
NSD1: A Lysine Methyltransferase between Developmental Disorders and Cancer


{kind=link}
{kind=link}
Abstract
1. Introduction
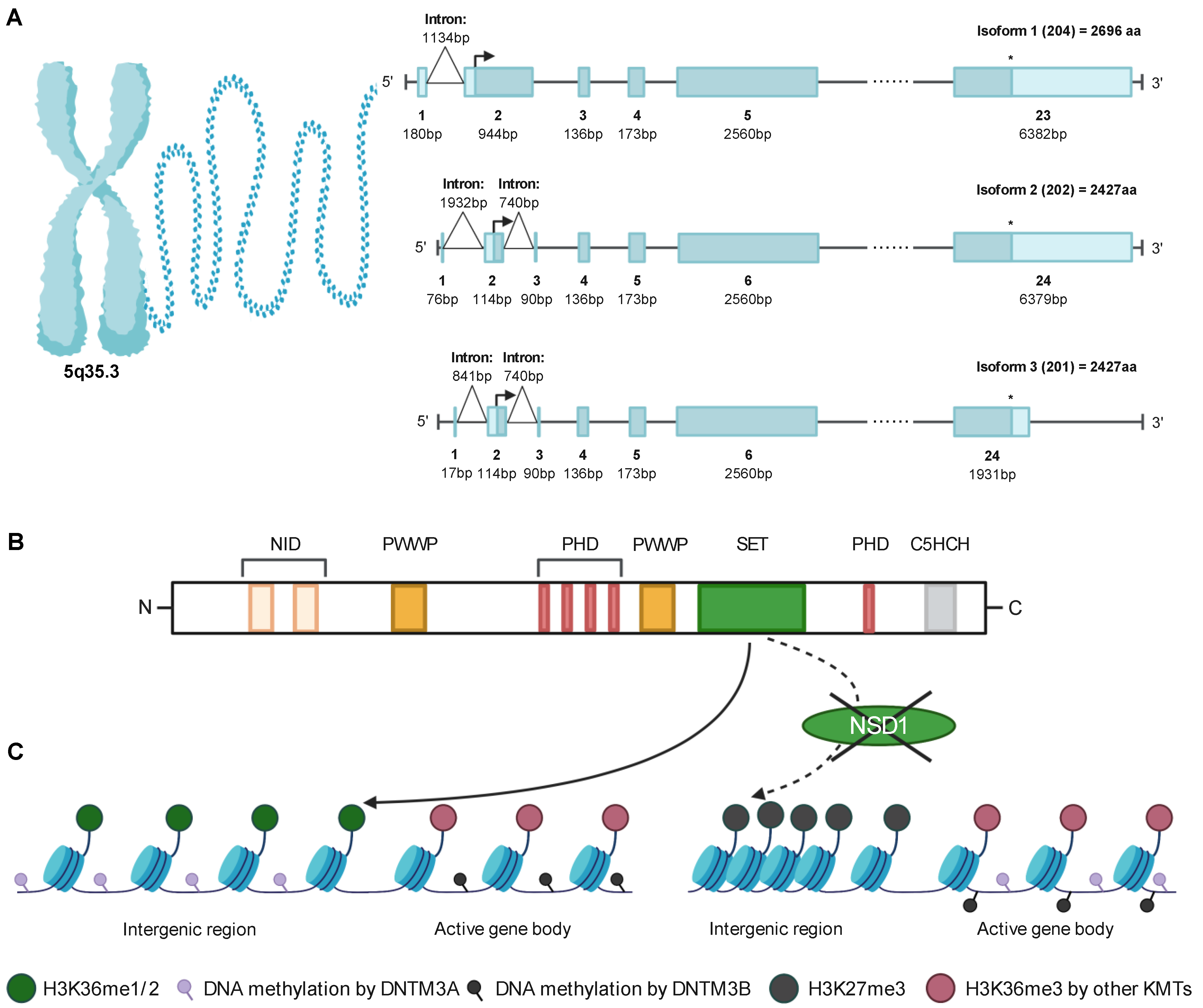
2. Identification and Structure of NSD1
3. NSD1 Is an Epigenetic Regulator Writing and Reading Chromatin Marks
3.1. The SET Domain Mediates the Catalytic Activity
3.2. NSD1 Chromatin Modification and Regulation
3.3. Functional Interaction with DNA Methyltransferases
3.4. Regulation of Gene Expression
4. Cellular Functions of NSD1
4.1. Modeling NSD1 Activity in the Fly
4.2. Modeling NSD1 Activity in the Mouse
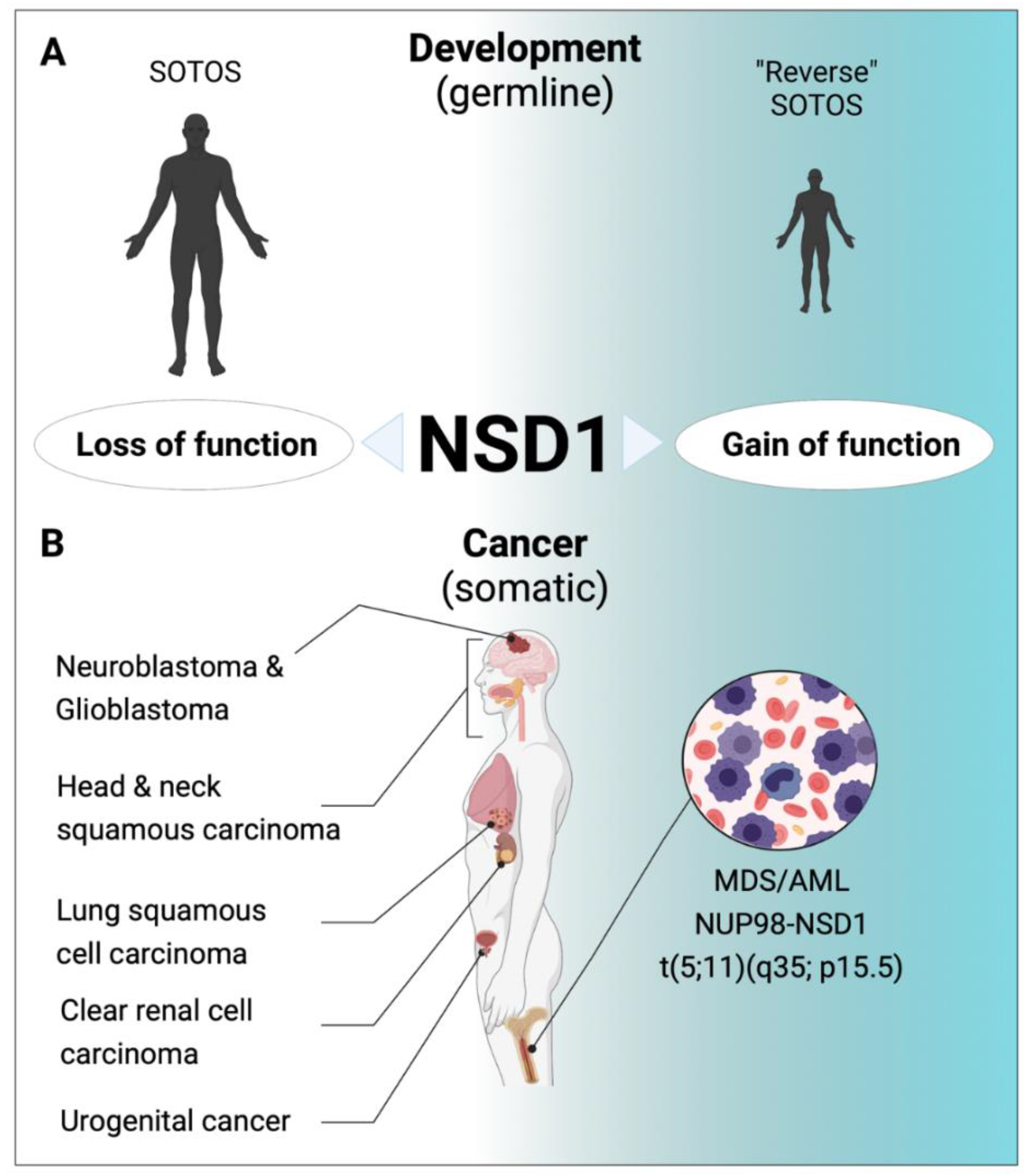
5. Role of NSD1 in Human Diseases
5.1. Aberrant NSD1 Activity Is a Hallmark of Developmental Syndromes
5.2. Aberrant NSD1 in Human Cancers
6. Therapeutic Interference with NSD1
7. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Bhat, K.P.; Kaniskan, H.Ü.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef]
- Huang, N.; Baur, E.V.; Garnier, J.; Lerouge, T.; Vonesch, J.; Lutz, Y.; Chambon, P.; Losson, R. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 1998, 17, 3398–3412. [Google Scholar] [CrossRef] [PubMed]
- Sampson, E.R.; Yeh, S.Y.; Chang, H.-C.; Tsai, M.Y.; Wang, X.; Ting, H.J.; Chang, C. Identification and characterization of androgen receptor associated coregulators in prostate cancer cells. J. Boil. Regul. Homeost. Agents 2001, 15, 123–129. [Google Scholar]
- Kurotaki, N.; Harada, N.; Yoshiura, K.-I.; Sugano, S.; Niikawa, N.; Matsumoto, N. Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 2001, 279, 197–204. [Google Scholar] [CrossRef]
- Ensembl Gene: NSD1 ENSG00000165671. Available online: http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000165671;r=5:177133025-177300213;t=ENST00000347982 (accessed on 17 June 2021).
- Wang, X.; Yeh, S.; Wu, G.; Hsu, C.-L.; Wang, L.; Chiang, T.; Yang, Y.; Guo, Y.; Chang, C. Identification and Characterization of a Novel Androgen Receptor Coregulator ARA267-α in Prostate Cancer Cells. J. Biol. Chem. 2001, 276, 40417–40423. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000165671-NSD1/tissue (accessed on 13 July 2021).
- GeneCards: The Human Gene Database: NSD1. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=NSD1#protein_expression (accessed on 13 July 2021).
- GeneCards the Human Gene Database. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=NSD1 (accessed on 17 June 2021).
- Rosati, R.; La Starza, R.; Veronese, A.; Aventin, A.; Schwienbacher, C.; Vallespi, T.; Negrini, M.; Martelli, M.F.; Mecucci, C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 2002, 99, 3857–3860. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef]
