vila, J.; Urrutia, A.; Díez, C.; Coll, R.; Altimir, S.; Lupón, J. Soluble ST2 serum concentration and renal function in heart failure. J. Card. Fail. 2013, 19, 768–775. [Google Scholar] [CrossRef]
- Aimo, A.; Vergaro, G.; Ripoli, A.; Bayes-Genis, A.; Pascual Figal, D.A.; de Boer, R.A.; Lassus, J.; Mebazaa, A.; Gayat, E.; Breidthardt, T.; et al. Meta-Analysis of Soluble Suppression of Tumorigenicity-2 and Prognosis in Acute Heart Failure. JACC Heart Fail. 2017, 5, 287–296. [Google Scholar] [CrossRef]
- Boutin, L.; Dépret, F.; Gayat, E.; Legrand, M.; Chadjichristos, C.E. Galectin-3 in Kidney Diseases: From an Old Protein to a New Therapeutic Target. Int. J. Mol. Sci. 2022, 23, 3124. [Google Scholar] [CrossRef]
- Caravaca Perez, P.; González-Juanatey, J.R.; Nuche, J.; Matute-Blanco, L.; Serrano, I.; Martínez Selles, M.; Vázquez García, R.; Martínez Dolz, L.; Gómez-Bueno, M.; Pascual Figal, D.; et al. Renal Function Impact in the Prognostic Value of Galectin-3 in Acute Heart Failure. Front. Cardiovasc. Med. 2022, 9, 861651. [Google Scholar] [CrossRef]
- Zamora, E.; Lupón, J.; de Antonio, M.; Galán, A.; Domingo, M.; Urrutia, A.; Troya, M.; Bayes-Genis, A. Renal function largely influences Galectin-3 prognostic value in heart failure. Int. J. Cardiol. 2014, 177, 171–177. [Google Scholar] [CrossRef] [PubMed]
- van Vark, L.C.; Lesman-Leegte, I.; Baart, S.J.; Postmus, D.; Pinto, Y.M.; de Boer, R.A.; Asselbergs, F.W.; Wajon, E.M.C.J.; Orsel, J.G.; Boersma, E.; et al. Prognostic Value of Serial Galectin-3 Measurements in Patients with Acute Heart Failure. J. Am. Heart Assoc. 2017, 6, e003700. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Quarles, L.D. The Role of Fibroblast Growth Factor-23 in Cardiorenal Syndrome. Nephron Clin. Pract. 2013, 123, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Navarro-García, J.A.; González-Lafuente, L.; Fernández-Velasco, M.; Ruilope, L.M.; Ruiz-Hurtado, G. Fibroblast Growth Factor-23-Klotho Axis in Cardiorenal Syndrome: Mediators and Potential Therapeutic Targets. Front. Physiol. 2021, 12, 775029. [Google Scholar] [CrossRef]
- Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; Pendon-Ruiz de Mier, M.V.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins 2020, 12, 185. [Google Scholar] [CrossRef]
- Leidner, A.S.; Cai, X.; Zelnick, L.R.; Lee, J.; Bansal, N.; Pasch, A.; Kansal, M.; Chen, J.; Anderson, A.H.; Sondheimer, J.H.; et al. Fibroblast Growth Factor 23 and Risk of Heart Failure Subtype: The CRIC (Chronic Renal Insufficiency Cohort) Study. Kidney Med. 2023, 5, 100723. [Google Scholar] [CrossRef]
- Kurpas, A.; Supeł, K.; Idzikowska, K.; Zielińska, M. FGF23: A Review of Its Role in Mineral Metabolism and Renal and Cardiovascular Disease. Dis. Markers 2021, 2021, 8821292. [Google Scholar] [CrossRef]
- Ivey-Miranda, J.B.; Stewart, B.; Cox, Z.L.; McCallum, W.; Maulion, C.; Gleason, O.; Meegan, G.; Amatruda, J.G.; Moreno-Villagomez, J.; Mahoney, D.; et al. FGF-23 (Fibroblast Growth Factor-23) and Cardiorenal Interactions. Circ. Heart Fail. 2021, 14, e008385. [Google Scholar] [CrossRef]
