
Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase



Abstract
1. Introduction
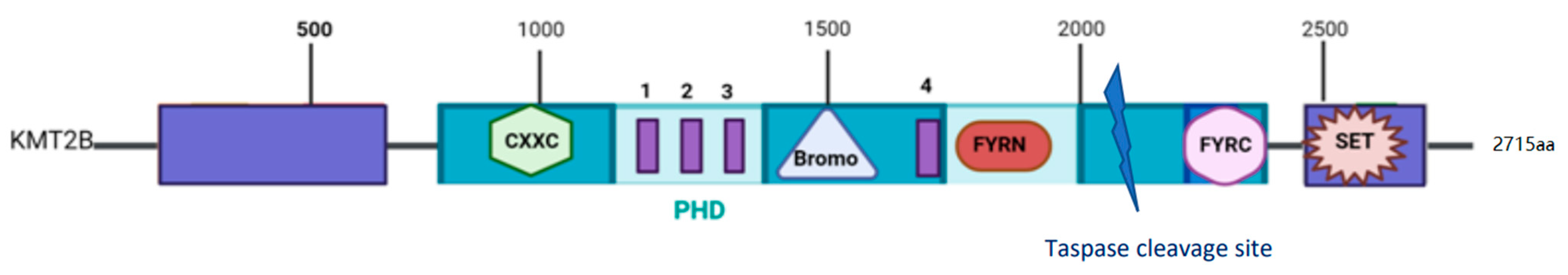
2. The MLL2 Protein
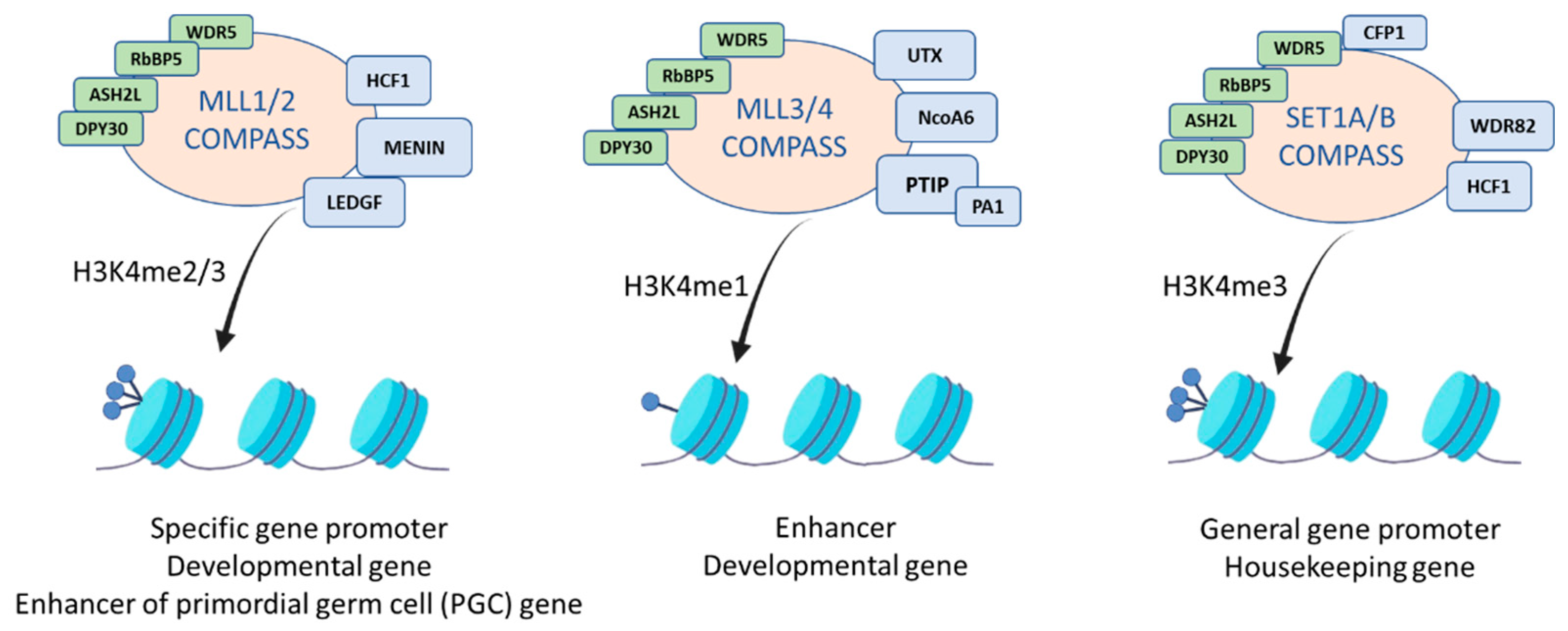
3. The MLL2 Protein Complex
4. Structural Nucleosome Recognition by MLL Complexes
5. MLL2 Role in Transcription Regulation
6. MLL2 Role in Human Physiology
7. MLL2 Implication in Diseases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gu, B.; Lee, M.G. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013, 3, 1–14. [Google Scholar] [CrossRef]
- Herz, H.M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.-M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 Branches of the COMPASS Family Function as Major Histone H3K4 Monomethylases at Enhancers. Mol. Cell. Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by polycomb and compass families. Science 2016, 352, 6290. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kim, J.A.; Kim, J. Transcriptional regulation by the KMT2 histone H3K4 methyltransferases. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194545. [Google Scholar] [CrossRef]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef]
- Fitzgerald, K.T.; Diaz, M.O. MLL2: A new mammalian member of the trx/MLL family of genes. Genomics 1999, 59, 187–192. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Li, Y.; Han, J.; Zhang, Y.; Cao, F.; Liu, Z.; Li, S.; Wu, J.; Hu, C.; Wang, Y.; Shuai, J.; et al. Structural basis for activity regulation of MLL family methyltransferases. Nature 2016, 530, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Grummitt, C.G.; Hilcenko, C.; Min, S.Y.; Tonkin, L.M.; Johnson, C.M.; Freund, S.M.; Bycroft, M.; Warren, A.J. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 2006, 25, 4503–4512. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Hom, R.A.; Blakeslee, W.; Ikenouye, L.; Kutateladze, T.G. Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 366–371. [Google Scholar] [CrossRef]
- Sanchez, R.; Zhou, M.M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, J.; Milne, T.A.; Wang, G.G.; Li, H.; Allis, C.D.; Patel, D.J. Pro isomerization in MLL1 PHD3-Bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell 2010, 141, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.D.; Cheng, E.H.Y.; Korsmeyer, S.J. Taspase1: A threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 2003, 115, 293–303. [Google Scholar] [CrossRef]
- Hsieh, J.J.-D.; Ernst, P.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Proteolytic Cleavage of MLL Generates a Complex of N- and C-Terminal Fragments That Confers Protein Stability and Subnuclear Localization. Mol. Cell. Biol. 2003, 23, 186–194. [Google Scholar] [CrossRef]
- Takeda, S.; Chen, D.Y.; Westergard, T.D.; Fisher, J.K.; Rubens, J.A.; Sasagawa, S.; Kan, J.T.; Korsmeyer, S.J.; Cheng, E.H.Y.; Hsieh, J.J.D. Proteolysis of MLL family proteins is essential for Taspase1-orchestrated cell cycle progression. Genes Dev. 2006, 20, 2397–2409. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kitabayashi, I.; Ayton, P.M.; Cleary, M.L.; Ohki, M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood 2002, 100, 3710–3718. [Google Scholar] [CrossRef]
