
The NLRP3 Inflammasome in Age-Related Cerebral Small Vessel Disease Manifestations: Untying the Innate Immune Response Connection

 , , , , , and
, , , , , and
Abstract
1. Introduction
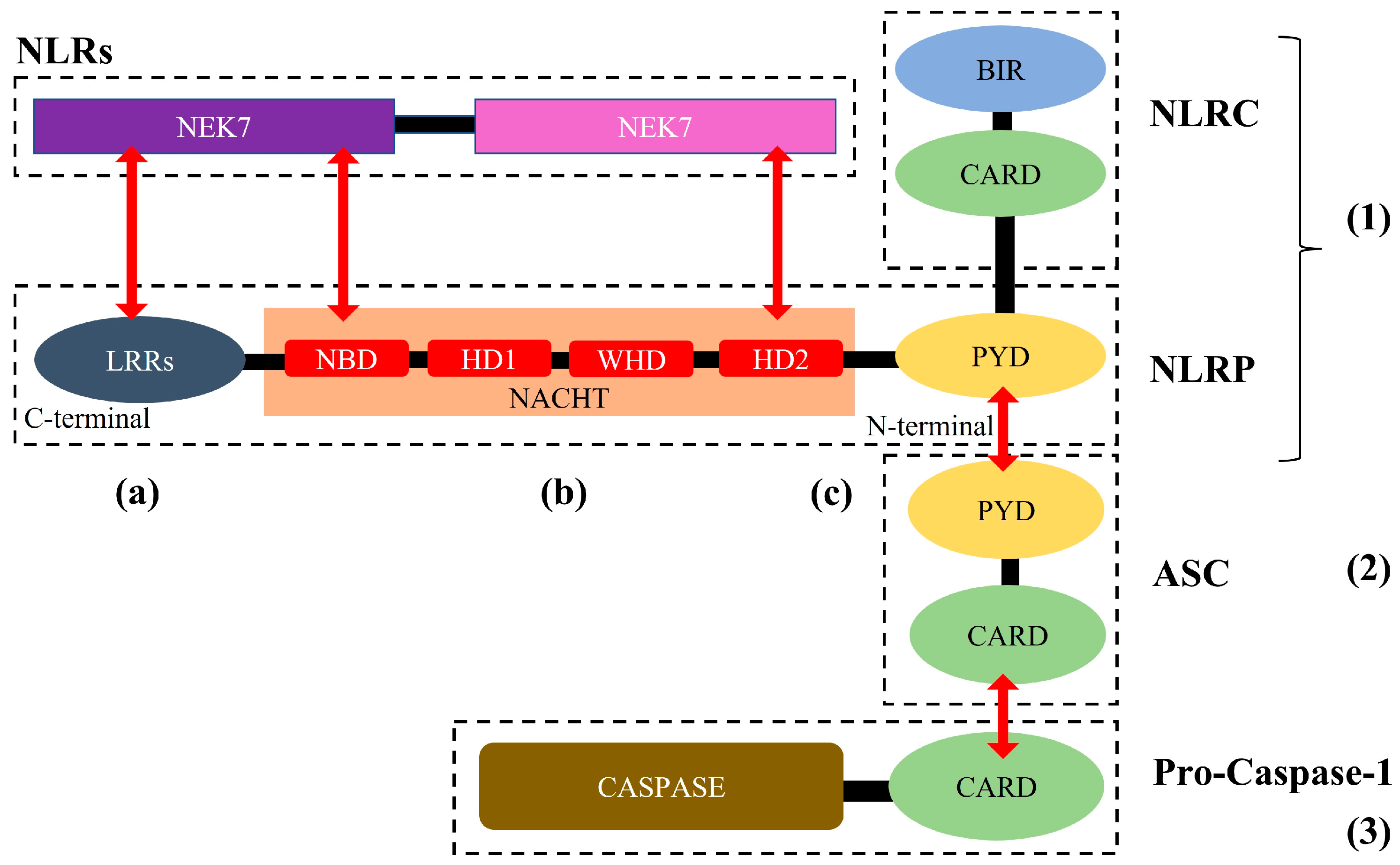
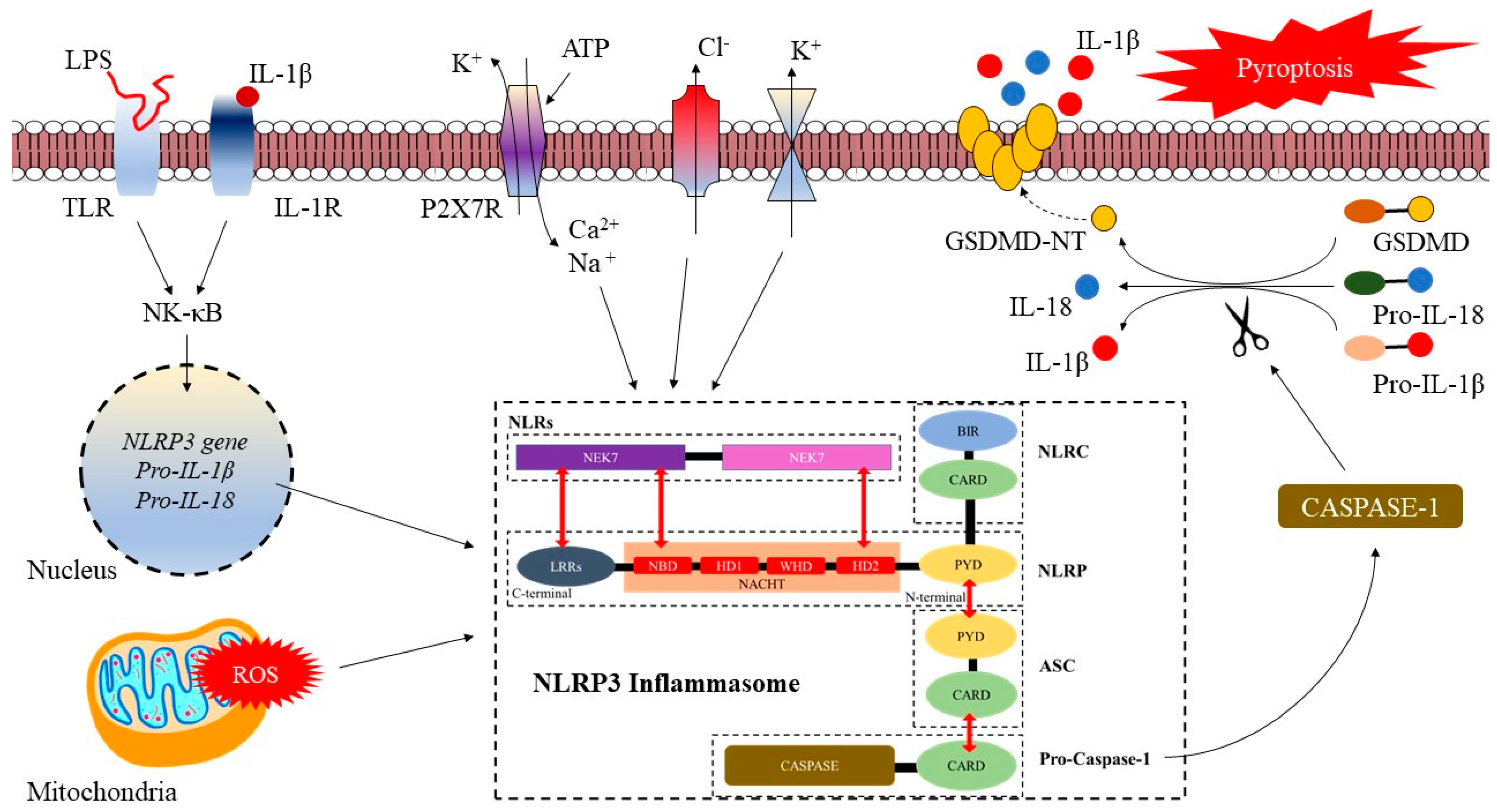
2. The NLRP3 Inflammasome: Structure, Activation, and Role in Cardio-Cerebrovascular Diseases
2.1. NLRP3 Inflammasome: Structure, Activation, and Role in Cardio-Cerebrovascular Diseases
2.2. Activation of the NLRP3 Inflammasome
2.3. The Role of NLRP3 Inflammasome in Cerebrovascular Diseases
2.4. The Mutation of nlrp3 Gene against NLRP3 Inflammasome Pathway
3. Cerebral Small Vessel Disease (CSVD) and NLRP3 Inflammasome
3.1. CSVD: Pathophysiological Mechanisms
3.2. The Hypothetical Link between NLRP3 Inflammasome and Manifestations of CSVD
3.3. Neuro-Thrombo-Inflammation: The Involvement of NLRP3 Inflammasome
3.4. The Progression of CSVD Heterogeneity Following Activation of NLRP3 Inflammasome
3.5. The NLRP3 Inflammasome Inhibition as Neuroprotective Potential for CSVD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Underlying Cause of Death, 1999–2020. CDC WONDER Online Database. 2018. Available online: https://wonder.cdc.gov/controller/datarequest/D76 (accessed on 12 March 2020).
