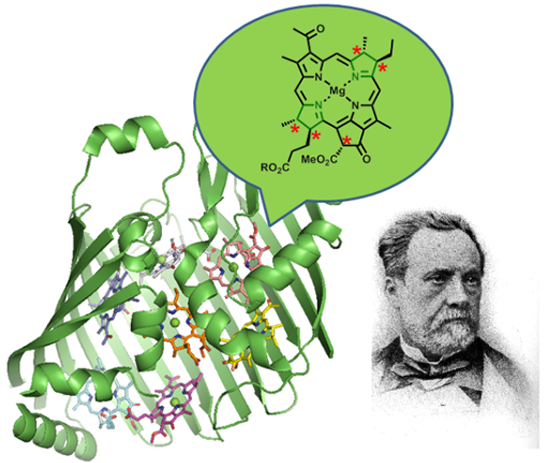
Chlorophylls, Symmetry, Chirality, and Photosynthesis †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Chlorophylls are a fundamental class of tetrapyrroles and function as the central reaction center, accessory and photoprotective pigments in photosynthesis. Their unique individual photochemical properties are a consequence of the tetrapyrrole macrocycle, the structural chemistry and coordination behavior of the phytochlorin system, and specific substituent pattern. They achieve their full potential in solar energy conversion by working in concert in highly complex, supramolecular structures such as the reaction centers and light-harvesting complexes of photobiology. The biochemical function of these structures depends on the controlled interplay of structural and functional principles of the apoprotein and pigment cofactors. Chlorophylls and bacteriochlorophylls are optically active molecules with several chiral centers, which are necessary for their natural biological function and the assembly of their supramolecular complexes. However, in many cases the exact role of chromophore stereochemistry in the biological context is unknown. This review gives an overview of chlorophyll research in terms of basic function, biosynthesis and their functional and structural role in photosynthesis. It highlights aspects of chirality and symmetry of chlorophylls to elicit further interest in their role in nature.1. Tetrapyrroles and Chirality
1.1. Basic Tetrapyrrole Types
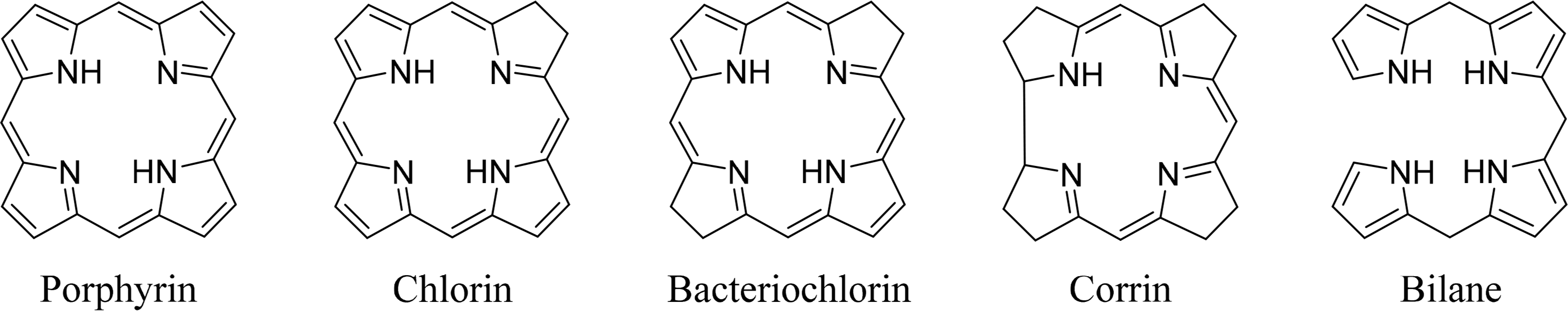
Tetrapyrroles are ubiquitous in nature. Their unique structure is essential for a number of critical biological processes including photosynthesis and respiration. The most commonly known tetrapyrroles are heme and chlorophyll, often referred to as “The Colors of Life” [1]. These pigments are responsible for the red color of the blood and the green color of leaves. In their cyclic form four pyrrole subunits are linked together via methine bridges, with the exception of corrins, where two pyrroles are directly linked to each other. The macrocycle describes an aromatic system where the different families can be determined using the oxidation state or degree of saturation. In addition to cyclic tetrapyrroles, linear tetrapyrroles (bilanes) function in cell metabolism, as regulators and as photoreceptors (biliverdins, phytochrome) (Figure 1).
Variations in the chemical structure open access to a multitude of functions, catalyzing an array of reactions [2]. Those variations include the mentioned changes in the oxidation state, different associated metals or substituents on the outer sphere of the tetrapyrroles. Interactions with the surrounding protein, such as hydrogen bonding, electrostatic interactions and also covalent binding are having further impact on the structure and therefore the observed function (Figures 2 and 3).
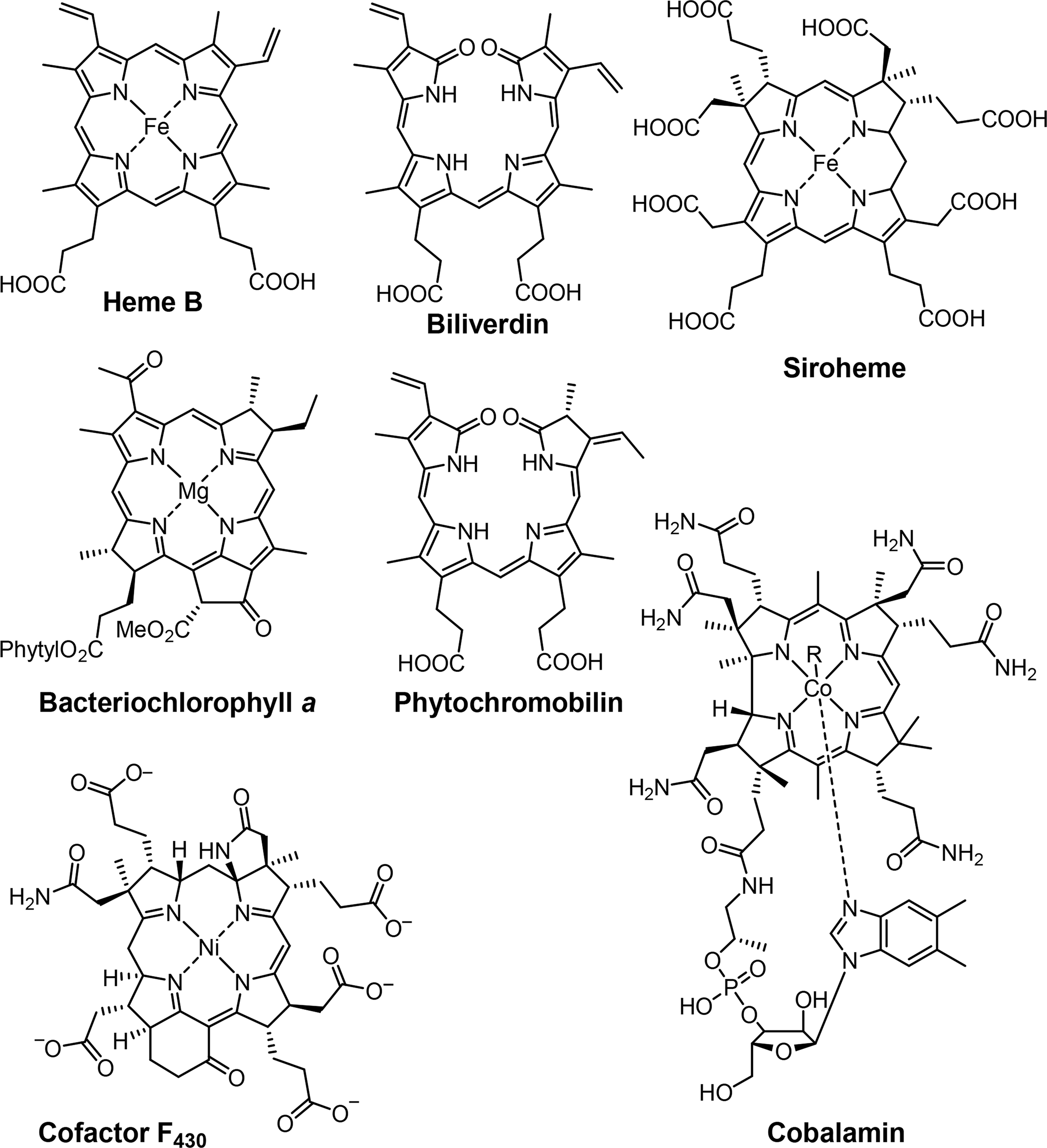
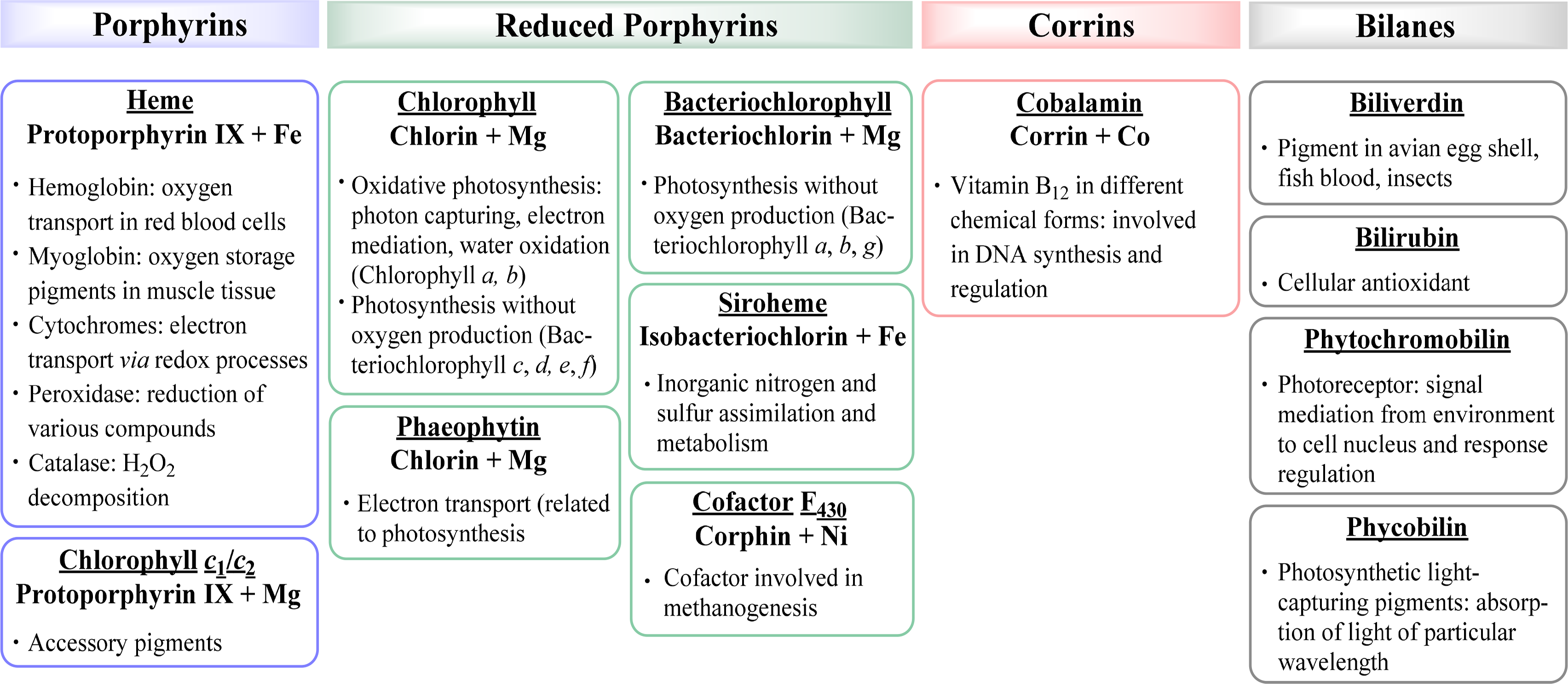
Associated with oxygen transport and storage as well as electron transfer are heme proteins [1,3]. These prosthetic groups contain iron porphyrins. While hemoglobin transports the oxygen from the lungs to the tissue cells, myoglobin, a structurally similar protein, manages the storage of oxygen in muscle cells. The protein environment mainly accounts for the differences in oxygen affinity and related properties [4]. Other important enzymes with heme cofactors are the cytochromes which are associated to various reactions occurring in nature. They facilitate electron transfer in photosynthesis, respiration, and cell metabolism. Due to the environmental influence of certain amino acids which directly link the cofactor to the protein backbone the redox potential of cytochromes is more favorable for reversible changes from the +II to the +III oxidation state of the iron [5]. Numerous peroxidases and catalases also utilize hemes as cofactors and form another link to the oxygen metabolism [6–8].
Most reduced porphyrins (hydroporphyrins) which coordinate magnesium are termed chlorophylls and are the most important cofactors in photosynthesis [9–11]. The formation of chlorophyll clusters, so-called antennas enhances the ability to trap energy from sunlight efficiently. Via highly energetic intermediates the electronic energy is converted into chemical energy and finally stored in the form of carbohydrates. The production of oxygen, which is essential for most species on this planet, occurs as a result of the oxidation of water. Other products of photosynthesis are adenosine triphosphate (ATP), the cellular fuel, and reduced nicotinamide adenine dinucleotide phosphate (NADPH).