- Zeleznik-Le, N.J.; Harden, A.M.; Rowley, J.D. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl. Acad. Sci. USA 1994, 91, 10610–10614. [Google Scholar] [CrossRef]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef]
- Ford, D.J.; Dingwall, A.K. The cancer COMPASS: Navigating the functions of MLL complexes in cancer. Cancer Genet. 2015, 208, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Dharmarajan, V.; Vought, V.E.; Cosgrove, M.S. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2009, 284, 24242–24256. [Google Scholar] [CrossRef]
- Cao, F.; Chen, Y.; Cierpicki, T.; Liu, Y.; Basrur, V.; Lei, M.; Dou, Y. An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS ONE 2010, 5, e14102. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Yao, T.; Cao, M.; Zhu, G.; Li, Y.; Yuan, G.; Chen, Y.; Lei, M.; Huang, J. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature 2019, 573, 445–449. [Google Scholar] [CrossRef]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin associates with a trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar] [CrossRef]
- Jiang, H.; Lu, X.; Shimada, M.; Dou, Y.; Tang, Z.; Roeder, R.G. Regulation of transcription by the MLL2 complex and MLL complex-associated AKAP95. Nat. Struct. Mol. Biol. 2013, 20, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Hong, T.; Hong, S.H.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef]
- Goo, Y.-H.; Sohn, Y.C.; Kim, D.-H.; Kim, S.-W.; Kang, M.-J.; Jung, D.-J.; Kwak, E.; Barlev, N.A.; Berger, S.L.; Chow, V.T.; et al. Activating Signal Cointegrator 2 Belongs to a Novel Steady-State Complex That Contains a Subset of Trithorax Group Proteins. Mol. Cell. Biol. 2003, 23, 140–149. [Google Scholar] [CrossRef]
- Patel, S.R.; Kim, D.; Levitan, I.; Dressler, G.R. The BRCT-Domain Containing Protein PTIP Links PAX2 to a Histone H3, Lysine 4 Methyltransferase Complex. Dev. Cell 2007, 13, 580–592. [Google Scholar] [CrossRef]
- Lee, J.H.; Tate, C.M.; You, J.S.; Skalnik, D.G. Identification and characterization of the human Set1B histone H3-Lys 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 13419–13428. [Google Scholar] [CrossRef]
- Mohan, M.; Herz, H.-M.; Smith, E.R.; Zhang, Y.; Jackson, J.; Washburn, M.P.; Florens, L.; Eissenberg, J.C.; Shilatifard, A. The COMPASS Family of H3K4 Methylases in Drosophila. Mol. Cell. Biol. 2011, 31, 4310–4318. [Google Scholar] [CrossRef]
- Poreba, E.; Lesniewicz, K.; Durzynska, J. Aberrant activity of histone–lysine n-methyltransferase 2 (Kmt2) complexes in oncogenesis. Int. J. Mol. Sci. 2020, 21, 9340. [Google Scholar] [CrossRef]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006, 7, 397–403. [Google Scholar] [CrossRef]