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, E153–E639. [Google Scholar]
- Lampe, L.; Kharabian-Masouleh, S.; Kynast, J.; Arelin, K.; Steele, C.; Löffler, M.; Witte, A.V.; Schroeter, M.L.; Villringer, A.; Bazin, P.-L. Lesion location matters: The relationships between white matter hyperintensities on cognition in the healthy elderly. J. Cereb. Blood Flow Metab. 2017, 39, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Markus, H.S. New Treatment Approaches to Modify the Course of Cerebral Small Vessel Diseases. Stroke 2020, 51, 38–46. [Google Scholar] [CrossRef]
- Nassir, C.M.N.C.M.; Ghazali, M.M.; Hashim, S.; Idris, N.S.; Yuen, L.S.; Hui, W.J.; Norman, H.H.; Gau, C.H.; Jayabalan, N.; Na, Y.; et al. Diets and Cellular-Derived Microparticles: Weighing a Plausible Link with Cerebral Small Vessel Disease. Front. Cardiovasc. Med. 2021, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.N.; Ishrat, T.; Fagan, S.C.; El-Remessy, A.B. Role of inflammasome activation in the pathophysiology of vascular diseases of the neurovascular unit. Antioxid. Redox Signal. 2015, 22, 1188–1206. [Google Scholar] [CrossRef] [PubMed]
- Sandercock, P.A.G.; Soane, T. Corticosteroids for acute ischaemic stroke. Cochrane. Database Syst. Rev. 2011, 2011, CD000064. [Google Scholar] [CrossRef]
- Kalra, L.; Irshad, S.; Hodsoll, J.; Simpson, M.; Gulliford, M.; Smithard, D.; Patel, A.; Rebollo-Mesa, I. Prophylactic antibiotics after acute stroke for reducing pneumonia in patients with dysphagia (STROKE-INF): A prospective, cluster-randomised, open-label, masked endpoint, controlled clinical trial. Lancet 2015, 386, 1835–1844. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2018, 113, 5. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Xia, M.; Gulbins, E.; Boini, K.M.; Li, P.L. Activation of Nlrp3 Inflammasomes Enhances Macrophage Lipid-Deposition and Migration: Implication of a Novel Role of Inflammasome in Atherogenesis. PLoS ONE 2014, 9, e87552. [Google Scholar] [CrossRef]
- Varghese, G.P.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart. Assoc. 2022, 5, e003031. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yin, R.; Yang, S.; Pan, X.; Ma, A. Rs4612666 polymorphism of the NLRP3 gene is associated with the occurrence of large artery atherosclerotic ischemic strokes and microembolic signals. Biomed. Res. Int. 2018, 2018, 6345805. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, W.; Lu, C.; Ma, Z.; Jiang, S.; Gu, C.; Acuña-Castroviejo, D.; Yang, Y. Targeting NLRP3 (nucleotide binding domain, leucine-rich–containing family, pyrin domain–containing-3) inflammasome in cardiovascular disorders. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2765–2779. [Google Scholar] [CrossRef]
- Shim, D.W.; Lee, K.H. Posttranslational regulation of the NLR family pyrin domain containing 3 inflammasome. Front. Immunol. 2018, 9, 1054. [Google Scholar] [CrossRef] [PubMed]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef]
- Ting, J.P.Y.; Davis, B.K. CATERPILLER: A novel gene family important in immunity, cell death, and diseases. Annu. Rev. Immunol. 2005, 23, 387. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Lammerding, L.; Slowik, A.; Johann, S.; Beyer, C.; Zendedel, A. Poststroke inflammasome expression and regulation in the peri-infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 2016, 103, 460–475. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Higa, N.; Koizumi, Y.; Nakasone, N.; Ogura, Y.; McCoy, A.J.; Franchi, L.; Uematsu, S.; Sagara, J.; Taniguchi, S.I.; et al. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-κB signaling. J. Immunol. 2010, 184, 5287–5297. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-κB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Herman, F.J.; Pasinetti, G.M. Principles of inflammasome priming and inhibition: Implications for psychiatric disorders. Brain Behav. Immun. 2018, 73, 66–84. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Hornung, V. Alternative inflammasome activation enables IL-1β release from living cells. Curr. Opin. Immunol. 2017, 44, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Li, T. Regulation of NLRP3 inflammasome by phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; de Andrade Mello, P.; Da Silva, C.G.; Coutinho-Silva, R. The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, H.; Jin, J.; Liu, Q.; Zhong, D.; Li, G. Inhibition of the NLRP3 inflammasome reduces brain edema and regulates the distribution of aquaporin-4 after cerebral ischaemia-reperfusion. Life Sci. 2020, 251, 117638. [Google Scholar] [CrossRef]
- Li, J.; Liu, Z.; Wang, L.; Xu, H.; Wang, Y. Thousand and one kinase 1 protects MCAO503 induced cerebral ischemic stroke in rats by decreasing apoptosis and pro-inflammatory factors. Biosci. Rep. 2019, 39, BSR20190749. [Google Scholar] [CrossRef]
- Friedlander, R.M.; Gagliardini, V.; Hara, H.; Fink, K.B.; Li, W.; MacDonald, G.; Fishman, M.C.; Greenberg, A.H.; Moskowitz, M.A.; Yuan, J. Expression of a Dominant Negative Mutant of Interleukin-1β Converting Enzyme in Transgenic Mice Prevents Neuronal Cell Death Induced by Trophic Factor Withdrawal and Ischemic Brain Injury. J. Exp. Med. 1997, 185, 933–940. [Google Scholar] [CrossRef]
- Caso, J.R.; Moro, M.A.; Lorenzo, P.; Lizasoain, I.; Leza, J.C. Involvement of IL-1β in acute stress-induced worsening of cerebral ischaemia in rats. Eur. Neuropsychopharmacol. 2007, 17, 600–607. [Google Scholar] [CrossRef]
- Liang, H.; Sun, Y.; Gao, A.; Zhang, N.; Jia, Y.; Yang, S.; Na, M.; Liu, H.; Cheng, X.; Fang, X.; et al. Ac-YVAD-cmk improves neurological function by inhibiting caspase-1-mediated inflammatory response in the intracerebral hemorrhage of rats. Int. Immunopharmacol. 2019, 75, 105771. Available online: https://www.sciencedirect.com/science/article/pii/S1567576919305867 (accessed on 15 May 2022). [CrossRef]
- Moriwaki, T.; Takagi, Y.; Sadamasa, N.; Aoki, T.; Nozaki, K.; Hashimoto, N. Impaired Progression of Cerebral Aneurysms in Interleukin-1-Deficient Mice. 2006. Available online: http://ahajournals.org (accessed on 20 September 2022).
- Li, F.; Chen, Y.; Li, Y.; Huang, M.; Zhao, W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-κB pathway. Eur. J. Pharmacol. 2020, 886, 173449. [Google Scholar] [CrossRef]
- Che Mohd Nassir, C.M.N.; Zolkefley, M.K.I.; Ramli, M.D.; Norman, H.H.; Abdul Hamid, H.; Mustapha, M. Neuroinflammation and COVID-19 Ischemic Stroke Recovery—Evolving Evidence for the Mediating Roles of the ACE2/Angiotensin-(1–7)/Mas Receptor Axis and NLRP3 Inflammasome. Int. J. Mol. Sci. 2022, 23, 3085. [Google Scholar] [CrossRef]
- Taoka, T.; Naganawa, S. Imaging for central nervous system (CNS) interstitial fluidopathy: Disorders with impaired interstitial fluid dynamics. Jpn. J. Radiol. 2021, 39, 1–14. [Google Scholar] [CrossRef]
- Gaberel, T.; Gakuba, C.; Goulay, R.; De Lizarrondo, S.M.; Hanouz, J.-L.; Emery, E.; Touze, E.; Vivien, D.; Gauberti, M. Impaired Glymphatic Perfusion After Strokes Revealed by Contrast-Enhanced MRI. Stroke 2014, 45, 3092–3096. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef] [PubMed]
- Song, H.L.; Zhang, S.B. Therapeutic effect of dexmedetomidine on intracerebral hemorrhage via regulating NLRP3. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 23–2612. [Google Scholar]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Yan, J.; Zhang, J.; Shao, A. Apelin-13/APJ system attenuates early brain injury via suppression of endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation and oxidative stress in a AMPK-dependent manner after subarachnoid hemorrhage in rats. J. Neuroinflammation 2019, 16, 247. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef]
- Italiani, P.; Puxeddu, I.; Napoletano, S.; Scala, E.; Melillo, D.; Manocchio, S.; Angiolillo, A.; Migliorini, P.; Boraschi, D.; Vitale, E.; et al. Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? J. Neuroinflammation 2018, 15, 1–12. [Google Scholar] [CrossRef]