- Moe, S.M.; Chertow, G.M.; Parfrey, P.S.; Kubo, Y.; Block, G.A.; Correa-Rotter, R.; Drüeke, T.B.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis. Circulation 2015, 132, 27–39. [Google Scholar] [CrossRef]
- Dörr, K.; Kammer, M.; Reindl-Schwaighofer, R.; Lorenz, M.; Prikoszovich, T.; Marculescu, R.; Beitzke, D.; Wielandner, A.; Erben, R.G.; Oberbauer, R. Randomized Trial of Etelcalcetide for Cardiac Hypertrophy in Hemodialysis. Circ. Res. 2021, 128, 1616–1625. [Google Scholar] [CrossRef]
- Kaur, N.; Gare, S.R.; Shen, J.; Raja, R.; Fonseka, O.; Liu, W. Multi-organ FGF21-FGFR1 signaling in metabolic health and disease. Front. Cardiovasc. Med. 2022, 9, 962561. [Google Scholar] [CrossRef]
- Prida, E.; Álvarez-Delgado, S.; Pérez-Lois, R.; Soto-Tielas, M.; Estany-Gestal, A.; Fernø, J.; Seoane, L.M.; Quiñones, M.; Al-Massadi, O. Liver Brain Interactions: Focus on FGF21 a Systematic Review. Int. J. Mol. Sci. 2022, 23, 13318. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Yuan, L.; Yang, F.; Wu, G.; Jiang, X. Emerging roles of fibroblast growth factor 21 in critical disease. Front. Cardiovasc. Med. 2022, 9, 1053997. [Google Scholar] [CrossRef] [PubMed]
- Płonka, J.; Olejnik, A.; Klus, A.; Gawrylak-Dryja, E.; Wężyk, N.; Rzepiela, L.; Dąbrowska, K.; Nalewajko, K.; Porażko, T.; Bil-Lula, I.; et al. Use of FGF-23 and sαKlotho for Risk Stratification in Patients with Acute Heart Failure. J. Clin. Med. 2025, 14, 860. [Google Scholar] [CrossRef] [PubMed]
- Lasaad, S.; Crambert, G. GDF15, an Emerging Player in Renal Physiology and Pathophysiology. Int. J. Mol. Sci. 2024, 25, 5956. [Google Scholar] [CrossRef]
- Płonka, J.; Klus, A.; Wężyk, N.; Dąbrowska, K.; Rzepiela, L.; Gawrylak-Dryja, E.; Nalewajko, K.; Feusette, P.; Gierlotka, M. Combined Use of GDF-15 and NT-Pro BNP for Outcome Prediction in Patients with Acute Heart Failure. J. Clin. Med. 2024, 13, 5936. [Google Scholar] [CrossRef]
- di Candia, A.M.; de Avila, D.X.; Moreira, G.R.; Villacorta, H.; Maisel, A.S. Growth differentiation factor-15, a novel systemic biomarker of oxidative stress, inflammation, and cellular aging: Potential role in cardiovascular diseases. Am. Heart J. Plus Cardiol. Res. Pract. 2021, 9, 100046. [Google Scholar] [CrossRef]
- Valiño-Rivas, L.; Cuarental, L.; Ceballos, M.I.; Pintor-Chocano, A.; Perez-Gomez, M.V.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Growth differentiation factor-15 preserves Klotho expression in acute kidney injury and kidney fibrosis. Kidney Int. 2022, 101, 1200–1215. [Google Scholar] [CrossRef]
- Bao, X.; Xu, B.; Borné, Y.; Orho-Melander, M.; Melander, O.; Nilsson, J.; Christensson, A.; Engström, G. Growth differentiation factor-15 and incident chronic kidney disease: A population-based cohort study. BMC Nephrol. 2021, 22, 351. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, H.; Ju, H.; Chen, H.; Jin, H.; Sun, M. Circulating GDF-15 in relation to the progression and prognosis of chronic kidney disease: A systematic review and dose-response meta-analysis. Eur. J. Intern. Med. 2023, 110, 77–85. [Google Scholar] [CrossRef]