- Musselman, C.A.; Khorasanizadeh, S.; Kutateladze, T.G. Towards understanding methyllysine readout. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 686–693. [Google Scholar] [CrossRef]
- Dreijerink, K.M.A.; Mulder, K.W.; Winkler, G.S.; Höppener, J.W.M.; Lips, C.J.M.; Timmers, H.T.M. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006, 66, 4929–4935. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Sindhu, K.V.; Li, S.; Nishio, H.; Stoller, J.Z.; Oishi, K.; Puttreddy, S.; Lee, T.J.; Epstein, J.A.; Walsh, M.J.; et al. Transcription factor Ap2δ associates with Ash2l and ALR, a trithorax family histone methyltransferase, to activate Hoxc8 transcription. Proc. Natl. Acad. Sci. USA 2008, 105, 7472–7477. [Google Scholar] [CrossRef]
- Ullius, A.; Lüscher-Firzlaff, J.; Costa, I.G.; Walsemann, G.; Forst, A.H.; Gusmao, E.G.; Kapelle, K.; Kleine, H.; Kremmer, E.; Vervoorts, J.; et al. The interaction of MYC with the trithorax protein ASH2L promotes gene transcription by regulating H3K27 modification. Nucleic Acids Res. 2014, 42, 6901–6920. [Google Scholar] [CrossRef] [PubMed]
- Demers, C.; Chaturvedi, C.P.; Ranish, J.A.; Juban, G.; Lai, P.; Morle, F.; Aebersold, R.; Dilworth, F.J.; Groudine, M.; Brand, M. Activator-Mediated Recruitment of the MLL2 Methyltransferase Complex to the β-Globin Locus. Mol. Cell 2007, 27, 573–584. [Google Scholar] [CrossRef]
- Deng, C.; Li, Y.; Liang, S.; Cui, K.; Salz, T.; Yang, H.; Tang, Z.; Gallagher, P.G.; Qiu, Y.; Roeder, R.; et al. USF1 and hSET1A Mediated Epigenetic Modifications Regulate Lineage Differentiation and HoxB4 Transcription. PLoS Genet. 2013, 9, e1003524. [Google Scholar] [CrossRef] [PubMed]
- Fossati, A.; Dolfini, D.; Donati, G.; Mantovani, R. NF-Y recruits Ash2L to impart H3K4 trimethylation on CCAAT promoters. PLoS ONE 2011, 6, e17220. [Google Scholar] [CrossRef]
- Tyagi, S.; Chabes, A.L.; Wysocka, J.; Herr, W. E2F Activation of S Phase Promoters via Association with HCF-1 and the MLL Family of Histone H3K4 Methyltransferases. Mol. Cell 2007, 27, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Madrigal, P.; Galli, A.; Hubner, N.C.; Moreno, I.; Burks, D.; Brown, S.; Pedersen, R.A.; Gaffney, D.; Mendjan, S.; et al. Activin/Nodal signaling and NANOG orchestrate human embryonic stem cell fate decisions by controlling the H3K4me3 chromatin mark. Genes Dev. 2015, 29, 702–717. [Google Scholar] [CrossRef]
- Kawabe, Y.I.; Wang, Y.X.; McKinnell, I.W.; Bedford, M.T.; Rudnicki, M.A. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 2012, 11, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Chen, W.Y.; Shimada, M.; Nguyen, U.T.T.; Kim, J.; Sun, X.J.; Sengoku, T.; McGinty, R.K.; Fernandez, J.P.; Muir, T.W.; et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell 2013, 154, 297. [Google Scholar] [CrossRef]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.; Hess, J.L. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.B.; Anderson, M.; Diaz, M.O.; Zeleznik-Le, N.J. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc. Natl. Acad. Sci. USA 2003, 100, 8342–8347. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Ayoub, A.; Lee, Y.T.; Xu, J.; Kim, H.; Zheng, W.; Zhang, B.; Sha, L.; An, S.; Zhang, Y.; et al. Cryo-EM structure of the human MLL1 core complex bound to the nucleosome. Nat. Commun. 2019, 5, 5540. [Google Scholar] [CrossRef]