- Antony, P.M.A.; Diederich, N.J.; Krüger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/febs.12335 (accessed on 20 September 2022). [CrossRef]
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein–antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, A.; Green, J.P.; Brough, D.; Lopez-Castejon, G. Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine Growth Factor Rev. 2020, 55, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Williams, M.R.; Ryffel, B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol. Metab. 2018, 29, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Marín-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gómez, E.; Castejón-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Pérez-Pulido, A.J.; Varela-López, A.; Quiles, J.L.; et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 2020, 19, e13050. [Google Scholar] [CrossRef]
- Navarro-Pando, J.M.; Alcocer-Gómez, E.; Castejón-Vega, B.; Navarro-Villarán, E.; Condés-Hervás, M.; Mundi-Roldan, M.; Muntané, J.; Pérez-Pulido, A.J.; Bullon, P.; Wang, C.; et al. Inhibition of the NLRP3 inflammasome prevents ovarian aging. Sci. Adv. 2021, 7, eabc7409. [Google Scholar] [CrossRef]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740. [Google Scholar] [CrossRef]
- Smith, E.E. Clinical presentations and epidemiology of vascular dementia. Clin. Sci. 2017, 131, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, S.; Naidech, A.M. Ischemic brain injury after intracerebral hemorrhage: A critical review. Stroke 2012, 43, 2258–2263. [Google Scholar] [CrossRef] [PubMed]
- Chojdak-Łukasiewicz, J.; Dziadkowiak, E.; Zimny, A.; Paradowski, B. Cerebral small vessel disease: A review. Adv. Clin. Exp. Med. 2021, 30, 349–356. [Google Scholar] [CrossRef]
- Nassir, C.; Ghazali, M.; Safri, A.; Jaffer, U.; Abdullah, W.; Idris, N.; Muzaimi, M. Elevated Circulating Microparticle Subpopulations in Incidental Cerebral White Matter Hyperintensities: A Multimodal Study. Brain Sci. 2021, 11, 133. [Google Scholar] [CrossRef]
- Li, X.; Yan, X.; Wang, Y.; Wang, J.; Zhou, F.; Wang, H.; Xie, W.; Kong, H. NLRP3 inflammasome inhibition attenuates silica-induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells. Exp. Cell Res. 2018, 362, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Ong, L.; Gyawali, P.; Nassir, C.; Mustapha, M.; Nandurkar, H.; Sashindranath, M. Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease. Biomolecules 2021, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mato, M.; Iglesias-Rey, R.; Vieites-Prado, A.; Dopico-López, A.; Argibay, B.; Fernández-Susavila, H.; da Silva-Candal, A.; Pérez-Díaz, A.; Correa-Paz, C.; Günther, A.; et al. Blood glutamate EAAT2-cell grabbing therapy in cerebral ischemia. EBioMedicine 2019, 39, 118–131. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, J. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Hung, W.; Ho, C.; Pan, M. Targeting the NLRP3 inflammasome in neuroinflammation: Health promoting effects of dietary phytochemicals in neurological disorders. Mol. Nutr. Food Res. 2020, 64, 1900550. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef] [PubMed]
- Rui, W.; Li, S.; Xiao, H.; Xiao, M.; Shi, J. Baicalein attenuates neuroinflammation by inhibiting NLRP3/caspase-1/GSDMD pathway in MPTP-induced mice model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2020, 23, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Ding, Z.-H.; Zhan, F.-X.; Cai, L.; Yin, X.; Ling, J.-L.; Ye, J.-J.; Hou, S.-Y.; Lu, Z.; Wang, Z.-H.; et al. The NLRP3 inflammasome and stroke. Int. J. Clin. Exp. Med. 2015, 8, 4787. [Google Scholar] [PubMed]
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The potential role of the NLRPinflammasome activation as a link between 609 mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Front Cell Neurosci. 2019, 13, 73. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Liu, Y.-Z.; Shen, X.-L.; Wu, T.-Y.; Zhang, T.; Wang, W.; Wang, Y.-X.; Jiang, C.-L. NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.Y. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflammation 2018, 15, 242. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef]