- Morishita, M.; di Luccio, E. Structural insights into the regulation and the recognition of histone marks by the SET domain of NSD1. Biochem. Biophys. Res. Commun. 2011, 412, 214–219. [Google Scholar] [CrossRef]
- Amin, N.; Nietlispach, D.; Qamar, S.; Coyle, J.; Chiarparin, E.; Williams, G. NMR backbone resonance assignment and solution secondary structure determination of human NSD1 and NSD2. Biomol. NMR Assign. 2016, 10, 315–320. [Google Scholar] [CrossRef][Green Version]
- Pasillas, M.P.; Shah, M.; Kamps, M.P. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum. Mutat. 2011, 32, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Ropero, S.; Setien, F.; Fraga, M.; Lapunzina, P.; Losson, R.; Alaminos, M.; Cheung, N.-K.; Rahman, N.; Esteller, M. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. USA 2009, 106, 21830–21835. [Google Scholar] [CrossRef]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Cytokine 2009, 48, 19–20. [Google Scholar] [CrossRef]
- Kudithipudi, S.; Lungu, C.; Rathert, P.; Happel, N.; Jeltsch, A. Substrate Specificity Analysis and Novel Substrates of the Protein Lysine Methyltransferase NSD1. Chem. Biol. 2014, 21, 226–237. [Google Scholar] [CrossRef]
- Li, J.; Ahn, J.H.; Wang, G.G. Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell. Mol. Life Sci. 2019, 76, 2899–2916. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhu, B. Roles of H3K36-specific histone methyltransferases in transcription: Antagonizing silencing and safeguarding transcription fidelity. Biophys. Rep. 2018, 4, 170–177. [Google Scholar] [CrossRef]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2007, 27, 406–420. [Google Scholar] [CrossRef]
- Qiao, Q.; Li, Y.; Chen, Z.; Wang, M.; Reinberg, D.; Xu, R.-M. The Structure of NSD1 Reveals an Autoregulatory Mechanism Underlying Histone H3K36 Methylation. J. Biol. Chem. 2011, 286, 8361–8368. [Google Scholar] [CrossRef]
- Li, W.; Tian, W.; Yuan, G.; Deng, P.; Sengupta, D.; Cheng, Z.; Cao, Y.; Ren, J.; Qin, Y.; Zhou, Y.; et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nat. Cell Biol. 2021, 590, 498–503. [Google Scholar]
- Yuan, G.; Ma, B.; Yuan, W.; Zhang, Z.; Chen, P.; Ding, X.; Feng, L.; Shen, X.; Chen, S.; Li, G.; et al. Histone H2A Ubiquitination Inhibits the Enzymatic Activity of H3 Lysine 36 Methyltransferases. J. Biol. Chem. 2013, 288, 30832–30842. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef]
- Streubel, G.; Watson, A.; Jammula, S.G.; Scelfo, A.; Fitzpatrick, D.J.; Oliviero, G.; McCole, R.; Conway, E.; Glancy, E.; Negri, G.L.; et al. The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol. Cell 2018, 70, 371–379.e5. [Google Scholar] [CrossRef]
- Hoang, N.-M.; Rui, L. DNA methyltransferases in hematological malignancies. J. Genet. Genom. 2020, 47, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP Domain Reads Histone 3 Lysine 36 Trimethylation and Guides DNA Methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef]