- Vedadi, M.; Blazer, L.; Eram, M.S.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Hajian, T. Targeting human SET1/MLL family of proteins. Protein Sci. 2017, 26, 662–676. [Google Scholar] [CrossRef]
- Lee, Y.T.; Ayoub, A.; Park, S.H.; Sha, L.; Xu, J.; Mao, F.; Zheng, W.; Zhang, Y.; Cho, U.S.; Dou, Y. Mechanism for DPY30 and ASH2L intrinsically disordered regions to modulate the MLL/SET1 activity on chromatin. Nat. Commun. 2021, 19, 2953. [Google Scholar] [CrossRef]
- An, W.; Roeder, R.G. Reconstitution and Transcriptional Analysis of Chromatin In vitro. Methods Enzymol. 2003, 377, 460–474. [Google Scholar] [CrossRef]
- Hu, D.; Gao, X.; Cao, K.; Morgan, M.A.; Mas, G.; Smith, E.R.; Volk, A.G.; Bartom, E.T.; Crispino, J.D.; Di Croce, L.; et al. Not All H3K4 Methylations Are Created Equal: Mll2/COMPASS Dependency in Primordial Germ Cell Specification. Mol. Cell 2017, 65, 460–475.e6. [Google Scholar] [CrossRef]
- Bach, C.; Mueller, D.; Buhl, S.; Garcia-Cuellar, M.P.; Slany, R.K. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene 2009, 28, 815–823. [Google Scholar] [CrossRef]
- Milne, T.A.; Kim, J.; Wang, G.G.; Stadler, S.C.; Basrur, V.; Whitcomb, S.J.; Wang, Z.; Ruthenburg, A.J.; Elenitoba-Johnson, K.S.J.; Roeder, R.G.; et al. Multiple Interactions Recruit MLL1 and MLL1 Fusion Proteins to the HOXA9 Locus in Leukemogenesis. Mol. Cell 2010, 38, 853–863. [Google Scholar] [CrossRef]
- Xu, C.; Liu, K.; Lei, M.; Yang, A.; Li, Y.; Hughes, T.R.; Min, J. DNA Sequence Recognition of Human CXXC Domains and Their Structural Determinants. Structure 2018, 26, 85–95.e3. [Google Scholar] [CrossRef]
- Tomizawa, S.I.; Kobayashi, Y.; Shirakawa, T.; Watanabe, K.; Mizoguchi, K.; Hoshi, I.; Nakajima, K.; Nakabayashi, J.; Singh, S.; Dahl, A.; et al. Kmt2b conveys monovalent and bivalent H3K4me3 in mouse spermatogonial stem cells at germline and embryonic promoters. Development 2018, 145, dev169102. [Google Scholar] [CrossRef]
- Denissov, S.; Hofemeister, H.; Marks, H.; Kranz, A.; Ciotta, G.; Singh, S.; Anastassiadis, K.; Stunnenberg, H.G.; Stewart, A.F. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 2014, 141, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Sze, C.C.; Cao, K.; Collings, C.K.; Marshall, S.A.; Rendleman, E.J.; Ozark, P.A.; Chen, F.X.; Morgan, M.A.; Wang, L.; Shilatifard, A. Histone H3K4 methylation-dependent and -independent functions of set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev. 2017, 31, 1732–1737. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Douillet, D.; Sze, C.C.; Ryan, C.; Piunti, A.; Shah, A.P.; Ugarenko, M.; Marshall, S.A.; Rendleman, E.J.; Zha, D.; Helmin, K.A.; et al. Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat. Genet. 2020, 52, 615–625. [Google Scholar] [CrossRef]