Coenzyme B12 (Cobalamin) is the only example of a corrin in nature [12,13]. It is the chemically most complex tetrapyrrole and is solely synthesized by prokaryotes (single-celled organisms, e.g., bacteria). It coordinates cobalt as the central metal and variations of B12 are distinguished by the exchangeable second axial ligand, e.g., cyano, adenosyl, hydroxyl and methyl groups. B12 is involved in the cell metabolism, specifically in DNA synthesis and regulation. Other essential tetrapyrroles in nature are the cofactor F430 and siroheme [14,15]. The former coordinates nickel and plays an important role in methanogenesis, whilst the iron coordinating bacteriochlorin siroheme is involved in the assimilation of inorganic sulfur and nitrogen in plants from sulfite and nitrite, respectively.
The linear tetrapyrroles in humans are categorized as bile pigments and are degradation products of hemoglobin and myoglobin and contain only three methine bridges [16,17]. Other species such as insects are able to use similar brightly colored pigments as parts of their appearance. However, linear tetrapyrroles play their most important roles in plants and bacteria where, e.g., phytochromobilin acts as a light sensor in phytochrome. This chromophore, as a member of red/far-red photoreceptors, mediates signals from the cell environment to the nucleus.
1.2. Scope of the Review
When asked to write a review on the “supramolecular chirality of porphyrins” we were faced with the need to focus on a manageable topic. The most logical one for an organic chemist would be to focus on the chiral properties of porphyrin arrays, e.g., the multitude of multiporphyrin systems which have been prepared for electron transfer studies [18,19]. However, in many of them aspects of chirality either do not arise or have been neglected in the various studies with the exception of systems with planar chirality [20–24]. Alternatively, many chiral porphyrin systems have been prepared for catalytic applications or as sensors, a topic dealt with in this issue nicely by Borovkov [25]. With their significant biological relevance any chiral effects in or of porphyrins must be looked at in their natural context, which are the ultimate supramolecular arrangements of porphyrin pigments. Likewise chiral effects in nature often go hand in hand with highly symmetric arrangements of the pigments, which appears to be counterintuitive. Natural porphyrin systems can broadly be separated into “red” and “green” biochemistry. The former is based on heme-type systems and they often participate in highly enantioselective reactions, i.e., involve “chirality” despite the basic porphyrin macrocycle being achiral. One only needs to recall the multitude of selective chemical reactions catalyzed by the various cytochrome P450s [26]. Often these are understood in quite some detail and a comprehensive analysis of chiral effects therein in one treatise is impossible below the level of a monograph.
A quite different situation is encountered in “green” biochemistry. Ignoring the heme complexes involved in the various biosynthetic pathways, the tetrapyrroles utilized are (bacterio)chlorophylls, which are intrinsically chiral. On the other hand, their main function in photosynthesis appears to not involve “chirality” and their spatial arrangement—more so than that of hemes—is often characterized by a high degree of symmetry. This intriguing dichotomy and our longstanding interest in structure-function relationships in photosynthetic pigments prompted the present attempt to take an initial look at aspects of chirality and symmetry of chlorophylls in photosynthesis to stimulate further interest in this area of research [27] (and [28–30]).
1.3. Origin of Chirality
For any compound three means exist to “generate” chirality. The molecule itself may contain chiral centers and thus will be optically active. Secondly, even if the molecule is achiral binding to a molecule which is chiral will result in the overall complex to be chiral. And, last but not least, changes in the three-dimensional structure of a molecule, for example through macrocycle distortion of planar systems or a helical twisting of linear molecules, can result in chiral systems.