- Deevy, O.; Bracken, A.P. PRC2 functions in development and congenital disorders. Development 2019, 146, dev181354. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Lu, C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep. 2021, 22, e51803. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K.N.; Horth, C.; McGuire, J.T.; Xu, X.; Nikbakht, H.; et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nat. Cell Biol. 2019, 573, 281–286. [Google Scholar] [CrossRef]
- Nielsen, A.L.; Jørgensen, P.; Lerouge, T.; Cerviño, M.; Chambon, P.; Losson, R. Nizp1, a Novel Multitype Zinc Finger Protein That Interacts with the NSD1 Histone Lysine Methyltransferase through a Unique C2HR Motif. Mol. Cell. Biol. 2004, 24, 5184–5196. [Google Scholar] [CrossRef]
- Berardi, A.; Ghitti, M.; Quilici, G.; Musco, G. In silico derived small molecules targeting the finger-finger interaction between the histone lysine methyltransferase NSD1 and Nizp1 repressor. Comput. Struct. Biotechnol. J. 2020, 18, 4082–4092. [Google Scholar] [CrossRef]
- Fang, Y.; Tang, Y.; Zhang, Y.; Pan, Y.; Jia, J.; Sun, Z.; Zeng, W.; Chen, J.; Yuan, Y.; Fang, D. The H3K36me2 methyltransferase NSD1 modulates H3K27ac at active enhancers to safeguard gene expression. Nucleic Acids Res. 2021, 49, 6281–6295. [Google Scholar] [CrossRef] [PubMed]
- Yamada-Okabe, T.; Matsumoto, N. Decreased serum dependence in the growth of NIH3T3 cells from the overexpression of human nuclear receptor-binding SET-domain-containing protein 1 (NSD1) or fission yeast su(var)3-9, enhancer-of-zeste, trithorax 2 (SET2). Cell Biochem. Funct. 2007, 26, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Kim, T.; Kim, S.; Hong, Y.-K.; Cho, K.S.; Lee, I.-S. Overexpression of histone methyltransferase NSD in Drosophila induces apoptotic cell death via the Jun-N-terminal kinase pathway. Biochem. Biophys. Res. Commun. 2018, 496, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, T.; Jeong, Y.; Choi, S.; Yamaguchi, M.; Lee, I.-S. The Drosophila histone methyltransferase NSD is positively regulated by the DRE/DREF system. Genes Genom. 2018, 40, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Shin, H.; Song, B.; Won, C.; Yoshida, H.; Yamaguchi, M.; Cho, K.S.; Lee, I. Overexpression of H3K36 methyltransferase NSD in glial cells affects brain development in Drosophila. Glia 2020, 68, 2503–2516. [Google Scholar] [CrossRef]
- Choi, S.; Song, B.; Shin, H.; Won, C.; Kim, T.; Yoshida, H.; Lee, D.; Chung, J.; Cho, K.S.; Lee, I.-S. Drosophila NSD deletion induces developmental anomalies similar to those seen in Sotos syndrome 1 patients. Genes Genom. 2021, 43, 737–748. [Google Scholar] [CrossRef]