- Benes, J.; Kotrc, M.; Wohlfahrt, P.; Conrad, M.J.; Franekova, J.; Jabor, A.; Lupinek, P.; Kautzner, J.; Melenovsky, V.; Jarolim, P. The Role of GDF-15 in Heart Failure Patients with Chronic Kidney Disease. Can. J. Cardiol. 2019, 35, 462–470. [Google Scholar] [CrossRef]
- Monzo, L.; Jarolim, P.; Borlaug, B.A.; Benes, J.; Jurcova, I.; Jenca, D.; Kroupova, K.; Wohlfahrt, P.; Kotrc, M.; Melenovsky, V. Growth Differentiation Factor–15 Is Associated with Congestion-Related Anorexia and Weight Loss in Advanced Heart Failure. JACC Heart Fail. 2025, 13, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, P.; Cunha, F.M.; Ferreira-Coimbra, J.; Barroso, I.; Guimarães, J.; Bettencourt, P. Dynamics of growth differentiation factor 15 in acute heart failure. ESC Heart Fail. 2021, 8, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, M.; de Poel, J.H.C.; de Jager, S.C.A. Growth differentiation factor 15 in adverse cardiac remodelling: From biomarker to causal player. ESC Heart Fail. 2020, 7, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, M.C.; Oprea, V.D.; Munteanu, S.N.; Nechita, A.; Tutunaru, D.; Nechita, L.C.; Romila, A. Carbohydrate Antigen 125 (CA 125): A Novel Biomarker in Acute Heart Failure. Diagnostics 2024, 14, 795. [Google Scholar] [CrossRef]
- Hung, C.-L.; Hung, T.-C.; Lai, Y.-H.; Lu, C.-S.; Wu, Y.-J.; Yeh, H.-I. Beyond malignancy: The role of carbohydrate antigen 125 in heart failure. Biomark. Res. 2013, 1, 25. [Google Scholar] [CrossRef]
- Núñez-Marín, G.; Palau, P.; Domínguez, E.; de la Espriella, R.; López, L.; Flor, C.; Marín, P.; Lorenzo, M.; Miñana, G.; Bodí, V.; et al. CA125 outperforms NT-proBNP in the prediction of maximum aerobic capacity in heart failure with preserved ejection fraction and kidney dysfunction. Clin. Kidney J. 2024, 17, sfae199. [Google Scholar] [CrossRef]
- Feng, R.; Zhang, Z.; Fan, Q. Carbohydrate antigen 125 in congestive heart failure: Ready for clinical application? Front. Oncol. 2023, 13, 1161723. [Google Scholar] [CrossRef]
- Miñana, G.; de la Espriella, R.; Lorenzo-Hernández, M.; Rodriguez-Borja, E.; Mollar, A.; Palau, P.; Fernández-Cisnal, A.; Valero, E.; Carratalá, A.; Santas, E.; et al. Changes in Antigen Carbohydrate 125 in Patients Receiving Dapagliflozin following an Admission for Acute Heart Failure. Cardiorenal Med. 2025, 15, 122–132. [Google Scholar] [CrossRef]
- Tuersun, R.; Abudouwayiti, A.; Li, Y.X.; Pan, Y.; Aimaier, S.; Wen, Z.-Y.; Gao, W.-T.; Ma, L.-J.; Mahemuti, A.; Zheng, Y.-Y. Serum CA125: A prognostic biomarker for mortality in chronic heart failure. BMC Cardiovasc. Disord. 2025, 25, 1–14. [Google Scholar] [CrossRef]
- Núñez, J.; Llàcer, P.; Bertomeu-González, V.; Bosch, M.J.; Merlos, P.; García-Blas, S.; Montagud, V.; Bodí, V.; Bertomeu-Martínez, V.; Pedrosa, V.; et al. Carbohydrate Antigen-125–Guided Therapy in Acute Heart Failure. JACC Heart Fail. 2016, 4, 833–843. [Google Scholar] [CrossRef]