1.3.1. Stereocenters in Chlorophylls
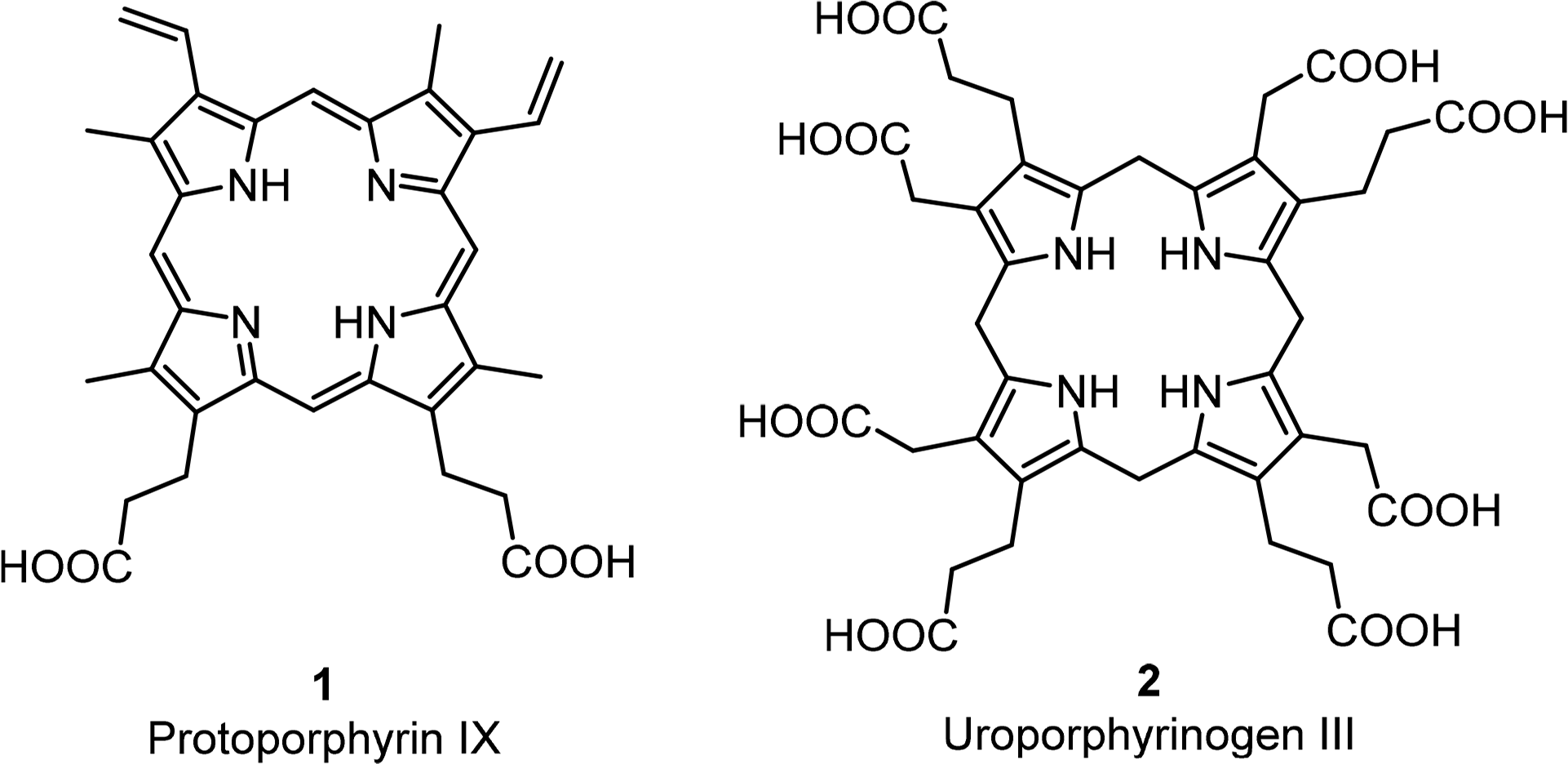
The basic porphyrin system found in nature is protoporphyrin IX 1. All “porphyrin-type” tetrapyrroles with a biological function are biosynthetically derived thereof. Siroheme, corrins and corphins (see cofactor F430, Figure 2) are derived from uroporphyrinogen III 2, a biosynthetic precursor of protoporphyrins IX (Figures 4 and 5) [31]. Neither of these biosynthetic branch point pigments is chiral. Thus, chirality is introduced “late” in the biosynthetic pathways. However, a compound such as protoporphyrin IX has facial enantiotopicity, depending on which face of the macrocycle (α or β) is used for coordination. A detailed description of the individual enzymatic reactions will be given below for the individual classes of compounds.
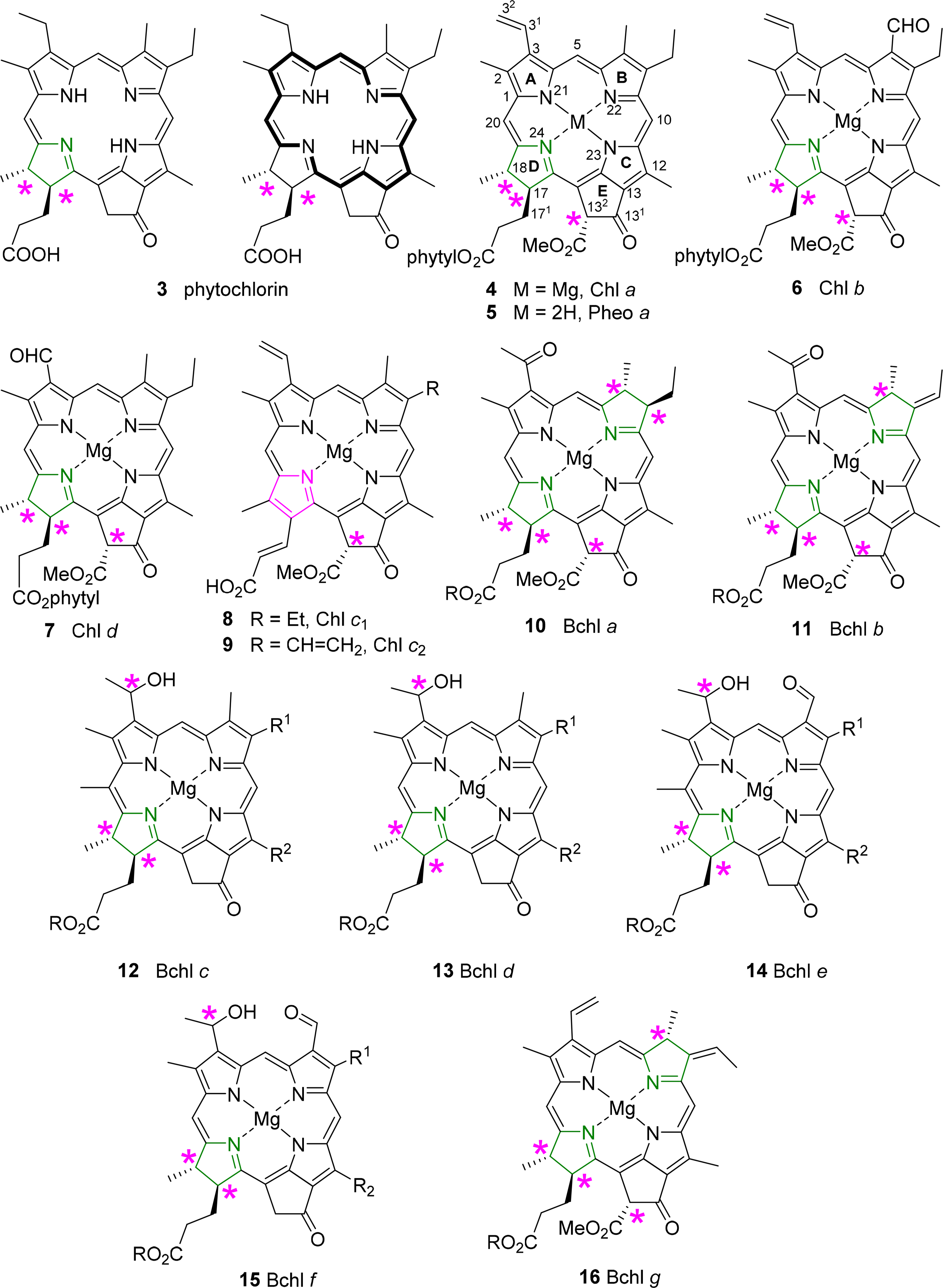
All chlorophylls involved in photosynthesis can be envisaged as derivatives of phytochlorin 3 (Figure 6). Phytochlorin has two chiral centers at the 17 and 18 position in ring D and their stereochemistry is 17S/18S [32]. The chlorophylls of Higher organisms, chlorophyll a and b (Chl a 4, Chl b 6) contain an additional chiral center in the isocyclic pentanone ring (V) at C132, which is R. This situation is retained in Chl d [33]. To make things not too easy, the chlorophylls c1 8 and c2 9 are actually porphyrins, i.e., contain only the chiral center at 132, with an additional trans-alkene stereocenter at 171 [34,35].
The exact determination of the absolute structure of Chl a 4 took several decades and was largely promoted by the important work by Willstätter in the early 1900s [36], whereby he isolated the pigment and proved that its molecular composition was C55H72O5N4Mg. The general structure of Chl a was proposed by Hans Fischer in 1940s, whereby it was suggested that (a) the “extra” hydrogen atoms of the chlorin macrocycle were placed on adjacent carbon atoms C17 and C18; (b) that the macrocycle retained its continuous conjugation (reflected through its absorption properties) and also (c) that the compound was optically active [37,38]. Chl a bears up to six asymmetric centers (depending on the ligation of the central magnesium [39]) and Ficken et al., determined the relative configuration of ring D, the C17 methyl and C18 propionic ester groups (and the extra hydrogen atoms of the reduced ring) to be trans relative to each other [40]. Woodward reaffirmed this structure to be correct in 1960 through its total synthesis, although the relative configurations of the side chains took nearly a decade to complete [41,42]. The relative configuration at C132 was established by Inhoffen [43], whereby the methyl ester side group is trans to the phytyl side chain of C17 [44]. The stereochemistry and absolute configuration of the phytyl chain (P) was elucidated by Burrell as P7′R, P11′R [45]. Completing the exact structure of chlorophyll a resulted from work by Fleming, whereby the absolute configuration at C17 and C18 was determined (S,S), giving an overall configuration of 17S, 18S, 132R, P7′R, P11′R for Chlorophyll a [32]. Ultimately, these studies were confirmed by various X-ray crystal structure determinations of chlorophyllides and pheophorbides [46].
Bacteriochlorins contain two reduced pyrrole rings and the archetypical examples are the bacteriochlorophylls a 10 and b 11. Thus, they contain the same stereocenters as in the chlorophyll series and two additional chiral centers at C7 and C8 in the case of BChl a 10 and BChl g 16 and one at C7 in BChl b 11 [44,47]. To make matters of nomenclature worse, the bacteriochlorophylls c–f (12–15) are actually chlorins, i.e., 17,18-dihydroporphyrins [48]. They also lack the carboxy methyl ester group at position C132, and thus have only two chiral centers in the macrocycle, similar to the parent phytochlorin. In addition, they contain a new chiral center in a side chain at C31 [49]. The latter is less well defined and depends on organism type and light conditions; typically mixtures of the enantiomers at this position are observed as will be discussed in more detail below.