- Rayasam, G.V.; Wendling, O.; Angrand, P.-O.; Mark, M.; Niederreither, K.; Song, L.; Lerouge, T.; Hager, G.L.; Chambon, P.; Losson, R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003, 22, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nat. Cell Biol. 2004, 429, 900–903. [Google Scholar] [CrossRef]
- Shirane, K.; Miura, F.; Ito, T.; Lorincz, M.C. NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat. Genet. 2020, 52, 1088–1098. [Google Scholar] [CrossRef]
- Leonards, K.; Almosailleakh, M.; Tauchmann, S.; Bagger, F.O.; Thirant, C.; Juge, S.; Bock, T.; Méreau, H.; Bezerra, M.F.; Tzankov, A.; et al. Nuclear interacting SET domain protein 1 inactivation impairs GATA1-regulated erythroid differentiation and causes erythroleukemia. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Amarilio, R.; Viukov, S.V.; Sharir, A.; Eshkar-Oren, I.; Johnson, R.; Zelzer, E. HIF1α regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development 2007, 134, 3917–3928. [Google Scholar] [CrossRef]
- Shao, R.; Zhang, Z.; Xu, Z.; Ouyang, H.; Wang, L.; Ouyang, H.; Zou, W. H3K36 methyltransferase NSD1 regulates chondrocyte differentiation for skeletal development and fracture repair. Bone Res. 2021, 9, 1–11. [Google Scholar]
- Oishi, S.; Zalucki, O.; Vega, M.S.; Harkins, D.; Harvey, T.J.; Kasherman, M.; Davila, R.A.; Hale, L.; White, M.; Piltz, S.; et al. Investigating cortical features of Sotos syndrome using mice heterozygous for Nsd1. Genes Brain Behav. 2020, 19, e12637. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, N.; Imaizumi, K.; Harada, N.; Masuno, M.; Kondoh, T.; Nagai, T.; Ohashi, H.; Naritomi, K.; Tsukahara, M.; Makita, Y.; et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002, 30, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Rahman, N. Sotos syndrome. Eur. J. Hum. Genet. 2006, 15, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Dikow, N.; Maas, B.; Gaspar, H.; Kreiss-Nachtsheim, M.; Engels, H.; Kuechler, A.; Garbes, L.; Netzer, C.; Neuhann, T.M.; Koehler, U.; et al. The phenotypic spectrum of duplication 5q35.2-q35.3 encompassing NSD1: Is it really a reversed sotos syndrome? Am. J. Med Genet. Part A 2013, 161, 2158–2166. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Loveday, C.; Yost, S.; Clarke, M.; Ramsay, E.; Zachariou, A.; Elliott, A.; Wylie, H.; Ardissone, A.; Rittinger, O.; et al. Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. Am. J. Hum. Genet. 2017, 100, 725–736. [Google Scholar] [CrossRef]
- Madsen, R.R.; Knox, R.G.; Pearce, W.; Lopez, S.; Mahler-Araujo, B.; McGranahan, N.; Semple, R.K. Oncogenic PIK3CA promotes cellular stemness in an allele dose-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 8380–8389. [Google Scholar] [CrossRef]