- Nikitiuk, B.E.; Rydzewska-Rosołowska, A.; Kakareko, K.; Głowińska, I.; Hryszko, T. On Whether Ca-125 Is the Answer for Diagnosing Overhydration, Particularly in End-Stage Kidney Disease Patients—A Systematic Review. Int. J. Mol. Sci. 2024, 25, 2192. [Google Scholar] [CrossRef]
- Bálint, L.; Nelson-Maney, N.P.; Tian, Y.; Serafin, S.D.; Caron, K.M. Clinical Potential of Adrenomedullin Signaling in the Cardiovascular System. Circ. Res. 2023, 132, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Czajkowska, K.; Zbroch, E.; Bielach-Bazyluk, A.; Mitrosz, K.; Bujno, E.; Kakareko, K.; Rydzewska-Rosolowska, A.; Hryszko, T. Mid-Regional Proadrenomedullin as a New Biomarker of Kidney and Cardiovascular Diseases—Is It the Future? J. Clin. Med. 2021, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Voordes, G.H.D.; Davison, B.; Biegus, J.; Edwards, C.; Damman, K.; ter Maaten, J.M.; Mebazaa, A.; Takagi, K.; Adamo, M.; Ambrosy, A.P.; et al. Biologically active adrenomedullin as a marker for residual congestion and early rehospitalization in patients hospitalized for acute heart failure: Data from STRONG-HF. Eur. J. Heart Fail. 2024, 26, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- ter Maaten, J.M.; Kremer, D.; Demissei, B.G.; Struck, J.; Bergmann, A.; Anker, S.D.; Ng, L.L.; Dickstein, K.; Metra, M.; Samani, N.J.; et al. Bio-adrenomedullin as a marker of congestion in patients with new-onset and worsening heart failure. Eur. J. Heart Fail. 2019, 21, 732–743. [Google Scholar] [CrossRef]
- Ansari Ramandi, M.M.; Hendriks, P.M.; Voors, A.A.; van den Bosch, A.E.; van Melle, J.P. Bioactive adrenomedullin as a marker of congestion and disease progression in patients with a systemic right ventricle. Int. J. Cardiol. 2024, 408, 132107. [Google Scholar] [CrossRef]
- Iglesias, P.; Silvestre, R.A.; Fernández-Reyes, M.J.; Díez, J.J. The role of copeptin in kidney disease. Endocrine 2023, 79, 420–429. [Google Scholar] [CrossRef]
- Mu, D.; Cheng, J.; Qiu, L.; Cheng, X. Copeptin as a Diagnostic and Prognostic Biomarker in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 901990. [Google Scholar] [CrossRef]
- Afsar, B. Pathophysiology of copeptin in kidney disease and hypertension. Clin. Hypertens. 2017, 23, 13. [Google Scholar] [CrossRef]
- Schneider, M.P.; Schmid, M.; Nadal, J.; Krane, V.; Saritas, T.; Busch, M.; Schultheiss, U.T.; Meiselbach, H.; Friedrich, N.; Nauck, M.; et al. Copeptin, Natriuretic Peptides, and Cardiovascular Outcomes in Patients with CKD: The German Chronic Kidney Disease (GCKD) Study. Kidney Med. 2023, 5, 100725. [Google Scholar] [CrossRef]
