
Role of Transposable Elements in Gene Regulation in the Human Genome



Abstract
1. Overview of Transposable Elements and Their Role in the Human Genome
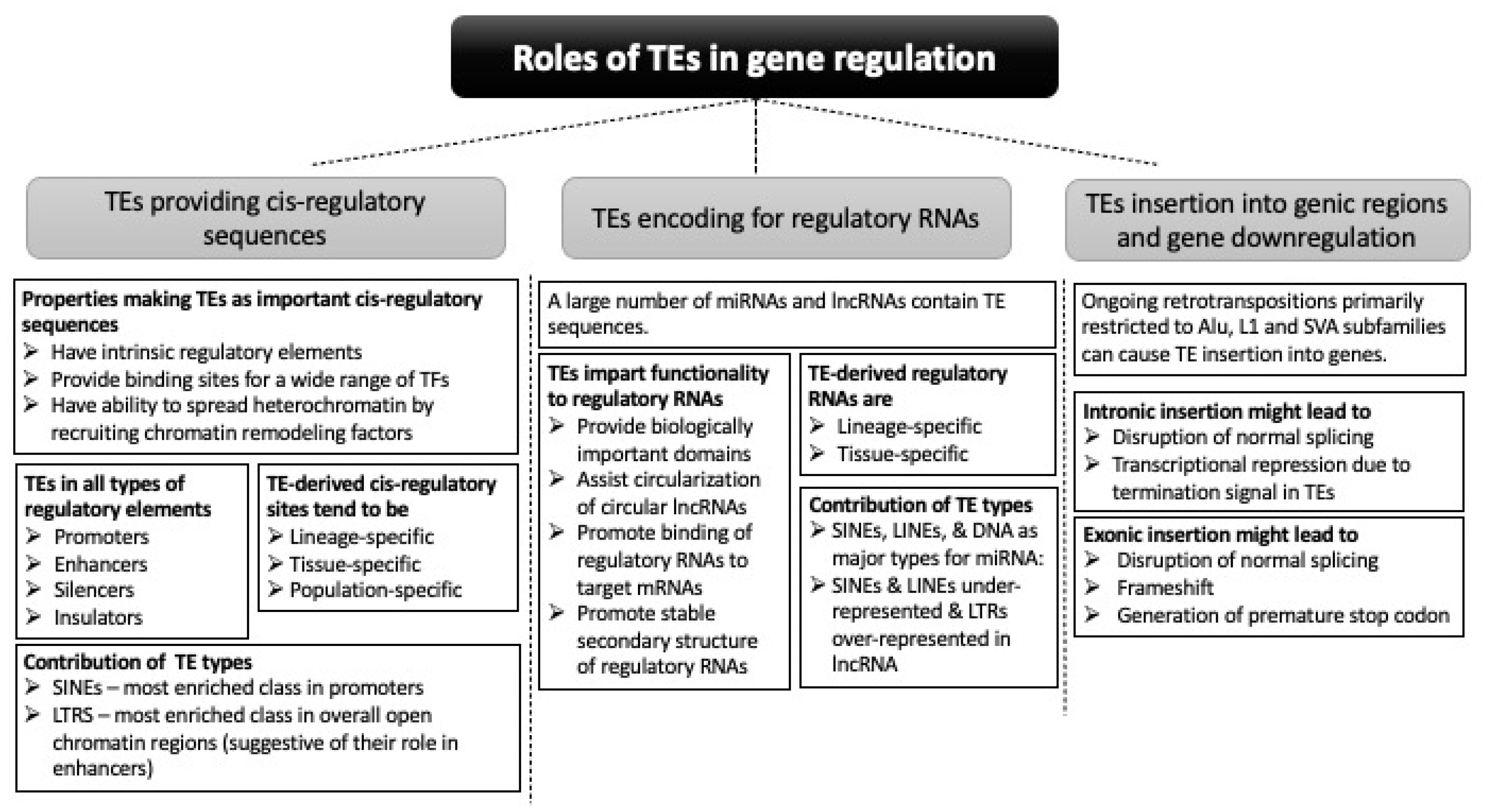
2. Cis-Regulatory Activities of TEs
2.1. Contribution of TEs in Different Regulatory Elements in the Genome
2.1.1. Regulatory Elements in the Genome
2.1.2. Intrinsic Regulatory Properties of TEs
2.1.3. TEs Contribute to Regulatory Elements in the Genome
2.1.4. Contribution of TEs to TFBSs
2.1.5. Differential Contribution of TEs by Type in Regulatory Regions
2.2. Genes Regulated by TE-Derived cis-Regulatory Sequences
2.3. Tissue-Specific Gene Regulation by TEs
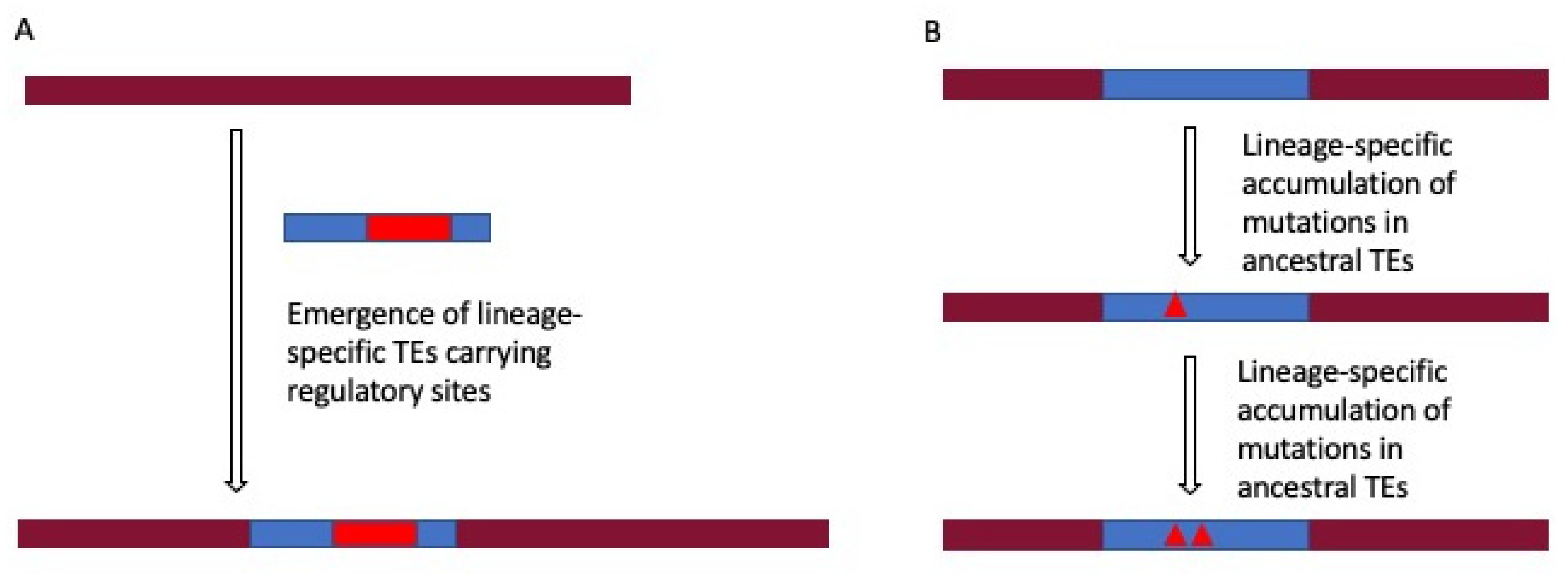
2.4. Lineage-Specific Gene Regulation by TEs
2.5. Population-Specific Gene Regulation by Polymorphic TEs
3. TEs Contribute to Non-Coding Regulatory RNAs
3.1. Contribution of TEs to the Makeup of Regulatory RNAs
3.2. Functional Significance of TEs in Regulatory RNA Sequences
3.3. Role of TEs in Lineage Specificity of Regulatory RNAs
3.4. Tissue-Specificity of TE-Derived Regulatory RNAs
3.5. Differential Contribution to Regulatory RNAs among TE Types
4. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tang, W.; Mun, S.; Joshi, A.; Han, K.; Liang, P. Mobile elements contribute to the uniqueness of human genome with 15,000 human-specific insertions and 14 Mbp sequence increase. DNA Res. 2018, 25, 521–533. [Google Scholar] [CrossRef]
- Stewart, C.; Kural, D.; Strömberg, M.P.; Walker, J.A.; Konkel, M.K.; Stütz, A.M.; Urban, A.E.; Grubert, F.; Lam, H.Y.K.; Lee, W.-P.; et al. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011, 7, e1002236. [Google Scholar] [CrossRef]
- Deininger, P.L.; Moran, J.V.; Batzer, M.A.; Kazazian, H.H. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 2003, 13, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef]
- Pace, J.K.; Feschotte, C. The evolutionary history of human DNA transposons: Evidence for intense activity in the primate lineage. Genome Res. 2007, 17, 422–432. [Google Scholar] [CrossRef]
- Allet, B. Mu insertion duplicates a 5 base pair sequence at the host inserted site. Cell 1979, 16, 123–129. [Google Scholar] [CrossRef]
- Grindley, N.D.F. IS1 insertion generates duplication of a nine base pair sequence at its target site. Cell 1978, 13, 419–426. [Google Scholar] [CrossRef]
- Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [CrossRef] [PubMed]
- Wang, H.; Xing, J.; Grover, D.; Hedges Kyudong Han, D.J.; Walker, J.A.; Batzer, M.A. SVA elements: A hominid-specific retroposon family. J. Mol. Biol. 2005, 354, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Ayarpadikannan, S.; Kim, H.-S. The Impact of Transposable Elements in Genome Evolution and Genetic Instability and Their Implications in Various Diseases. Genom. Inform. 2014, 12, 98. [Google Scholar] [CrossRef]
- Britten, R.J. Transposable element insertions have strongly affected human evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 19945–19948. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Liu, G.; Eichler, E.E. An Alu Transposition Model for the Origin and Expansion of Human Segmental Duplications. Am. J. Hum. Genet. 2003, 73, 823–834. [Google Scholar] [CrossRef]
- Han, K. Genomic rearrangements by LINE-1 insertion-mediated deletion in the human and chimpanzee lineages. Nucleic Acids Res. 2005, 33, 4040–4052. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human Genomic Deletions Mediated by Recombination between Alu Elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef]
- Kelkar, Y.D.; Tyekucheva, S.; Chiaromonte, F.; Makova, K.D. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2007, 18, 30–38. [Google Scholar] [CrossRef]
- Ahmed, M.; Liang, P. Transposable elements are a significant contributor to tandem repeats in the human genome. Comp. Funct. Genom. 2012, 2012, 947089. [Google Scholar] [CrossRef] [PubMed]
- Elisaphenko, E.A.; Kolesnikov, N.N.; Shevchenko, A.I.; Rogozin, I.B.; Nesterova, T.B.; Brockdorff, N.; Zakian, S.M. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS ONE 2008, 3, e2521. [Google Scholar] [CrossRef]
- Sha, M.; Lee, X.; Li, X.P.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Ono, R.; Kobayashi, S.; Wagatsuma, H.; Aisaka, K.; Kohda, T.; Kaneko-Ishino, T.; Ishino, F. A Retrotransposon-Derived Gene, PEG10, Is a Novel Imprinted Gene Located on Human Chromosome 7q21. Genomics 2001, 73, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Nakamura, K.; Inoue, K.; Naruse, M.; Usami, T.; Wakisaka-Saito, N.; Hino, T.; Suzuki-Migishima, R.; Ogonuki, N.; Miki, H.; et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet. 2006, 38, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Szak, S.T.; Boeke, J.D. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 2004, 429, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Stacey, S.N.; Kehr, B.; Gudmundsson, J.; Zink, F.; Jonasdottir, A.; Gudjonsson, S.A.; Sigurdsson, A.; Halldorsson, B.V.; Agnarsson, B.A.; Benediktsdottir, K.R.; et al. Insertion of an SVA-E retrotransposon into the CASP8 gene is associated with protection against prostate cancer. Hum. Mol. Genet. 2016, 25, 1008–1018. [Google Scholar] [CrossRef]
- Vidaud, D.; Vidaud, M.; Bahnak, B.R.; Siguret, V.; Gispert Sanchez, S.; Laurian, Y.; Meyer, D.; Goossens, M.; Lavergne, J.M. Haemophilia B Due to a De Novo Insertion of a Human-Specific Alu Subfamily Member within the Coding Region of the Factor IX Gene. Eur. J. Hum. Genet. 1993, 1, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.É.; Jeyakani, J.; Bourque, G. The Majority of Primate-Specific Regulatory Sequences Are Derived from Transposable Elements. PLoS Genet. 2013, 9, e1003504. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Conley, A.B.; Piriyapongsa, J.; Jordan, I.K. Retroviral promoters in the human genome. Bioinformatics 2008, 24, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.F.; López-Leal, R.; Nasif, S.; Beati, P.; Gelman, D.M.; Low, M.J.; De Souza, F.J.S.; Rubinstein, M. Convergent evolution of two mammalian neuronal enhancers by sequential exaptation of unrelated retroposons. Proc. Natl. Acad. Sci. USA 2011, 108, 15270–15275. [Google Scholar] [CrossRef]
- Gao, T.; Qian, J. EnhancerAtlas 2.0: An updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2019, 48, D58–D64. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Kernaleguen, M.; Daviaud, C.; Shen, Y.; Bonnet, E.; Renault, V.; Deleuze, J.-F.; Mauger, F.; Tost, J. Whole-Genome Bisulfite Sequencing for the Analysis of Genome-Wide DNA Methylation and Hydroxymethylation Patterns at Single-Nucleotide Resolution. In Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 311–349. [Google Scholar]
- The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [CrossRef] [PubMed]
- Martens, J.H.A.; Stunnenberg, H.G. BLUEPRINT: Mapping human blood cell epigenomes. Haematologica 2013, 98, 1487–1489. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef]
- Genomics of Gene Regulation. Available online: https://www.genome.gov/Funded-Programs-Projects/Genomics-of-Gene-Regulation (accessed on 29 January 2021).