- Xiao-Yu, Q.U.; Zhang, Y.M.; Li-Na, T.A.O.; Huan, G.A.O.; Jing-Hui, Z.H.A.I.; Jing-Meng, S.U.N.; Yan-Qing, S.O.N.G.; Zhang, S.X. XingNaoJing injections protect against cerebral ischemia/reperfusion injury and alleviate blood-brain barrier disruption in rats, through an underlying mechanism of NLRP3 inflammasomes suppression. Chin. J. Nat. Med. 2019, 17, 498–505. [Google Scholar]
- Zhang, Y.; Li, X.; Qiao, S.; Yang, D.; Li, Z.; Xu, J.; Li, W.; Su, L.; Liu, W. Occludin degradation makes brain microvascular endothelial cells more vulnerable to reperfusion injury in vitro. J. Neurochem. 2021, 156, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yao, Q.; Wan, Y.; Wang, J.; Huang, C.; Li, D.; Yang, B. Adiponectin reduces brain injury after intracerebral hemorrhage by reducing NLRP3 inflammasome expression. Int. J. Neurosci. 2020, 130, 301–308. [Google Scholar] [CrossRef]
- Ward, R.; Li, W.; Abdul, Y.; Jackson, L.; Dong, G.; Jamil, S.; Filosa, J.; Fagan, S.C.; Ergul, A. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol. Res. 2019, 142, 237–250. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Xie, J.; Qiu, S.; Liu, Y.; Wang, J.; Xiu, X.; Li, L.; Tang, M. Hispidulin exhibits neuroprotective activities against cerebral ischemia reperfusion injury through suppressing NLRP3-mediated pyroptosis. Life Sci. 2019, 232, 116599. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yu, S.; Chen, X.; Zheng, P.; Hu, T.; Duan, Z.; Liu, X.; Liu, Q.; Ye, R.; Zhu, W.; et al. Suppression of NLRP3 attenuates hemorrhagic transformation after delayed rtPA treatment in thromboembolic stroke rats: Involvement of neutrophil recruitment. Brain Res. Bull. 2018, 137, 229–240. [Google Scholar] [CrossRef]
- Xiao, L.; Zheng, H.; Li, J.; Wang, Q.; Sun, H. Neuroinflammation mediated by NLRP3 inflammasome after intracerebral hemorrhage and potential therapeutic targets. Mol. Neurobiol. 2020, 57, 5130–5149. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Kim, H.J.; Fox, Z.; Jäger, H.R.; Wilson, D.; Charidimou, A.; Na, H.K.; Na, D.L.; Seo, S.W.; Werring, D.J. MRI-visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain 2017, 140, 1107–1116. [Google Scholar] [CrossRef]
- Wharton, S.B.; Simpson, J.E.; Brayne, C.; Ince, P.G. Age- Associated White Matter Lesions: The MRC C ognitive F unction and A geing S tudy. Brain Pathol. 2015, 25, 35–43. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Haley, K.; Auriel, E.; van Etten, E.S.; Fotiadis, P.; Reijmer, Y.; Ayres, A.; Vashkevich, A.; Dipucchio, Z.Y.; et al. White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2016, 86, 505–511. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Pasi, M.; Auriel, E.; van Etten, E.S.; Haley, K.; Ayres, A.; Schwab, K.M.; Martinez-Ramirez, S.; Goldstein, J.N.; et al. MRI visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2017, 88, 1157–1164. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. 659 Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol. 2013, 12, 483–497. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- She, Y.; Shao, L.; Zhang, Y.; Hao, Y.; Cai, Y.; Cheng, Z.; Deng, C.; Liu, X. Neuroprotective effect of glycosides in Buyang Huanwu Decoction on pyroptosis following cerebral ischemia reperfusion injury in rats. J. Ethnopharmacol. 2019, 242, 112051. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Z.; Jia, L.Q.; Zhan, K.X.; Wang, L.J.; Song, N.; Liu, Y.; Cheng, Y.Y.; Yang, Y.J.; Guan, L.; et al. Valproic acid attenuates global cerebral ischemia/reperfusion injury in gerbils via anti-pyroptosis pathways. Neurochem. Int. 2019, 124, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Aspects Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- El-Sharkawy, L.Y.; Brough, D.; Freeman, S. Inhibiting the NLRP3 inflammasome. Molecules 2020, 25, 5533. [Google Scholar] [CrossRef]