- Ren, Y.; Lin, H.; Guo, J.; Su, X.; Wang, L.; Qiao, X. Roles of microRNAs in cardiorenal syndrome. Mol. Cell. Biochem. 2025, 480, 4069–4078. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, A.; Prosperi, S.; Severino, P.; Myftari, V.; Correale, M.; Perrone Filardi, P.; Badagliacca, R.; Fedele, F.; Vizza, C.D.; Palazzuoli, A. MicroRNA and Heart Failure: A Novel Promising Diagnostic and Therapeutic Tool. J. Clin. Med. 2024, 13, 7560. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-K.; Bär, C.; Thum, T. miR-21, Mediator, and Potential Therapeutic Target in the Cardiorenal Syndrome. Front. Pharmacol. 2020, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Sessa, F.; Salerno, M.; Esposito, M.; Cocimano, G.; Pomara, C. miRNA Dysregulation in Cardiovascular Diseases: Current Opinion and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 5192. [Google Scholar] [CrossRef]
- West, M.; Kirby, A.; Stewart, R.A.; Blankenberg, S.; Sullivan, D.; White, H.D.; Hunt, D.; Marschner, I.; Janus, E.; Kritharides, L.; et al. Circulating Cystatin C Is an Independent Risk Marker for Cardiovascular Outcomes, Development of Renal Impairment, and Long-Term Mortality in Patients with Stable Coronary Heart Disease: The LIPID Study. J. Am. Heart Assoc. 2022, 11, e020745. [Google Scholar] [CrossRef]
- Kramer, T.; Brinkkoetter, P.; Rosenkranz, S. Right Heart Function in Cardiorenal Syndrome. Curr. Heart Fail. Rep. 2022, 19, 386–399. [Google Scholar] [CrossRef]
- Mullens, W.; Damman, K.; Harjola, V.-P.; Mebazaa, A.; Brunner-La Rocca, H.-P.; Martens, P.; Testani, J.M.; Tang, W.H.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef]
- Metra, M.; Tomasoni, D.; Adamo, M.; Bayes-Genis, A.; Filippatos, G.; Abdelhamid, M.; Adamopoulos, S.; Anker, S.D.; Antohi, L.; Böhm, M.; et al. Worsening of chronic heart failure: Definition, epidemiology, management and prevention. A clinical consensus statement by the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2023, 25, 776–791. [Google Scholar] [CrossRef]
- Bozkurt, B.; Rossignol, P.; Vassalotti, J.A. Albuminuria as a diagnostic criterion and a therapeutic target in heart failure and other cardiovascular disease. Eur. J. Heart Fail. 2025. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Martin-Cleary, C.; Sanz, A.B.; Avello, A.; Sanchez-Niño, M.D.; Ortiz, A. NephroCheck at 10: Addressing unmet needs in AKI diagnosis and risk stratification. Clin. Kidney J. 2023, 16, 1359–1366. [Google Scholar] [CrossRef]
- Brazzelli, M.; Aucott, L.; Aceves-Martins, M.; Robertson, C.; Jacobsen, E.; Imamura, M.; Poobalan, A.; Manson, P.; Scotland, G.; Kaye, C.; et al. Biomarkers for assessing acute kidney injury for people who are being considered for admission to critical care: A systematic review and cost-effectiveness analysis. NIHR J. Libr. 2022, 26, 1–286. [Google Scholar] [CrossRef]
- Ilaria, G.; Kianoush, K.; Ruxandra, B.; Francesca, M.; Mariarosa, C.; Davide, G.; Claudio, R. Clinical adoption of Nephrocheck® in the early detection of acute kidney injury. Ann. Clin. Biochem. 2021, 58, 6–15. [Google Scholar] [CrossRef]
- Ronco, C.; Rizo-Topete, L.; Serrano-Soto, M.; Kashani, K. Pro: Prevention of acute kidney injury: Time for teamwork and new biomarkers. Nephrol. Dial. Transplant. 2017, 32, 408–413. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).