- Shooshtari, P.; Feng, S.; Nelakuditi, V.; Foong, J.; Brudno, M.; Cotsapas, C. OCHROdb: A comprehensive, quality checked database of open chromatin regions from sequencing data. bioRxiv 2018, 484840. [Google Scholar] [CrossRef]
- Czipa, E.; Schiller, M.; Nagy, T.; Kontra, L.; Steiner, L.; Koller, J.; Pálné-Szén, O.; Barta, E. ChIPSummitDB: A ChIP-seq-based database of human transcription factor binding sites and the topological arrangements of the proteins bound to them. Database 2020, 2020. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef]
- Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Zheng, R.; Zang, C.; Zhu, M.; Wu, J.; Shi, X.; Taing, L.; et al. Cistrome Data Browser: A data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017, 45, D658–D662. [Google Scholar] [CrossRef]
- Samuelson, L.C.; Wiebauer, K.; Snow, C.M.; Meisler, M.H. Retroviral and pseudogene insertion sites reveal the lineage of human salivary and pancreatic amylase genes from a single gene during primate evolution. Mol. Cell. Biol. 1990, 10, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Brini, A.T.; Lee, G.M.; Kinet, J.P. Involvement of Alu sequences in the cell-specific regulation of transcription of the γ chain of Fc and T cell receptors. J. Biol. Chem. 1993, 268, 1355–1361. [Google Scholar] [CrossRef]
- Hambor, J.E.; Mennone, J.; Coon, M.E.; Hanke, J.H.; Kavathas, P. Identification and characterization of an Alu-containing, T-cell-specific enhancer located in the last intron of the human CD8 alpha gene. Mol. Cell. Biol. 1993, 13, 7056–7070. [Google Scholar] [CrossRef] [PubMed]
- Swergold, G.D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol. Cell. Biol. 1990, 10, 6718–6729. [Google Scholar] [CrossRef]
- van Regenmortel, M.H.; Mahy, B.W. (Eds.) Desk Encyclopedia of General Virology; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Roy, A.M.; West, N.C.; Rao, A.; Adhikari, P.; Alemán, C.; Barnes, A.P.; Deininger, P.L. Upstream flanking sequences and transcription of SINEs. J. Mol. Biol. 2000, 302, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.; Gruss, P. Enhancer elements. Cell 1983, 33, 313–314. [Google Scholar] [CrossRef][Green Version]
- Ono, M.; Kawakami, M.; Takezawa, T. A novel human nonviral retroposon derived from an endogenous retrovirus. Nucleic Acids Res. 1987, 15, 8725–8737. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H. SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol. 2010, 20, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Speek, M. Antisense Promoter of Human L1 Retrotransposon Drives Transcription of Adjacent Cellular Genes. Mol. Cell. Biol. 2001, 21, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Brosius, J.; Gould, S.J. On “genomenclature”: A comprehensive (and respectful) taxonomy for pseudogenes and other “junk DNA”. Proc. Natl. Acad. Sci. USA 1992, 89, 10706–10710. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Vrba, E.S. Exaptation—A Missing Term in the Science of Form. Paleobiology 1982, 8, 4–15. [Google Scholar] [CrossRef]
- Bejerano, G.; Lowe, C.B.; Ahituv, N.; King, B.; Siepel, A.; Salama, S.R.; Rubin, E.M.; James Kent, W.; Haussler, D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 2006, 441, 87–90. [Google Scholar] [CrossRef]
- Lamprecht, B.; Walter, K.; Kreher, S.; Kumar, R.; Hummel, M.; Lenze, D.; Köchert, K.; Bouhlel, M.A.; Richter, J.; Soler, E.; et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med. 2010, 16, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xie, G.; Singh, M.; Ghanbarian, A.T.; Raskó, T.; Szvetnik, A.; Cai, H.; Besser, D.; Prigione, A.; Fuchs, N.V.; et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 2014, 516, 405–409. [Google Scholar] [CrossRef]
- Lu, X.; Sachs, F.; Ramsay, L.; Jacques, P.-É.; Göke, J.; Bourque, G.; Ng, H.-H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014, 21, 423–425. [Google Scholar] [CrossRef]
- Wu, J.; Grindlay, G.J.; Bushel, P.; Mendelsohn, L.; Allan, M. Negative regulation of the human epsilon-globin gene by transcriptional interference: Role of an Alu repetitive element. Mol. Cell. Biol. 1990, 10, 1209–1216. [Google Scholar] [CrossRef]