- Varon, D.; Shai, E. Platelets and their microparticles as key players in pathophysiological responses. J. Thromb. Haemost. 2015, 13, S40–S46. [Google Scholar] [CrossRef]
- Badimon, L.; Suades, R.; Fuentes, E.; Palomo, I.; Padró, T. Role of platelet-derived microvesicles as crosstalk mediators in atherothrombosis and future pharmacology targets: A link between inflammation, atherosclerosis, and thrombosis. Front. Pharmacol. 2016, 7, 293. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Dai, Z.; Chen, X.Y.; An, L.Y.; Li, C.C.; Zhao, N.; Yang, F.; You, S.T.; Hou, C.Z.; Li, K.; Jiang, C.; et al. Development of novel tetrahydroquinoline inhibitors of NLRP3 inflammasome for potential treatment of DSS-induced mouse colitis. J. Med. Chem. 2020, 64, 871–889. [Google Scholar] [CrossRef]
- Mahmoud, T.N.; El-Maadawy, W.H.; Kandil, Z.A.; Khalil, H.; El-Fiky, N.M.; El Alfy, T.S.M.A. Canna x generalis LH Bailey rhizome extract ameliorates dextran sulfate sodium induced colitis via modulating intestinal mucosal dysfunction, oxidative stress, inflammation, and TLR4/NF-ҡB and NLRP3 inflammasome pathways. J. Ethnopharmacol. 2021, 269, 113670. [Google Scholar] [CrossRef] [PubMed]
- Tomani, J.C.D.; Kagisha, V.; Tchinda, A.T.; Jansen, O.; Ledoux, A.; Vanhamme, L.; Frederich, M.; Muganga, R.; Souopgui, J. The inhibition of NLRP3 inflammasome and IL-6 production by hibiscus noldeae baker f. Derived constituents provides a link to its anti-Inflammatory therapeutic potentials. Molecules 2020, 25, 4693. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-H.; Park, Y.-L.; Song, A.-Y.; Kim, W.-G.; Je, C.-Y.; Jung, D.-H.; Kim, Y.-J.; Oh, J.; Cho, J.-Y.; Kim, D.-J.; et al. Water extract of Artemisia scoparia Waldst. & Kitam suppresses LPS-induced cytokine production and NLRP3 inflammasome activation in macrophages and alleviates carrageenan-induced acute inflammation in mice. J. Ethnopharmacol. 2021, 268, 113606. [Google Scholar]
- Zhao, J.; Wang, Z.; Yuan, Z.; Lv, S.; Su, Q. Baicalin ameliorates atherosclerosis by inhibiting NLRP3 inflammasome in apolipoprotein E-deficient mice. Diabetes Vasc. Dis. Res. 2020, 17, 1479164120977441. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.C.; Chang, Y.H. Comparison of inhibitory capacities of 6-, 8- and 10-Gingerols/shogaols on the canonical NLRP3 inflammasome-mediated IL-1beta secretion. Molecules 2018, 23, 466. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; de Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pé-rez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Zhou, S.; Xu, X.; Gao, W.; Li, F.; Ma, Y.; Sun, D.; Wu, Y.; Guo, Q.; Liu, H.; et al. Dexmedetomidine attenuated early brain injury in rats with subarachnoid haemorrhage by suppressing the inflammatory response: The TLR4/NF-κB pathway and the NLRP3 inflammasome may be involved in the mechanism. Brain Res. 2018, 1698, 1–10. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, H.; Zheng, M.; Xu, W.; Yang, Y.; Shi, F. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J. 2020, 34, 208–221. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong antiinflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, T.; Ma, X.; Jiang, K.; Wu, H.; Qiu, C.; Guo, M.; Deng, G. Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264. 7 cells and acute lung injury. Oncotarget 2017, 8, 68153. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Peng, M.; Xu, P.; Huang, F.; Xie, Y.; Li, J.; Hong, Y.; Guo, H.; Liu, Q.; Zhu, W. Low-density lipoprotein receptor (LDLR) regulates NLRP3-mediated neuronal pyroptosis following cerebral ischemia/reperfusion injury. J. Neuroinflammation 2020, 17, 330. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 Inflammasome Inhibitor OLT1177 Rescues Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. Available online: www.pnas.org/cgi/doi/10.1073/pnas.2009680117 (accessed on 20 September 2022).