- Sze, C.C.; Ozark, P.A.; Cao, K.; Ugarenko, M.; Das, S.; Wang, L.; Marshall, S.A.; Rendleman, E.J.; Ryan, C.A.; Zha, D.; et al. Coordinated regulation of cellular identity–associated H3K4me3 breadth by the COMPASS family. Sci. Adv. 2020, 6, eaaz4764. [Google Scholar] [CrossRef]
- Margaritis, T.; Oreal, V.; Brabers, N.; Maestroni, L.; Vitaliano-Prunier, A.; Benschop, J.J.; van Hooff, S.; van Leenen, D.; Dargemont, C.; Géli, V.; et al. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3’-end antisense transcription. PLoS Genet. 2012, 8, e1002952. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Webb, S.; Bird, A. Cfp1 is required for gene expression-dependent H3K4 trimethylation and H3K9 acetylation in embryonic stem cells. Genome Biol. 2014, 15, 451. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Ladopoulos, V.; Hofemeister, H.; Hoogenkamp, M.; Riggs, A.D.; Stewart, A.F.; Bonifer, C. The Histone Methyltransferase KMT2B Is Required for RNA Polymerase II Association and Protection from DNA Methylation at the MagohB CpG Island Promoter. Mol. Cell. Biol. 2013, 33, 1383–1393. [Google Scholar] [CrossRef]
- Hanna, C.W.; Taudt, A.; Huang, J.; Gahurova, L.; Kranz, A.; Andrews, S.; Dean, W.; Stewart, A.F.; Colomé-Tatché, M.; Kelsey, G. MLL2 conveys transcription-independent H3K4 trimethylation in oocytes. Nat. Struct Mol. Biol. 2018, 25, 73–82. [Google Scholar] [CrossRef]
- Guo, C.; Chang, C.C.; Wortham, M.; Chen, L.H.; Kernagis, D.N.; Qin, X.; Cho, Y.W.; Chi, J.T.; Grant, G.A.; McLendon, R.E.; et al. Global identification of MLL2-targeted loci reveals MLL2′s role in diverse signaling pathways. Proc. Natl. Acad. Sci. USA 2012, 109, 17603–17608. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.; Tian, X.; Peng, C.; Gong, F.; Chen, Y. Crystal Structure of MLL2 Complex Guides the Identification of a Methylation Site on P53 Catalyzed by KMT2 Family Methyltransferases. Structure 2020, 28, 1141–1148.e4. [Google Scholar] [CrossRef]
- Crump, N.T.; Milne, T.A. Why are so many MLL lysine methyltransferases required for normal mammalian development? Cell. Mol. Life Sci. 2019, 76, 2885–2898. [Google Scholar] [CrossRef]
- Glaser, S.; Schaft, J.; Lubitz, S.; Vintersten, K.; van der Hoeven, F.; Tuftteland, K.R.; Aasland, R.; Anastassiadis, K.; Ang, S.L.; Stewart, A.F. Multiple epigenetic maintenance factors implicated by the loss of MII2 in mouse development. Development 2006, 133, 1423–1432. [Google Scholar] [CrossRef]
- Glaser, S.; Lubitz, S.; Loveland, K.L.; Ohbo, K.; Robb, L.; Schwenk, F.; Seibler, J.; Roellig, D.; Kranz, A.; Anastassiadis, K.; et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin 2009, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Vieyra, C.V.; Chen, R.; Agno, J.E.; Glaser, S.; Anastassiadis, K.; Stewart Francis, A.; Matzuk, M.M. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010, 8, 53–54. [Google Scholar] [CrossRef]