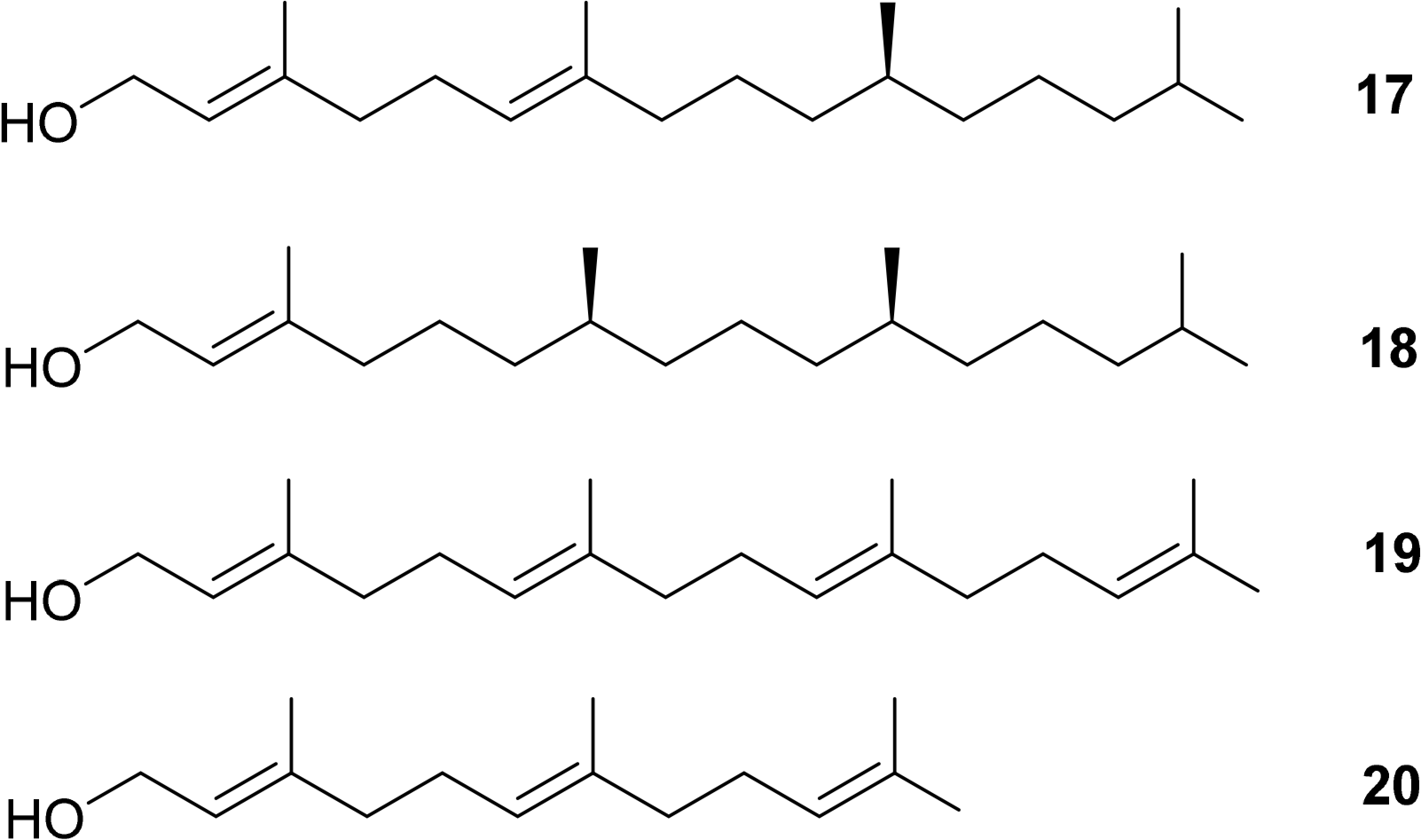
Additionally, the esterified alcohols which are present at the C17 side chain, in most but not all chlorophylls, contain chiral centers. Typical examples are Δ2,6-phytadienol 17 and phytol 18. Other esterified alcohols such as geranylgeraniol 19 and farnesol 20 do not contain chiral centres but variances in the number and positioning of the double bond stereogenic groups do occur (Figure 7) [50–54].
1.3.2. Environmental (Apoprotein and Axial Ligand Effects)
Typically (bacterio)chlorophylls do not occur as “isolated” pigments in monomeric and/or unbound form. In their functional state they are often bound to side chains of the apoproteins. This is facilitated through axial ligation, i.e., the coordination of a suitable amino acid heteroatom (or water) to the central metal to yield penta- or hexacoordinated metals [55]. As (most) amino acids and proteins are chiral, any complex of a tetrapyrrole—even an achiral one—with a protein is optically active (Figure 8). These ligations enable important interactions with the apoprotein environment, assisting chlorophyll protein organization and modulating their electronic properties.
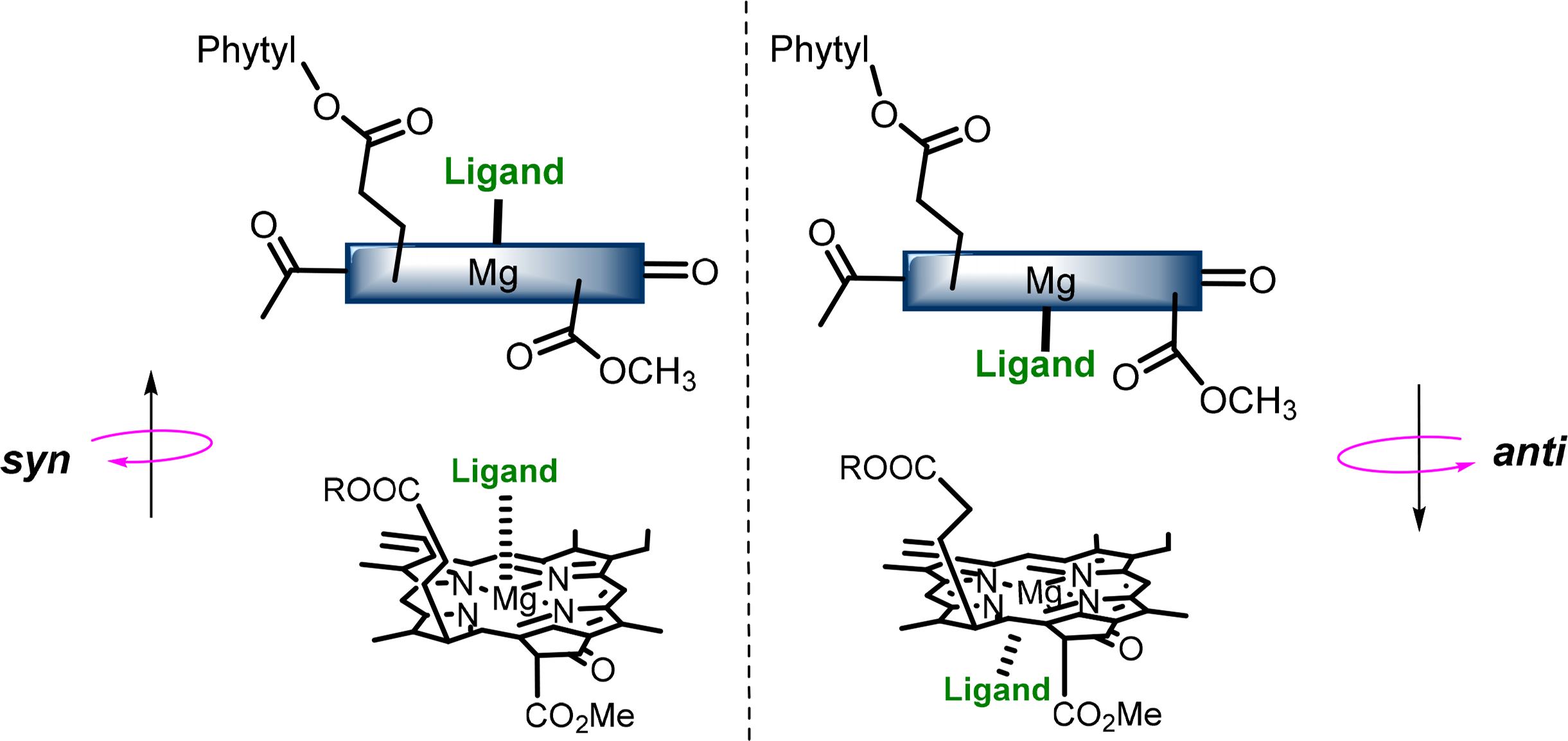
The central magnesium atoms of Chls and BChls are in most instances penta-coordinated resulting in the formation of a new stereochemical center and the possibility of two different types of chlorophyll-ligand interaction [56–58]. The two “faces” of the chlorophyll macrocycle are diastereotopic, referred to as syn- and anti- (Figure 9), which arise from the three chiral centers on the ring. Syn ligation occurs when an amino acid coordinates the Mg atom the same side as the C17 phytyl chain, whereby the hydrophobic chain bends over the back face of the Chl and forces the syn-ligation of polar residues at the opposite side (also referred to as “face” complexes or β-ligation from the top face). This geometry only accounts for a small percentage of those found in natural systems, although it is proposed to play an important role in energy trapping in light harvesting complexes (LHCs). The anti geometries (“back” or α-ligated complexes where coordination occurs from below) are more energetically favored by a factor of ∼4 kJ·mol−1 as determined by computational calculations of model pigments [59].