- Kim, D.S.; Hahn, Y. Identification of human-specific transcript variants induced by DNA insertions in the human genome. Bioinformatics 2011, 27, 14–21. [Google Scholar] [CrossRef]
- Cohen, C.J.; Lock, W.M.; Mager, D.L. Endogenous retroviral LTRs as promoters for human genes: A critical assessment. Gene 2009, 448, 105–114. [Google Scholar] [CrossRef] [PubMed]
- de Souza, F.S.J.; Franchini, L.F.; Rubinstein, M. Exaptation of Transposable Elements into Novel Cis-Regulatory Elements: Is the Evidence Always Strong? Mol. Biol. Evol. 2013, 30, 1239–1251. [Google Scholar] [CrossRef]
- Kellner, M.; Makałowski, W. Transposable elements significantly contributed to the core promoters in the human genome. Sci. China Life Sci. 2019, 62, 489–497. [Google Scholar] [CrossRef]
- Simonti, C.N.; Pavličev, M.; Capra, J.A. Transposable Element Exaptation into Regulatory Regions Is Rare, Influenced by Evolutionary Age, and Subject to Pleiotropic Constraints. Mol. Biol. Evol. 2017, 34, 2856–2869. [Google Scholar] [CrossRef]
- Zeng, L.; Pederson, S.M.; Cao, D.; Qu, Z.; Hu, Z.; Adelson, D.L.; Wei, C. Genome-Wide Analysis of the Association of Transposable Elements with Gene Regulation Suggests that Alu Elements Have the Largest Overall Regulatory Impact. J. Comput. Biol. 2018, 25, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef]
- Brattås, P.L.; Jönsson, M.E.; Fasching, L.; Nelander Wahlestedt, J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 Controls a Gene Regulatory Network Based on Endogenous Retroviruses in Human Neural Progenitor Cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis -regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Leong, B.; Vega, V.B.; Chen, X.; Lee, Y.L.; Srinivasan, K.G.; Chew, J.-L.; Ruan, Y.; Wei, C.-L.; Ng, H.H.; et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008, 18, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Nishihara, H. Retrotransposons spread potential cis-regulatory elements during mammary gland evolution. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef] [PubMed]
- Zemojtel, T.; Kiebasa, S.M.; Arndt, P.F.; Behrens, S.; Bourque, G.; Vingron, M. CpG deamination creates transcription factor-binding sites with high efficiency. Genome Biol. Evol. 2011, 3, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Rayan, N.A.; del Rosario, R.C.H.; Prabhakar, S. Massive contribution of transposable elements to mammalian regulatory sequences. Semin. Cell Dev. Biol. 2016, 57, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef]
- Testori, A.; Caizzi, L.; Cutrupi, S.; Friard, O.; De Bortoli, M.; Cora’, D.; Caselle, M. The role of Transposable Elements in shaping the combinatorial interaction of Transcription Factors. BMC Genom. 2012, 13, 400. [Google Scholar] [CrossRef] [PubMed]
- Marnetto, D.; Mantica, F.; Molineris, I.; Grassi, E.; Pesando, I.; Provero, P. Evolutionary Rewiring of Human Regulatory Networks by Waves of Genome Expansion. Am. J. Hum. Genet. 2018, 102, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, D.; Garazha, A.; Sorokin, M.; Penzar, D.; Tkachev, V.; Markov, A.; Gaifullin, N.; Borger, P.; Poltorak, A.; Buzdin, A. Retroelement-Linked Transcription Factor Binding Patterns Point to Quickly Developing Molecular Pathways in Human Evolution. Cells 2019, 8, 130. [Google Scholar] [CrossRef]
- Polavarapu, N.; Mariño-Ramírez, L.; Landsman, D.; McDonald, J.F.; King, I.K. Evolutionary rates and patterns for human transcription factor binding sites derived from repetitive DNA. BMC Genom. 2008, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-C.; Upton, K.R. Human transposons are an abundant supply of transcription factor binding sites and promoter activities in breast cancer cell lines. Mob. DNA 2019, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, D.; Penzar, D.; Garazha, A.; Sorokin, M.; Tkachev, V.; Borisov, N.; Poltorak, A.; Prassolov, V.; Buzdin, A.A. Profiling of human molecular pathways affected by retrotransposons at the level of regulation by transcription factor proteins. Front. Immunol. 2018, 9, 30. [Google Scholar] [CrossRef]