- Antunes, E.T.B.; Ottersbach, K. The MLL/SET family and haematopoiesis. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194579. [Google Scholar] [CrossRef]
- Yang, W.; Ernst, P. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2017, 24, 322–328. [Google Scholar] [CrossRef]
- Sierra, J.; Yoshida, T.; Joazeiro, C.A.; Jones, K.A. The APC tumor suppressor counteracts β-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006, 20, 586–600. [Google Scholar] [CrossRef]
- Zech, M.; Boesch, S.; Maier, E.M.; Borggraefe, I.; Vill, K.; Laccone, F.; Pilshofer, V.; Ceballos-Baumann, A.; Alhaddad, B.; Berutti, R.; et al. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. Am. J. Hum. Genet. 2016, 99, 1377–1387. [Google Scholar] [CrossRef]
- Meyer, E.; Carss, K.J.; Rankin, J.; Nichols, J.M.E.; Grozeva, D.; Joseph, A.P.; Mencacci, N.E.; Papandreou, A.; Ng, J.; Barral, S.; et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet. 2017, 49, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Ng, A.; Galosi, S.; Salz, L.; Wong, T.; Schwager, C.; Amudhavalli, S.; Gelineau-Morel, R.; Chowdhury, S.; Friedman, J.; et al. Failure to thrive—An overlooked manifestation of KMT2B-related dystonia: A case presentation. BMC Neurol. 2020, 20, 1–6. [Google Scholar] [CrossRef]
- Takahashi, S.; Yokoyama, A. The molecular functions of common and atypical MLL fusion protein complexes. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2020, 1863, 194548. [Google Scholar] [CrossRef]
- Risner, L.E.; Kuntimaddi, A.; Lokken, A.A.; Achille, N.J.; Birch, N.W.; Schoenfelt, K.; Bushweller, J.H.; Zeleznik-Le, N.J. Functional specificity of CpG DNA-binding CXXC domains in mixed lineage leukemia. J. Biol. Chem. 2013, 288, 29901–29910. [Google Scholar] [CrossRef]
- Chen, Y.; Anastassiadis, K.; Kranz, A.; Stewart, A.F.; Arndt, K.; Waskow, C.; Yokoyama, A.; Jones, K.; Neff, T.; Lee, Y.; et al. MLL2, Not MLL1, Plays a Major Role in Sustaining MLL-Rearranged Acute Myeloid Leukemia. Cancer Cell 2017, 31, 755–770.e6. [Google Scholar] [CrossRef]
- Thiel, A.T.; Blessington, P.; Zou, T.; Feather, D.; Wu, X.; Yan, J.; Zhang, H.; Liu, Z.; Ernst, P.; Koretzky, G.A.; et al. MLL-AF9-Induced Leukemogenesis Requires Coexpression of the Wild-Type Mll Allele. Cancer Cell 2010, 17, 148–159. [Google Scholar] [CrossRef]
- Lu, H.; Yang, S.; Zhu, H.; Tong, X.; Xie, F.; Qin, J.; Han, N.; Wu, X.; Fan, Y.; Shao, Y.W.; et al. Targeted next generation sequencing identified clinically actionable mutations in patients with esophageal sarcomatoid carcinoma. BMC Cancer 2018, 18, 1–7. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Genomic Alterations in Advanced Gastric Cancer Endoscopic Biopsy Samples Using Targeted Next-Generation Sequencing—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/28744403/ (accessed on 28 June 2021).