The uneven distribution of the two ligation states within chlorophyll proteins has been strongly conserved during evolution, and this phenomenon plays a critical part in light-harvesting function and energy transfer. The one exception here is the bacteriochlorophylls c–e which form self-organized aggregates without direct incorporation into a porphyrin backbone (see Section 7).
1.3.3. Macrocycle Distortion
Less obvious but more important for the natural function are three-dimensional structural effects. For example, an unsymmetrically substituted achiral porphyrin can become chiral upon out-of-plane distortion of the macrocycle. Thus, the conformation of the porphyrin, chlorin or bacteriochlorin ring system must be considered [60]. Likewise, linear tetrapyrroles may exist in different enantiomeric helical conformations.
Whilst the aromatic π-system of porphyrins may at first imply planar compounds, it has long been known that the macrocycle possesses considerable conformational flexibility (Figure 10) [61]. Indeed, extended aromatic systems such as those of Chl or BChl must be conformationally flexible and numerous structural and spectroscopic studies since have led to a deeper understanding of the importance of this phenomenon for the functional properties of porphyrins [62,63]. By now it is established that the macrocycle conformation can control the physicochemical properties of the tetrapyrrole to a significant extent. Increasing nonplanarity with its destabilization of the π-system leads to bathochromically shifted absorption bands, easier oxidation, lower fluorescence yields, larger Stokes shifts, shorter lifetimes of the lowest excited states, faster intersystem crossing and internal conversion.
In the biological context the macrocycle flexibility has been used to develop the concept of “conformational control”, wherein variation of the macrocycle conformation via steric interactions with the surrounding protein side chain in pigment-protein complexes and/or axial coordination of the central metal and/or π–π-interactions can be used to fine-tune the physicochemical properties of the macrocycle [64,65]. This presents an intriguing rationale for explaining the often diverse (physico)chemical reactivity of the same chromophore in different protein environments. The presence of the reduced pyrrole rings in chlorins and bacteriochlorins leads to even more conformational flexibility than is observed in porphyrins. For a more detailed discussion of this concept and related studies see references [45,63,65,66].
2. Chemistry and Structure of Isolated (Bacterio)chlorophylls
2.1. Fundamental Aspects of Chlorophyll Chemistry
Chlorophyll chemistry is governed by the aromatic character of the underlying tetrapyrrole moiety and functional group transformations in the side chains (Figure 11) [67]. As a cyclic tetrapyrrole with a fused five-membered cyclopentanone ring, the ring system exhibits the typical features of a heteroaromatic compound. Their unique biological functions derive from the extended π-system with its ability to coordinate metal ions with the core nitrogen atoms, the conformational flexibility of the macrocycle and the side chains. The aromatic system of pheophorbide 5 can undergo electrophilic reactions with a preference for reaction at C20, the meso position closest to the reduced pyrrole ring. The C7–C8 double bond in phytochlorin, which is reduced in the bacteriochlorophylls, is not part of the aromatic delocalization pathway and thus offers a convenient point for modification of the chlorophylls via addition and oxidation reactions. The side chain substituents undergo standard transformations, such as addition and oxidation reactions at vinyl groups and the ester groups at C173 and C132 can be saponified or transesterified. The chemistry of the isocyclic pentanone ring often involves enolization and follow-up chemistry of the β-keto ester system. Examples are oxidative degradation reactions, hydroxylation, ring-opening or decarboxylations. These and other peripheral substituent modifications also present the bulk of synthetic chemistry developments in the context of biosynthetic studies and the use of chlorophyll derivatives in photodynamic cancer therapy [68].
A special chemical aspect is related to the stereochemistry at position C132. In chlorophyll a 4 this is R. However, the hydrogen atom is highly acidic due to its β-dicarbonyl pattern. Thus, it can easily undergo enolization to 21 followed by reprotonation either from the re or si face. Upon epimerization this results in the formation of the 132S-stereoisomer, which is termed Chl a′ 22 (Figure 12). This reaction occurs quite easily in aqueous media, as do the related allomerization reactions [69–72]. The different enantiomers have distinct properties, mostly as a result of steric effects [73,74]. A functional role as one of the pigments in the photosynthetic reactions center has been suggested for the a′ form [75,76], but the ease of their non-enzymatic formation has allowed no independent verification yet. Chlorophyllide a′ is not a substrate of the chlorophyllase [77] and no structural evidence for the natural occurrence has yet been derived from protein structures [78].
For in depth reviews on chlorophyll chemistry see the book by Scheer and articles therein [11]. The newer literature is accessible through works by Pavlov and Ponomarev [79], Nyman and Hynninen [68], Pandey [80,81], Mironov [82] and the excellent contributions from Tamiaki’s group [83].
2.2. Spectroscopy and Symmetry
The absorption properties of chlorophylls provides key information about their overall structure and symmetry thereof, and it can be used instantaneously to distinguish the three macrocycle types related to the chlorophylls, depending on their degree of unsaturation, namely porphyrin, chlorin and bacteriochlorin derivatives. As will be discussed below, these electronic properties are largely determined by the symmetry of the macrocycle, in addition to ligation effects on the central metal and/or the presence of specific substituents around the macrocycle of chlorophylls at specific sites, all characteristics of which are vital to their specific ecological function.
The large aromatic nature of tetrapyrroles provides the basis for their unique absorption properties e.g., the typical UV/Vis spectra of porphyrins exhibit absorptions in two regions termed the Soret or B-band (∼380–420 nm) and the Q-bands (∼500–800 nm) [84]. This is a result of the splitting of the main frontier molecular orbitals and these aspects of the electronic structure of porphyrins are well described by the semi-quantitative MO-based “four-orbital model” of Gouterman (Figure 13), a mixture of Hückel and Configuration Interaction (CI) theory [85,86]. The four-orbital model considers the two HOMOs (labelled a1u and a2u) and LUMOs (labelled eg) of the porphyrin derived from Hückel theory and mixes the four possible excitations between them using configuration interaction theory to account for electron interaction.
The one electron transitions (along the x- an y-axes through opposing central nitrogen atoms as shown in Figure 14) between these molecular orbitals generate four excited states and thus causes them to overlap and interact with one another, both constructively and destructively. Whilst the LUMOs are formally degenerate, comparison of the nodal patterns of the Hückel HOMOs to the ±4 orbitals from both free-electron and cyclic polyene models suggested that they may be considered “accidently” degenerate. The result of this modification and subsequent application of charge interaction between the single excitations gives rise to two sets of x- and y-polarized degenerate excitations, one set of which are strongly allowed and at higher energy (referred to as 1Eu state, shorter wavelength B band) than the other formally forbidden set. Excited states arise from these forbidden transitions due to vibrations within the molecule and these have a lower energy and low oscillator strength (referred to as 1Eu state, Q band).
A key-difference between the spectra of metalloporphyrins and their free-base counterparts is the number of Q-bands. This results from the reduced symmetry of the free-base (D2h) so that the two axes defined by opposite pyrrole nitrogen atoms (Nopp–Nopp) are no longer equivalent affecting further splitting of the transitions. The generalized metalloporphyrin (Figure 14) belongs to the D4h molecular point group and exhibits the highest symmetry possible for a porphyrin derivative. β-Pyrrolic reductions decrease the molecular symmetry so that both free base and metallated forms of chlorins are of C2v symmetry and likewise bacteriochlorins are D2h in either form. Regarding the multiplicities of the Q-bands each degenerate excitation has a transition dipole orientated with one or the other axis (x- or y-) and are so termed the Qx- and Qy-excitations. Therefore, in metal complexes, where these axes are equivalent, the Qx and Qy pair is degenerate. However, for free base porphyrins, there is a reduced symmetry (D2h) in that the two axes defined by opposite pyrrole nitrogen atoms are no longer equivalent, due to the presence of two hydrogens on these opposing nitrogen atoms. This reduction in symmetry means that the excited states resulting from transitions between the molecular orbitals are not degenerate and results in further splitting of the transitions [88].
This is even more evident with the chlorin macrocycles (e.g., Chl a) and their metal complexes which have the lowest symmetry (C2v) due to the reduction of one β–β double bond which destabilizes the MOs that have a significant amount of electron density located at this reduced bond. This results in the raising of the energy of these orbitals, causing a red shift in absorption on the Qy band, blue shift of B band due to increase in the HOMO–LUMO gap for this transition and increased Qy band intensity due to a higher oscillator strength of these excited states. Although bacteriochlorins can be considered to have D2h symmetry, the conjugation of the macrocycle is longer along the y-axis than the x-axis resulting in an intense Qy absorption at lower energy (red shift to approx. 800 nm), in addition to a split B band as here the Bx,y bands no longer overlap (Bx and By, Figure 14). Obviously, the orbital situation in these usymmetrical systems is much more complicated than in Figure 13 and peripheral modification to the macrocycles at specific positions and axial ligations can account for other changes in the absorption spectra of chlorophylls due to effects on MOs and the HOMO–LUMO gap.
2.3. Structural Chemistry
A detailed description of this topic has recently been given by us [46]. Despite their importance only a limited number of small molecule single crystal X-ray determinations are available. None of these structures are of the—phyll type, i.e., contain the natural long chain esters at C17. For reasons of solubility, synthetic access and chemical stability the available set of structures encompasses only pheophorbides, chlorophyllides, mostly as 173 methyl or ethyl esters, and various metal complexes or synthetic derivatives thereof (Figure 15).