- Thornburg, B.G.; Gotea, V.; Makałowski, W. Transposable elements as a significant source of transcription regulating signals. Gene 2006, 365, 104–110. [Google Scholar] [CrossRef]
- Sultana, T.; Zamborlini, A.; Cristofari, G.; Lesage, P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017, 18, 292–308. [Google Scholar] [CrossRef]
- Raviram, R.; Rocha, P.P.; Luo, V.M.; Swanzey, E.; Miraldi, E.R.; Chuong, E.B.; Feschotte, C.; Bonneau, R.; Skok, J.A. Analysis of 3D genomic interactions identifies candidate host genes that transposable elements potentially regulate. Genome Biol. 2018, 19, 216. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, T.; Preissl, S.; Amaral, M.L.; Grinstein, J.D.; Farah, E.N.; Destici, E.; Qiu, Y.; Hu, R.; Lee, A.Y.; et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 2019, 51, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Pehrsson, E.C.; Choudhary, M.N.K.; Sundaram, V.; Wang, T. The epigenomic landscape of transposable elements across normal human development and anatomy. Nat. Commun. 2019, 10, 5640. [Google Scholar] [CrossRef] [PubMed]
- Pavlicev, M.; Hiratsuka, K.; Swaggart, K.A.; Dunn, C.; Muglia, L. Detecting endogenous retrovirus-driven tissue-specific gene transcription. Genome Biol. Evol. 2015, 7, 1082–1097. [Google Scholar] [CrossRef]
- Trizzino, M.; Kapusta, A.; Brown, C.D. Transposable elements generate regulatory novelty in a tissue-specific fashion. BMC Genom. 2018, 19, 468. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.G.; Ouyang, N.; Boyle, A.P. Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes. Nat. Commun. 2020, 11, 1796. [Google Scholar] [CrossRef]
- Trizzino, M.; Park, Y.S.; Holsbach-Beltrame, M.; Aracena, K.; Mika, K.; Caliskan, M.; Perry, G.H.; Lynch, V.J.; Brown, C.D. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 2017, 27, 1623–1633. [Google Scholar] [CrossRef]
- Lynch, V.J.; Leclerc, R.D.; May, G.; Wagner, G.P. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet. 2011, 43, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Kunarso, G.; Chia, N.Y.; Jeyakani, J.; Hwang, C.; Lu, X.; Chan, Y.S.; Ng, H.H.; Bourque, G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010, 42, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Pontis, J.; Planet, E.; Offner, S.; Turelli, P.; Duc, J.; Coudray, A.; Theunissen, T.W.; Jaenisch, R.; Trono, D. Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell 2019, 24, 724–735.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zeng, J.; Lowe, C.B.; Sellers, R.G.; Salama, S.R.; Yang, M.; Burgess, S.M.; Brachmann, R.K.; Haussler, D. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. USA 2007, 104, 18613–18618. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Batzer, M.A.; Deininger, P.L. A human-specific subfamily of Alu sequences. Genomics 1991, 9, 481–487. [Google Scholar] [CrossRef]
- Brouha, B.; Schustak, J.; Badge, R.M.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kazazian, H.H. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 2003, 100, 5280–5285. [Google Scholar] [CrossRef]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Devine, S.E. Which transposable elements are active in the human genome? Trends Genet. 2007, 23, 183–191. [Google Scholar] [CrossRef]
- Kahyo, T.; Yamada, H.; Tao, H.; Kurabe, N.; Sugimura, H. Insertionally polymorphic sites of human endogenous retrovirus-K (HML-2) with long target site duplications. BMC Genom. 2017, 18, 487. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.F.; Coffin, J.M. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: Implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 1668–1672. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Watkins, W.S.; Feusier, J.E.; Thomas, J.; Goubert, C.; Mallick, S.; Jorde, L.B. The Simons Genome Diversity Project: A Global Analysis of Mobile Element Diversity. Genome Biol. Evol. 2020, 12, 779–794. [Google Scholar] [CrossRef]
- Kazazian, H.H.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Spirito, G.; Mangoni, D.; Sanges, R.; Gustincich, S. Impact of polymorphic transposable elements on transcription in lymphoblastoid cell lines from public data. BMC Bioinform. 2019, 20, 495. [Google Scholar] [CrossRef]