- Wong, W.H.; Junck, L.; Druley, T.E.; Gutmann, D.H. NF1 glioblastoma clonal profiling reveals KMT2B mutations as potential somatic oncogenic events. Neurology 2019, 93, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Huntsman, D.G.; Chin, S.F.; Muleris, M.; Batley, S.J.; Collins, V.P.; Wiedemann, L.M.; Aparicio, S.; Caldas, C. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene 1999, 18, 7975–7984. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Wheeler, D.A.; Roberts, L.R. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Kishimoto, W.; Nishikori, M. Molecular pathogenesis of follicular lymphoma. J. Clin. Exp. Hematop. 2014, 54, 23–30. [Google Scholar] [CrossRef]
- Mountzios, G.; Rampias, T.; Psyrri, A. The mutational spectrum of squamous-cell carcinoma of the head and neck: Targetable genetic events and clinical impact. Ann. Oncol. 2014, 25, 1889–1900. [Google Scholar] [CrossRef]
- Chan, A.K.N.; Chen, C.-W. Rewiring the Epigenetic Networks in MLL-Rearranged Leukemias: Epigenetic Dysregulation and Pharmacological Interventions. Front. Cell Dev. Biol. 2019, 7, 81. [Google Scholar] [CrossRef]
- Zhu, S.; Cheng, X.; Wang, R.; Tan, Y.; Ge, M.; Li, D.; Xu, Q.; Sun, Y.; Zhao, C.; Chen, S.; et al. Restoration of microRNA function impairs MYC-dependent maintenance of MLL leukemia. Leukemia 2020, 34, 2484–2488. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Writers | Readers | Erasers |
|---|---|---|
| MLL1 MLL2 MLL3 MLL4 SET1A SET1B | BPTF (Bromodomain PHD Finger Transcription Factor) INGs (inhibitor of growth) RAG2 (Recombination Activating 2) TAF3 (TATA-Box Binding Protein Associated Factor 3) CHD1 (Chromodomain Helicase DNA Binding Protein 1) | JARID1A-D (Lysine-specific demethylase 5A, KDM5A) LSD1 (Lysine-specific histone demethylase 1A)/KDM1A (me1/2) JMJD2A (Jumonji domain-containing 2A)/KDM4A (Lysine Demethylase 4A) LSD1/KDM1B (me1/2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klonou, A.; Chlamydas, S.; Piperi, C. Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase. Life 2021, 11, 823. https://doi.org/10.3390/life11080823
Klonou A, Chlamydas S, Piperi C. Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase. Life. 2021; 11(8):823. https://doi.org/10.3390/life11080823
Chicago/Turabian StyleKlonou, Alexia, Sarantis Chlamydas, and Christina Piperi. 2021. "Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase" Life 11, no. 8: 823. https://doi.org/10.3390/life11080823
APA StyleKlonou, A., Chlamydas, S., & Piperi, C. (2021). Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase. Life, 11(8), 823. https://doi.org/10.3390/life11080823