The classic structure here is that of ethyl chlorophyllide a 23 which was reported in 1975 by Kratky and Dunitz [89] and Strouse and coworkers [90]. Subsequent studies reported on chlorophyllide b 24 and compounds with different C173 esters. Common features for chlorophyllides, i.e., magnesium chelates, were a penta-coordinated metal center (with Mg–Oax ∼2.03 Å); nonequivalent but typical Mg–N bond lengths (∼2.03–2.18 Å), with the shorter bonds to N21 and N23, and the longest one found for Mg–N24, i.e., involving the reduced pyrrole ring; similar crystal packing; and an almost planar macrocycle conformation. A simple means to compare the latter is the Δ26 value (defined as the average deviation of the 26 macrocycle atoms from their least-squares-plane), which was found to be 0.04–0.062 Å for the chlorophyllides [46]. Removal of the 132 ester group to yield the pyro-chlorophyllide a derivative 25 had no significant effect on the conformation [91].
Other studies reported on a number on “unnatural” metal complexes with Cd(II) [92], Fe(III)Cl [93], Zn(II) [94] and Ni(II) [95], and these illustrate the range of possible conformations which can be imposed on the macrocycle through metal effects with Δ26 values reaching 0.32 Å [96]. Free base derivatives, e.g., pheophorbides a 27 [97,98] or b [99], typically exhibit a planar aromatic system, a zig-zag conformation of the cyclopentanone ring and the standard differences between the reduced and aromatic pyrrole quadrants and conjugation of the vinyl group with the aromatic system.
The synthetic and structural chemistry of the bacteriochlorophylls is more limited. While similar to those of the chlorophylls they are more prone to oxidation to the chlorin or porphyrin level and less stable. As a result most structure-function correlations have been performed with more stable synthetic analogs, e.g., geminal dialkylated bacteriochlorins [100]. The basic reference structure is that of methyl bacteriopheophorbide a 29 which has a planar macrocycle akin to that of the chlorins of the pheophorbide series [101,102].
More data are available for the chlorin-type bacteriochlorophylls c–e [46]. As phytochlorin derivatives they are more stable and synthetically more easily accessible than the bacteriochlorins. They also show significant variability of the C8 and C12 substituents, i.e., do exist as homologous mixtures [103]. This allowed one of the first comparative analyses of closely related photosynthetic molecules derived from Chlorobium vibrioforme [104]. Intriguingly, these chemically very similar compounds exhibited a wide range of different conformations; one of the key points of departure for the concept of conformational flexibility.
A detailed comparative analysis of the tetrapyrrole conformation and core stereochemistry requires the determination of specific geometrical parameters and appropriate visual illustrations [46]. Simple conformational parameters such as atomic displacements from the 4N, 24-macrocycle or 26-phorbin system least-squares-plane and individual displacements of the Cm and Cb atoms serve as rough general descriptors. This can easily be visualized using skeletal deviation plots [105]. More stringent criteria for the analysis of the metal coordination, axial ligand stereochemistry and π–π-interactions have been provided by Scheidt and Lee [106]. They also developed the basic terminology to describe various tetrapyrrole conformations, e.g., through the use of the terms ruffled (ruf) and saddled (sad). ruf indicates a twisted macrocycle with meso carbons alternately displaced above and below the mean-plane while sad, obtained by a 45° rotation of the ruf conformer around the major C2-axis, describes a similar alternate displacement of the pyrrole rings relative to the mean-plane. Additionally, the by then well-known domed (dom), waved (wav; called “stepping” by Scheidt) and “roof” type conformations were also noted.
Additionally, steric strain often affects not only out-of-plane displacements but the in-plane distortions modes as well. Here, the best analysis utilizes the normal structural decomposition method (NSD) introduced by Shelnutt [107,108]. This method analyzes all out-of-plane and in-plane distortion modes based on experimental or calculated positional atomic data. NSD is a conceptually simple method that employs the decomposition of the conformation of the macrocycle by a basis set composed of its various normal modes of vibration. This technique affords clear separation of the contributing distortions to the macrocycle conformation in a quantitative fashion, often of reduced dimension and allows the large scale comparative analysis of porphyrinoid crystallographic data. The individual displacement parameters for each distortion mode can easily be plotted and used to illustrate the mix and interplay of the various distortions.
A typical example for such an analysis is shown in Figures 16 and 17. It is based on an investigation of the steric influence of the 20-methyl substituent in nickel(II) phytochlorins [95,96]. The structure of a Ni(II) 20-methylphytochlorin methyl ester 33 exhibited a highly ruffled conformation with a local increase in distortion in the vicinity of the 20-substituent when compared to the reference compound without the meso methyl group 26. This was indicated by larger Cm displacements and a smaller Ca–C20m–Ca angle (120.5°) compared to the phytochlorin without the methyl group (123.8°). Such compounds are related to naturally occurring BChl c and d and the results indicate that the larger distortion in the 20-methyl derivative might account for the 10 nm bathochromic shift in the absorption maxima of the BChl c compared to d [109]. The various conformational distortions in the different metal complexes are clearly evident from the illustrations. The magnesium derivative 25 exhibits an almost planar macrocycle while the nickel(II) derivatives are characterized by large out-of-plane displacements for the Cm carbon atoms (and larger ones in the C20-methylated from 33) and a rotation of the pyrrole rings about their β–β axes indicating the typical ruf distortion due to Ni–N bond contraction.
Studies such as these confirm that conformationally distorted systems, no matter how large or small the distortion, manifest a mixture of distortion modes, i.e., present unsymmetrically distorted macrocycles. Indeed, the degree and the relative contributions of individual distortion modes are indicative for specific physiological functions (see below).
3. Biosynthesis and Generation of Chiral Centers in Chlorophylls
3.1. Formation of Protoporphyrin IX
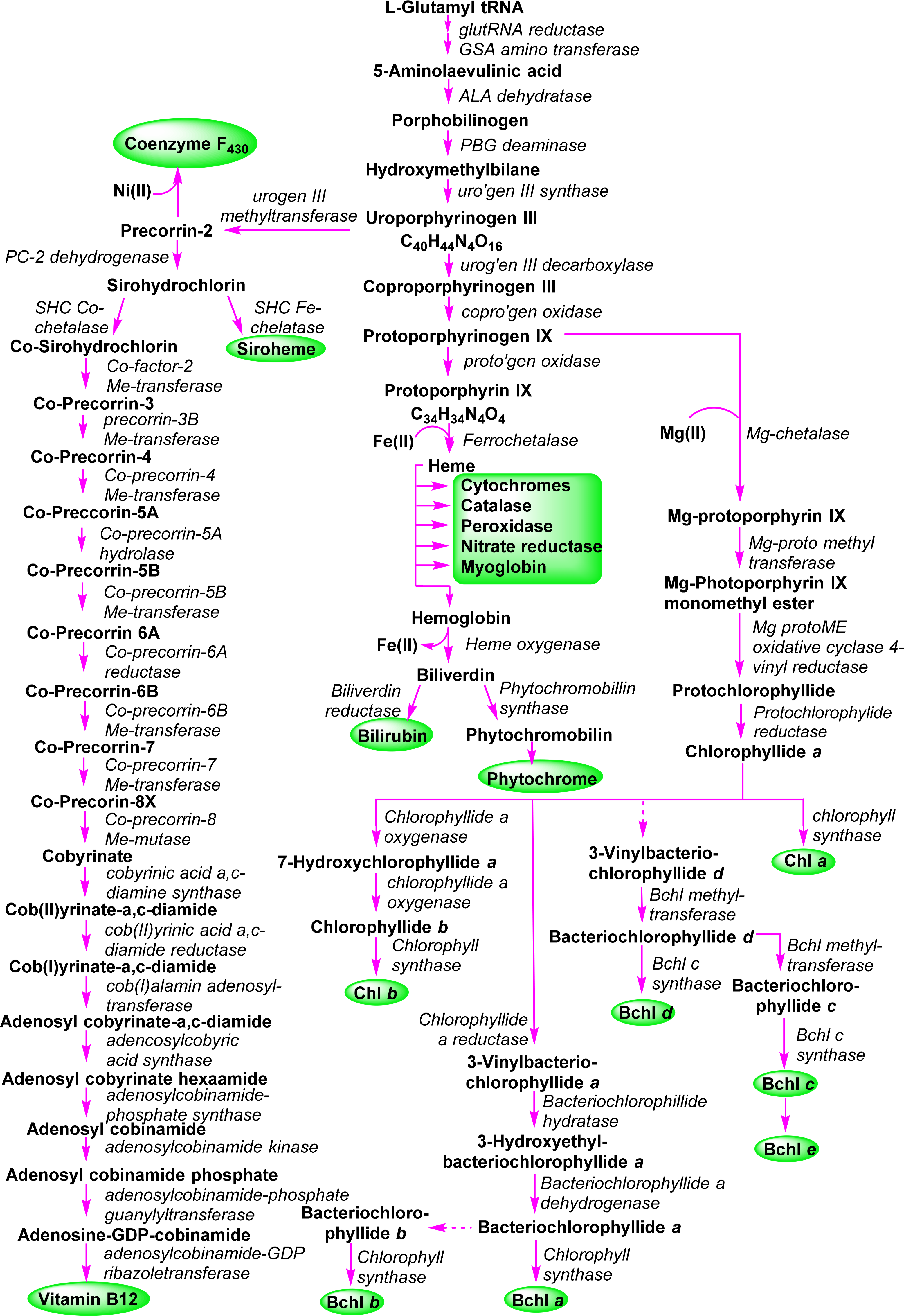
A general outline of the biosynthesis of the chlorophylls was given in Figure 5 [110]. The field has matured significantly in the past decades with now many enzymes being structurally characterized and the molecular genetics unraveled in many organisms. As a result the subsequent discussion highlights only the main chemical conversion steps without discussion of regulatory aspects or alternative pathways. Brevity also prevents a look at the highly intriguing chemistry of senescence and chlorophyll catabolism [111,112]. Contemporary reviews on this topic can be found in the book edited by Grimm et al. [113] and Volume 20 of the Handbook of Porphyrin Science [114]. All tetrapyrroles are formed in a complex reaction sequence starting with small molecules. The first intermediate which is common to all tetrapyrrole biosynthesis is 5-aminolevulinic acid 38 (ALA) (Figure 18). Depending on the organism two pathways are possible: the ALA-synthase pathway (C5) or the Shemin pathway (C5 + C1) [115,116]. The former is relevant to most bacteria and plants while the latter operates in purple photosynthetic bacteria, yeast and animals.