- Wang, L.; Rishishwar, L.; Mariño-Ramírez, L.; Jordan, I.K. Human population-specific gene expression and transcriptional network modification with polymorphic transposable elements. Nucleic Acids Res. 2017, 45, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; ‘t Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Habegger, L.; Sboner, A.; Gianoulis, T.A.; Rozowsky, J.; Agarwal, A.; Snyder, M.; Gerstein, M. RSEQtools: A modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics 2011, 27, 281–283. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Jalali, S.; Gandhi, S.; Scaria, V. Navigating the dynamic landscape of long noncoding RNA and protein-coding gene annotations in GENCODE. Hum. Genom. 2016, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Azlan, A.; Dzaki, N.; Azzam, G. Argonaute: The executor of small RNA function. J. Genet. Genom. 2016, 43, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.; Meister, G. Argonaute Proteins: Mediators of RNA Silencing. Mol. Cell 2007, 26, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Hadjiargyrou, M.; Delihas, N. The intertwining of transposable elements and non-coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307–13328. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE 2007, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Sun, X.; Jiang, D.; Ding, Y.; Lu, Z.; Gong, L.; Liu, H.; Xie, J. Origin and evolution of a placental-specific microRNA family in the human genome. BMC Evol. Biol. 2010, 10, 346. [Google Scholar] [CrossRef]
- Ahn, K.; Gim, J.-A.; Ha, H.-S.; Han, K.; Kim, H.-S. The novel MER transposon-derived miRNAs in human genome. Gene 2013, 512, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.; Brattås, P.L.; Sharma, Y.; Jonsson, M.E.; Pircs, K.; Bengzon, J.; Jakobsson, J. LINE-2 transposable elements are a source of functional human microRNAs and target sites. PLoS Genet. 2019, 15, e1008036. [Google Scholar] [CrossRef]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Sun, X.; Liu, H.; Xie, J. MicroRNA genes derived from repetitive elements and expanded by segmental duplication events in mammalian genomes. PLoS ONE 2011, 6, e17666. [Google Scholar] [CrossRef]
- Qin, S.; Jin, P.; Zhou, X.; Chen, L.; Ma, F. The role of transposable elements in the origin and evolution of microRNAs in human. PLoS ONE 2015, 10, e0131365. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Qin, S.; Li, W.; Chen, L.; Ma, F. MDTE DB: A Database for MicroRNAs Derived from Transposable Element. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.A.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable Elements Are Major Contributors to the Origin, Diversification, and Regulation of Vertebrate Long Noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kim, Y.J.; Hong, K.; Han, K. TE composition of human long noncoding RNAs and their expression patterns in human tissues. Genes Genom. 2015, 37, 87–95. [Google Scholar] [CrossRef]
- Ramakrishnaiah, Y.; Kuhlmann, L.; Tyagi, S. Computational approaches to functionally annotate long noncoding RNA (lncRNA). Preprints 2020. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Hon, C.-C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef] [PubMed]
- The RNA Central Consortium; Petrov, A.I.; Kay, S.J.E.; Kalvari, I.; Howe, K.L.; Gray, K.A.; Bruford, E.A.; Kersey, P.J.; Cochrane, G.; Finn, R.D.; et al. RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef]
- Kannan, S.; Chernikova, D.; Rogozin, I.B.; Poliakov, E.; Managadze, D.; Koonin, E.V.; Milanesi, L. Transposable element insertions in long intergenic non-coding RNA genes. Front. Bioeng. Biotechnol. 2015, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef]
- Spengler, R.M.; Oakley, C.K.; Davidson, B.L. Functional microRNAs and target sites are created by lineage-specific transposition. Hum. Mol. Genet. 2014, 23, 1783–1793. [Google Scholar] [CrossRef]