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Chen, Y.; Li, R.; Wang, Z.; Hou, X.; Wang, C.; Ai, Y.; Shi, W.; Zhan, X.; Wang, J.-B.; Xiao, X.; et al. Dehydrocostus lactone inhibits NLRP3 inflammasome activation by blocking ASC oligomerization and prevents LPS744 mediated inflammation in vivo. Cell Immunol. 2020, 349, 104046. [Google Scholar] [CrossRef]
- Inoue, M.; Shinohara, M.L. Nlrp3 inflammasome and MS/EAE. Autoimmune Dis. 2013, 2013, 859145. [Google Scholar] [CrossRef]
- Kuo, P.C.; Weng, W.T.; Scofield, B.A.; Furnas, D.; Paraiso, H.C.; Intriago, A.J.; Bosi, K.D.; Yu, I.C.; Yen, J.H. Interferon-β alleviates delayed tPA-induced adverse effects via modulation of MMP3/9 production in ischemic stroke. Blood Adv. 2020, 4, 4366–4381. [Google Scholar] [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Findings | Refs. |
|---|---|---|
| Ethanol extract of Canna x generalis rhizome |
| [82,103] |
| ER2.4 and ER2.7 from Hibiscus noldeae |
| [104] |
| Water extract of Artemisia scoparia |
| [105] |
| Baicalin |
| [106] |
| 6-shogaol, 8-shogaol, and 10-gingerol from ginger plant |
| [107] |
| MCC950 (or CP-456773 or CRID3) |
| [94,101,108,109,110] |
| Ginsenoside, Rg3 |
| [111] |
| Oridonin |
| [112,113] |
| CY-09 |
| [101,114] |
| Dapansutrile or OLT1177 |
| [115,116] |
| Tranilast |
| [117] |
| Dehydrocostus Lactone |
| [118] |
| Z-YVAD-FMK |
| [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che Mohd Nassir, C.M.N.; Damodaran, T.; Ismail, N.I.; Hashim, S.; Jaffer, U.; Hamid, H.A.; Mehat, M.Z.; Norazit, A.; Mustapha, M. The NLRP3 Inflammasome in Age-Related Cerebral Small Vessel Disease Manifestations: Untying the Innate Immune Response Connection. Life 2023, 13, 216. https://doi.org/10.3390/life13010216
Che Mohd Nassir CMN, Damodaran T, Ismail NI, Hashim S, Jaffer U, Hamid HA, Mehat MZ, Norazit A, Mustapha M. The NLRP3 Inflammasome in Age-Related Cerebral Small Vessel Disease Manifestations: Untying the Innate Immune Response Connection. Life. 2023; 13(1):216. https://doi.org/10.3390/life13010216
Chicago/Turabian StyleChe Mohd Nassir, Che Mohd Nasril, Thenmoly Damodaran, Nurul Iman Ismail, Sabarisah Hashim, Usman Jaffer, Hafizah Abdul Hamid, Muhammad Zulfadli Mehat, Anwar Norazit, and Muzaimi Mustapha. 2023. "The NLRP3 Inflammasome in Age-Related Cerebral Small Vessel Disease Manifestations: Untying the Innate Immune Response Connection" Life 13, no. 1: 216. https://doi.org/10.3390/life13010216
APA StyleChe Mohd Nassir, C. M. N., Damodaran, T., Ismail, N. I., Hashim, S., Jaffer, U., Hamid, H. A., Mehat, M. Z., Norazit, A., & Mustapha, M. (2023). The NLRP3 Inflammasome in Age-Related Cerebral Small Vessel Disease Manifestations: Untying the Innate Immune Response Connection. Life, 13(1), 216. https://doi.org/10.3390/life13010216