Next, two ALA molecules are condensed to the first pyrrole compound, porphobilinogen 39. This reaction is catalyzed by ALA-dehydratase (porphobilinogen synthase) [117]. It involves the formation of a Schiff base with an amino group of the enzyme. Formally a stabilized carbanion, it then reacts with a second molecule of ALA and after an aldol condensation the enzyme facilitates the abstraction of water. A nucleophilic attack of the amino group at the C-atom of the Schiff base in the second ALA molecule then results in a transamination and formation of the pyrrole. Thus, the mechanism resembles that of (trans)aldolases [118,119].
The next sequence is highly complex and involves the formation of uroporphyrinogen III 2 from four molecules of porphobilinogen. In a first step porphobilinogen deaminase (hydroxymethylbilane I synthase) links four porphobilinogen molecules into the linear tetrapyrrole (a bilane) 40. Intriguingly, this enzyme utilizes a dipyrromethane primer as a cofactor and thus in fact, first generates a hexapyrrolic species which then is cleaved to hydroxymethylbilane [120–123]. In a sense this bilane is a symmetric arrangement of four pyrroles and can spontaneously cyclize to the symmetric uroporphyrinogen I [124]. Under enzymatic control a desymmetrization occurs whereby uroporphyrinogen III synthase cyclizes and isomerizes hydroxymethylbilane to the spiro intermediate 41 under inversion of ring D [120,125,126] which then yields uroporphyrinogen III 2. This is the first true cyclic tetrapyrrole and the first branch point for different porphyrins. It can either lead to protoheme or (bacterio)chlorophylls or, via C-methylation to vitamin B12 and sirohemes. For the chlorophylls, uroporphyrinogen III decarboxylase decarboxylates four acetate residues under formation of coproporphyrinogen III 42. This, in turn, is oxidatively decarboxylated to protoporphyrinogen IX 43 [127]. Finally, protoporphyrinogen oxidase oxidizes this intermediate through abstraction of six hydrogen atoms to protoporphyrin IX 1, the first cyclic tetrapyrrole with a conjugated (porphyrin) ring system [128].
Protoporphyrin IX presents the next branch point in the biosynthesis of tetrapyrroles. Depending on the type of metal ion inserted this intermediate leads to the heme porphyrins and subsequently open-chain tetrapyrroles, while the Mg-complexes yield the chlorophyll-type pigments.
3.2. Formation of Chlorophyllide a
The first committed step for chorophylls is the incorporation of magnesium in protoporphyrin IX 1 (Figure 19) by the ATP-dependent Mg-chelatase [129]. The enzyme function is highly complex with different subunits having to act in concert and sequence. The magnesium porphyrin 44 is now converted into the monomethyl ester 45 by a S-adenosyl-l-methionine:Mg-protoporphyrin IX methyl transferase. The enzyme first binds the porphyrin, then S-adenosylmethionine, from which a methyl group is transferred to the C13 propionic acid group. Formation of the methyl ester is a prerequisite for the complex reaction that forms the isocyclic ring V and thus also the first stereo center in the chlorophylls. Mechanistically Mg-protoporphyrin IX monomethylester cyclase first hydroxylates then oxidizes the C131 position to ketone. This activates the C132 position which now is connected to the C15 meso position to yield [8-vinyl]-protochlorophyllide a 46 [130–132]. In plants this reaction sequence is oxygen dependent while in bacteria alternative enzymes use water as oxygen source under participation of a vitamin B12 cofactor and a [Fe–S] cluster [133]. Here, a SAM radical abstracts a hydrogen atom from C131 and the radical thus generated is then converted into a putative carbocation which is then hydroxylated from water. A second radical generation at C131 allows the abstraction of the proton from the hydroxy group and loss of an electron yields the keto species. Finally, a third radical formation at C132 was proposed to then react with C15 [133]. Thus far no protein structural data are available and the exact stereochemical mechanism is unknown for both pathways.
The 3,8-divinyl compound is now reduced to the monovinyl protochlorophyllide a 47. While shown here to occur at a specific point in the biosynthesis the vinyl reductase can act at various points in the biosynthesis after formation of Mg-protoporphyrin IX up to the -phyll level (i.e., from compound of type 46 onwards) and some organisms are known to contain only divinyl chlorophylls [134,135].
The last step of direct macrocycle modification in chlorophylls is the formation of chlorophyllide a 48, i.e., the reduction of the porphyrin to a chlorin under generation of two new chiral centers at C17 and C18. In plants this is a light-dependent reaction catalyzed by single subunit NADPH: protochlorophyllide oxidoreductases, however, some organisms use a so-called dark variant of this process incorporating a light-independent enzyme with ATP for this reduction [136,137]. The enzyme binds the substrate and co-substrate in the dark under participation of cysteine residues [138]. Light activation then converts the enzyme-substrate complex in an active form where hydride and proton transfer can occur. This process requires two photons. The first induces the conformation change in the enzyme-substrate complex, while the second induces the actual chemical reaction [139]. Despite the absence of a 3D structure the participating amino acid residues are known based on site-directed mutagenesis studies and the stereochemical aspects can be inferred to some extent (Figure 20). The new hydrogen atom on C17 is derived from the pro-S hydride of NADPH and the one on C18 from a tyrosine residue whose pKa is lowered by a neighboring lysine [140,141]. Intriguingly, the wheat enzyme requires a substrate with 132R configuration and did not tolerate any modifications of the 132 position, possibly indicating an involvement of this part in the photo-reduction [142].
In anoxygenic bacteria, and other organisms, a light-independent, dark-operative pathway is used for the formation of the chlorophyllides. Genetically the genes involved in this conversion show a high degree of amino acid conservation to those of nitrogenase [144]. Detailed structural information on the ternary dark-protochlorophyllide oxidoreductase complex and the involvement of [4Fe–4S] centers is now available from protein structures [145,146]. The critical components are two [4Fe–4S] clusters and two protochlorophyllide binding sites. In the substrate bound structure [147] the C17 propionate side chain has a distorted conformation, which can indicate a trans-specific substrate assisted protonation mechanism involving a neighboring aspartate residue [148].
3.3. Chlorophylls a and b
For chlorophyll a 4 the next step is simple; it involves the esterification of the C17 propionic acid group in 48 with phytyl pyrophosphate or geranylgeranyl pyrophosphate by the Chl synthase (Figure 21) [149,150]. The enzyme is specific for chlorophyllides and cannot convert bacteriochlorophyllides. For Chl b the final step utilizes the same enzyme as for Chl a and converts chlorophyllide b 50 to chlorophyll b 6. However, prior to that, chlorophyllide a oxygenase (a Rieske-type monooxygenase) introduces two oxygen atoms at the C71 position. First the 71-hydroxy derivative 49 can be detected from which an intermediate geminal diol forms and spontaneously loses water during the formation of chlorophyllide b 50 [151,152]. This pathway is also reversible using the chlorophyllide b reductase and 7-hydroxymethyl chlorophyll a reductase which allows a conversion of Chl b into Chl a in what is known as the “chlorophyll cycle” [153–155].
3.4. Bacteriochlorophylls a and b (Bacteriochlorins)
A central intermediate for chlorophylls and bacteriochlorophylls is chlorophyllide a (Figure 4). Nevertheless, it should be remembered that despite the chemical schemes shown here implying one pathway for the individual pigments, the biological reality is quite different. Depending on organism, cellular compartment and environmental conditions (light or dark, aerobic or anaerobic, autotroph or heterotroph) different enzymes and pathways are operative for a given target system. Thus, the formation of the tetrahydroporphyrin bacteriochlorophylls starts with this dihydroporphyrin, too (Figure 22).
Conversion to the bacteriochlorin 51 is accomplished by the chlorophyllide oxidoreductases, which are thought to operate similarly to the dark-protochlorophyllide oxidoreductases [156,157]. The 3-vinyl group is next hydrated by a 3-vinyl hydratase to 52, similar to the reaction involved in the formation of the BChls c, d, and e (vide infra) [144]. The OH group is then oxidized to a carbonyl unit to yield bacteriochlorophyllide a 53 [158]. Lastly, esterification with a long chain alcohol then yields bacteriochlorophyll a 10. It is not yet clear whether first geranylgeranyl pyrophosphate is attached followed by reduction to phytyl or whether the reduction step precedes the esterification [150,159].