- Gong, C.; Maquat, L.E. ALUstrious long ncRNAs and their roles in shortening mRNA half-lives. Cell Cycle 2011, 10, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Erratum: Circular RNAs are abundant, conserved, and associated with ALU repeats (RNA (156)). RNA 2013, 19, 426. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Guigó, R. The RIDL hypothesis: Transposable elements as functional domains of long noncoding RNAs. RNA 2014, 20, 959–976. [Google Scholar] [CrossRef]
- Mattick, J.S. Deconstructing the dogma: A new view of the evolution and genetic programming of complex organisms. Ann. N. Y. Acad. Sci. 2009, 1178, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Chishima, T.; Iwakiri, J.; Hamada, M. Identification of transposable elements contributing to tissue-specific expression of long non-coding RNAs. Genes 2018, 9, 23. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Primary Databases | |||
|---|---|---|---|
| Database | Brief Description | Specie | Reference |
| Encyclopedia of DNA Elements (ENCODE) | Provides following functional genomics data for the diverse range of tissues and cell lines: DNase-seq data, FAIRE-seq data, Histone ChIP-seq data, TF ChIP-seq data | Human | [36] |
| Roadmap Epigenomics Mapping Consortium (REMC) | Provides following functional genomics data for the diverse range of tissues and cell lines: DNase-seq data. Histone ChIP-seq data, WGBS data, RRBS data | Human | [30] |
| Genomics of Gene Regulation (GGR) | The database is limited to only A549 cell lines and few primary cells. Provides following functional genomics data: DNase-seq data, Histone ChIP-seq data, TF ChIP-seq data | Human, mouse | [39] |
| Blueprint epigenome project | Provides reference epigenomes of distinct types of hematopoietic cells. Includes following functional genomics data: DNase-seq data, Histone ChIP-seq data, WGBS data | Human | [37] |
| Secondary Databases | |||
| Database | Brief Description | Specie | Reference |
| Open Chromatin Database (OCHROdb) | Integrates DNase seq data from ENCODE, Roadmap Epigenomics, Genomics of Gene Regulation and Blueprint Epigenome to provide a comparison of open chromatin regions across multiple samples | Human | [40] |
| ChIPSummitDB | Determines cistrome of TFs by analyzing TF ChIP-seq data from primary databases | Human | [41] |
| Super-enhancer database (SEdb) | Maps super-enhancer regions in the genome by analyzing ChIP-seq data of H3K27ac. The current version documents a total of 331,601 super-enhancers from 542 samples | Human | [38] |
| EnhancerAtlas | Identifies enhancer region by integrating datasets of 12 high-throughput methods. In contrast to other enhancer databases (SEdb, HACER, REdb, HEDD, DiseaseEnhancer, TiED, GeneHancer, SEA, DENdb and dbSUPER), it combines a versatile and most comprehensive set of annotations | 9 species, including human | [29] |
| Genome Segmentations from ENCODE data | Identifies functional regulatory elements in the genome by integrating ChIP-seq data for 8 chromatin marks, RNA polymerase II, the CTCF transcription factor. It involves the application of two unsupervised machine learning techniques (ChromHMM and Segway) to assign genomic states to disjoint segments in the genome | Human | [42,43] |
| Cistrome Data Browser (Cistrome DB) | Combines raw ChIP-seq and chromatin accessibility data from ENCODE, Roadmap and few other resources and process it through the same pipeline and quality control metrics to achieve consistency and provides a dataset with standardized curation, quality control and analysis procedures | Human, mouse | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Han, K.; Liang, P. Role of Transposable Elements in Gene Regulation in the Human Genome. Life 2021, 11, 118. https://doi.org/10.3390/life11020118
Ali A, Han K, Liang P. Role of Transposable Elements in Gene Regulation in the Human Genome. Life. 2021; 11(2):118. https://doi.org/10.3390/life11020118
Chicago/Turabian StyleAli, Arsala, Kyudong Han, and Ping Liang. 2021. "Role of Transposable Elements in Gene Regulation in the Human Genome" Life 11, no. 2: 118. https://doi.org/10.3390/life11020118
APA StyleAli, A., Han, K., & Liang, P. (2021). Role of Transposable Elements in Gene Regulation in the Human Genome. Life, 11(2), 118. https://doi.org/10.3390/life11020118