- Madsen, R.R. PI3K in stemness regulation: From development to cancer. Biochem. Soc. Trans. 2020, 48, 301–315. [Google Scholar] [CrossRef]
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.Y.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.A.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6, 10207. [Google Scholar] [CrossRef]
- Martin-Herranz, D.E.; Aref-Eshghi, E.; Bonder, M.J.; Stubbs, T.M.; Choufani, S.; Weksberg, R.; Stegle, O.; Sadikovic, B.; Reik, W.; Thornton, J.M. Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jaju, R.J. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 2001, 98, 1264–1267. [Google Scholar] [CrossRef]
- Hollink, I.H.I.M.; Heuvel-Eibrink, M.M.V.D.; Arentsen-Peters, S.T.C.J.M.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; Van Galen, J.F.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.-J.J.L.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Thanasopoulou, A.; Tzankov, A.; Schwaller, J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica 2014, 99, 1465–1471. [Google Scholar] [CrossRef]
- Dolnik, A.; Engelmann, J.C.; Scharfenberger-Schmeer, M.; Mauch, J.; Kelkenberg-Schade, S.; Haldemann, B.; Fries, T.; Krönke, J.; Kühn, M.W.M.; Paschka, P.; et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 2012, 120, e83–e92. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Zhang, J.; Mouawad, R.; Compérat, E.; Rouprêt, M.; Allanic, F.; Malouf, G.G. NSD1 Inactivation and SETD2 Mutation Drive a Convergence toward Loss of Function of H3K36 Writers in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017, 77, 4835–4845. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Mohammad, F.; Helin, K. Oncohistones: Drivers of pediatric cancers. Genes Dev. 2017, 31, 2313–2324. [Google Scholar] [CrossRef]
- Schuhmacher, M.; Kusevic, D.; Kudithipudi, S.; Jeltsch, A. Kinetic Analysis of the Inhibition of the NSD1, NSD2 and SETD2 Protein Lysine Methyltransferases by a K36M Oncohistone Peptide. ChemistrySelect 2017, 2, 9532–9536. [Google Scholar] [CrossRef]
- Farhangdoost, N.; Horth, C.; Hu, B.; Bareke, E.; Chen, X.; Li, Y.; Coradin, M.; Garcia, B.A.; Lu, C.; Majewski, J. Chromatin dysregulation associated with NSD1 mutation in head and neck squamous cell carcinoma. Cell Rep. 2021, 34, 108769. [Google Scholar] [CrossRef]
- Rajagopalan, K.N.; Chen, X.; Weinberg, D.N.; Chen, H.; Majewski, J.; Allis, C.D.; Lu, C. Depletion of H3K36me2 recapitulates epigenomic and phenotypic changes induced by the H3.3K36M oncohistone mutation. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Brumbaugh, J.; Kim, I.S.; Ji, F.; Huebner, A.J.; Di Stefano, B.; Schwarz, B.A.; Charlton, J.; Coffey, A.; Choi, J.; Walsh, R.M.; et al. Inducible histone K-to-M mutations are dynamic tools to probe the physiological role of site-specific histone methylation in vitro and in vivo. Nat. Cell Biol. 2019, 21, 1449–1461. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global Levels of Specific Histone Modifications and an Epigenetic Gene Signature Predict Prostate Cancer Progression and Development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Bakardjieva-Mihaylova, V.; Kramarzova, K.S.; Slamova, M.; Svaton, M.; Rejlova, K.; Zaliova, M.; Dobiasova, A.; Fiser, K.; Stuchly, J.; Grega, M.; et al. Molecular Basis of Cisplatin Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 1316. [Google Scholar] [CrossRef]
- Lee, S.-T.; Wiemels, J.L. Genome-wide CpG island methylation and intergenic demethylation propensities vary among different tumor sites. Nucleic Acids Res. 2015, 44, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Supek, F.; Lehner, B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Spurr, L.; Li, M.; Alomran, N.; Zhang, Q.; Restrepo, P.; Movassagh, M.; Trenkov, C.; Tunnessen, N.; Apanasovich, T.; Crandall, K.A.; et al. Systematic pan-cancer analysis of somatic allele frequency. Sci. Rep. 2018, 8, 7735. [Google Scholar] [CrossRef]
- Chang, Y.; Zhang, X.; Horton, J.; Upadhyay, A.K.; Spannhoff, A.; Liu, J.; Snyder, J.P.; Bedford, M.T.; Cheng, X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Mevius, D.E.H.F.; Shen, Y.; Zhao, S.; di Luccio, E. BIX-01294 inhibits oncoproteins NSD1, NSD2 and NSD3. Med. Chem. Res. 2017, 26, 2038–2047. [Google Scholar] [CrossRef]
- Drake, K.M.; Watson, V.G.; Kisielewski, A.; Glynn, R.; Napper, A.D. A Sensitive Luminescent Assay for the Histone Methyltransferase NSD1 and Other SAM-Dependent Enzymes. ASSAY Drug Dev. Technol. 2014, 12, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Castellar, A.; Oyarzabal, J. Novel pharmacological maps of protein lysine methyltransferases: Key for target deorphanization. J. Chem. 2018, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Howard, C.A.; Zari, S.; Cho, H.J.; Shukla, S.; Li, H.; Ndoj, J.; González-Alonso, P.; Nikolaidis, C.; Abbott, J.; et al. Covalent inhibition of NSD1 histone methyltransferase. Nat. Chem. Biol. 2020, 16, 1403–1410. [Google Scholar] [CrossRef]
- Rahman, N. Mechanisms predisposing to childhood overgrowth and cancer. Curr. Opin. Genet. Dev. 2005, 15, 227–233. [Google Scholar] [CrossRef]
- Cytrynbaum, C.; Choufani, S.; Weksberg, R. Epigenetic signatures in overgrowth syndromes: Translational opportunities. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 491–501. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.; Lo, C.C.; McDonald, E.R., III; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Garraway, L.A. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Brennan, K.; Shin, J.H.; Tay, J.K.; Prunello, M.; Gentles, A.J.; Sunwoo, J.B.; Gevaert, O. NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Saloura, V.; Izumchenko, E.; Zuo, Z.; Bao, R.; Korzinkin, M.; Ozerov, I.; Zhavoronkov, A.; Sidransky, D.; Bedi, A.; Hoque, M.O.; et al. Immune profiles in primary squamous cell carcinoma of the head and neck. Oral Oncol. 2019, 96, 77–88. [Google Scholar] [CrossRef]
- Han, J.; Jun, Y.; Kim, S.H.; Hoang, H.-H.; Jung, Y.; Kim, S.; Kim, J.; Austin, R.H.; Lee, S.; Park, S. Rapid emergence and mechanisms of resistance by U87 glioblastoma cells to doxorubicin in an in vitro tumor microfluidic ecology. Proc. Natl. Acad. Sci. USA 2016, 113, 14283–14288. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.M.; Sims, D.; Dexter, T.; Fenwick, K.; Assiotis, I.; Kozarewa, I.; Mitsopoulos, C.; Hakas, J.; Zvelebil, M.; Lord, C.; et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc. Natl. Acad. Sci. USA 2012, 109, 2730–2735. [Google Scholar] [CrossRef]
- McNeer, N.; Philip, J.; Geiger, H.; Ries, R.E.; Lavallee, V.-P.; Walsh, M.; Shah, M.; Arora, K.; Emde, A.-K.; Robine, N.; et al. Abstract 2870: Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Tumor Biol. 2019, 33, 1934–1943. [Google Scholar]
- Barresi, V.; Di Bella, V.; Andriano, N.; Privitera, A.; Bonaccorso, P.; La Rosa, M.; Iachelli, V.; Spampinato, G.; Pulvirenti, G.; Scuderi, C.; et al. NUP-98 Rearrangements Led to the Identification of Candidate Biomarkers for Primary Induction Failure in Pediatric Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 4575. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; McCloskey, A.; Shokhirev, M.N.; Benner, C.; Rathore, A.; Hetzer, M.W. Nup98 recruits the Wdr82–Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev. 2017, 31, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.; Hu, W.; Chen, C.-W.; Chu, S.H.; Brien, G.; Park, C.Y.; et al. NUP98 Fusion Proteins Interact with the NSL and MLL1 Complexes to Drive Leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tauchmann, S.; Schwaller, J. NSD1: A Lysine Methyltransferase between Developmental Disorders and Cancer. Life 2021, 11, 877. https://doi.org/10.3390/life11090877
Tauchmann S, Schwaller J. NSD1: A Lysine Methyltransferase between Developmental Disorders and Cancer. Life. 2021; 11(9):877. https://doi.org/10.3390/life11090877
Chicago/Turabian StyleTauchmann, Samantha, and Juerg Schwaller. 2021. "NSD1: A Lysine Methyltransferase between Developmental Disorders and Cancer" Life 11, no. 9: 877. https://doi.org/10.3390/life11090877
APA StyleTauchmann, S., & Schwaller, J. (2021). NSD1: A Lysine Methyltransferase between Developmental Disorders and Cancer. Life, 11(9), 877. https://doi.org/10.3390/life11090877