For bacteriochlorophyll b 11 it was initially assumed that it is somehow formed from bacteriochlorophyllide a or that, e.g., a 3-hydroxyethyl-8-vinyl-chlorophyllide a would undergo reduction to the respective 8-vinyl bacteriochlorin, which after migration of the double bond would yield the 8-ethylidene bacteriochlorin followed by oxidation at C31 and esterification with an isoprenoid alcohol [119]. Recent work by Tsukatani et al., suggests instead that chlorophyllide a oxidoreductases, the nitrogenase-like enzyme mentioned above, are capable of directly converting 8-vinyl-chlorophyllide a 54 to bacteriochlorophyllide g 55 in a 1,4-trans hydrogenation reaction [160]. This would then be followed by hydratation and oxidation at the 3-vinyl group to yield bacteriochlorophyllide b 56 and bacteriochlorophyll b 11.
3.5. Bacteriochlorophylls c, d, and e (Chlorins)
The bacteriochlorophyll c, d and e present a complex group of photosynthetic pigments as, unlike the other chlorophylls, often more than one chemically defined species is present in an organism [103]. They act as the main antenna pigments in the green sulfur bacteria (Chlorobiaceae and Chloroflexaceae) [119,161]. Note, in these bacteriochlorophylls only one pyrrole ring is reduced, i.e., they are chlorins (dihydroporphyrins). Thus, they are quite different from the bacteriochlorophylls a and b, which are tetrahydroporphyrins. Spectroscopically they are similar to chlorophylls a and b and both exhibit the same stereochemistry at the chiral centers in the macrocycle. They lack the C132 methoxy carbonyl group and at position 3 they carry a hydroxyethyl group, which give rise to a new chiral center [162]. Bacteriochlorophylls c and e differ from bacteriochlorophyll d by an additional methyl group at the meso carbon 20 and bacteriochlorophyll d has an aldehyde group at C7 and thus is similar to chlorophyll b [49]. All three bacteriochlorophylls occur in nature as so-called homologue mixtures. Individual components of these mixtures differ in the substituents at C8 and C12, the stereochemistry at C31, and the nature of the esterified alcohol. Bacteriochlorophyll c often carries an ethyl group at C8, as is the case in most natural tetrapyrroles. Other possibilities are n-propyl or iso-butyl residues or neopentyl in BChl e. C12 mostly carries ethyl or methyl groups. Depending on the organism the long chain alcohol can be farnesol, geranylgeraniol, phytol, 2,6-phytadienol, hexadecanol and octadecanol.
New derivatives with alternative groups are discovered from time to time. However, often these are derived from genetically altered organism and thus, while intellectually interesting, their biological relevance is open [163]. A case in point is bacteriochlorophyll f 15 [164], which has not yet been identified in nature. However, inactivation of the gene encoding bacteriochlorophyll C20 methyltransferase (see below) in Chlorobaculum limnaeum resulted in a photosynthetically active mutant containing this pigment [165].
The biosynthesis of this class of pigments most likely branches off at the chlorophyllide a stage (Figure 23) [166]. The critical point here is the removal of the C132 methoxy carbonyl group, which is the main chemical difference to the other chlorin-type chlorophylls. While the bacterial enzyme(s) are still unknown [167] formation of 57 sets the stage for the action of methyl transferases which can modify the substituents at C8 and C12 through single or multiple methylation reactions to yield 58. The enzymes involved are of the radical-SAM protein type which probably catalyzes a hydrogen abstraction at the alkyl residues on C8 and C12 followed by methyl transfer involving S-adenosylmethione and a [4Fe–4S] cluster [133,168,169]. A combination of labelling and mutation studies indicates that the C8/C12 methylation reactions precede the C31 hydration, which occurs before C20 methylation and esterification [170].
Thus, bacteriochlorophyllide d 59 appears to be a common intermediate for the BChls c–e. Bacteriochlorophylls c are derived by action of a C20 methyl transferase followed by esterification [171]. The enzyme responsible for the last step seems to be rather promiscuous. Thus far, only one gene encoding for such an activity has been identified and studies suggest that any system without the C132–COOMe group and with a 3-hydroxyethyl unit can act as substrate [172]. The biosynthesis of bacteriochlorophyll e is not yet understood. Most likely it involves a transformation of bacteriochlorophyllide c 60 into bacteriochlorophyllide e 61 followed by esterification [173].
Hydration at C31 is catalyzed by a vinyl hydratase and generates a new chiral center [174,175]. As noted above, both the R and S epimers are present in a given organism and thus the enzyme is not diastereoselective [169,170]. On the other hand a clear correlation between the degree of C8/C12 methylation and the R/S ratio has been shown, with the R epimer being the principal component in [8-Et,12-Me] derivatives and increasing alkylation at C8 favoring S chirality [168].
4. Photosynthesis
4.1. Basic Principles
Photosynthesis is a complex biochemical process that can be described simply as the absorption of light by a collection of photosynthetic pigments. The pigments are highly organized to form a light-harvesting complex consisting mainly of chlorophylls or bacteriochlorophylls. Dyes such as these have strong absorptions in the visible region of the spectrum, an important characteristic in the process of natural photosynthesis. Bacteriochlorophylls are the main absorbing pigment in photosynthetic bacteria and their absorption maxima exhibits a bathochromic shift relative to other chlorin-based pigments. The process of light absorption leads to the step-wise generation of a number of reduction equivalents by a series of redox processes with a final result being the production of ATP. Carotenoids and open-chain tetrapyrroles are examples of important accessory pigments that optimize the process of light absorption by complementing the function of absorbing pigments throughout the photosynthetic process.
The light-harvesting complex maximizes the potential for light absorption within organisms by functioning as an antennae system. Pigments are arranged with a high absorption cross-section to increase the number of photons that can be absorbed at any time. A greater range of absorption across the visible spectrum is achieved by using complexes with a variety of pigments. Following photon absorption, antenna pigments channel excitation energy via exciton transfer to a closely arranged pair of (bacterio)chlorophylls in the photochemical reaction center (Figure 24). The reaction center is an integral membrane pigment protein and functions by carrying out a series of photo-induced electron transfer (PET) reactions. These excited (bacterio)chlorophylls transfer an electron to an acceptor molecule, resulting in a charge separated state between the reduced acceptor and the oxidized chlorophyll molecule. Structurally, a system which harvests lights and performs a charge-separation is called a photosystem (PS).
After the initial electron transfer event, a series of electron transfer reactions takes place that eventually stabilizes the stored energy in reduction equivalents and ATP. Higher plants have two different reaction center complexes that work together in sequence, with the reduced acceptors of one photoreaction (photosystem II) serving as the electron donor for photosystem I. Here, the ultimate electron donor is water, liberating molecular oxygen, and the final electron acceptor is carbon dioxide, which is reduced to carbohydrates. More simple and evolutionary older types of photosynthetic organisms contain only a single photosystem, either similar to photosystem II or photosystem I [176–179].
Following the initial photo-induced event, many different donor and acceptor molecules are employed in the transfer of an electron across a photosynthetic membrane. Finally, a quinone acceptor is reduced to a semiquinone and subsequently to a hydroquinone. This process is accompanied by the uptake of two protons from the cytoplasm. Hydroquinone migrates to a cytochrome bc complex where it becomes reoxidized. This complex is a proton pump and generates a transmembrane proton gradient by the translocation of protons across the membrane. The proton gradient is translated to chemical energy via the enzyme ATP synthase and is subsequently used to generate ATP. This highly organized, complex process is maintained by the specific location and arrangement of different structural components throughout the membrane (Figure 25).
Tetrapyrrole based pigments function mainly as electron donors and quinones as electron acceptors within natural photosynthetic processes. Donor-acceptor systems such as these are often employed in biomimetic systems that incorporate a range of variations to mimic these processes. Modulations in spatial arrangement, structural components—i.e., donor/acceptor, linker, and bridging groups—and changes in solvents and/or environmental factors are some of the few changes that can be made in efforts to optimize physicochemical properties of such systems. Countless systems have been developed in order to investigate photo-induced electron transfer (PET) interactions with many reviews being published on work in this area [18,19,180,181]. Porphyrins are often the main compound used in this area of study due to the wide scope of structural modulations that can be made to optimize and vary their function as donor-acceptor groups within electron transfer systems. Phthalocyanine compounds may be used; however, the lack of known synthetic routes for the development of unsymmetrically substituted derivatives and a low solubility makes them less desirable. However, interest regarding their function and development has recently been restored by applications involving solar energy conversion systems [182].
4.2. Oxygenic Photosynthesis
Oxygenic photosynthesis provides an abundant source of energy and essentially all of the oxygen on the planet. In obligate photoautotrophs (i.e., organisms that acquire energy from light capture to produce organic compounds) light energy is converted to energy (ATP) and reduction equivalents (NADPH) which then facilitate the chemical reduction of CO2 to carbohydrates. These in turn serve as a source for atoms for the chemical building blocks for the biosynthesis of other primary and secondary metabolites and constituent molecules [183]. Overall, oxygenic photosynthesis in plants, cyanobacteria and algae can be looked at as a means whereby electrons as reduction equivalents are generated via water splitting and release of hydrogen or oxygen from water and used for the reduction of carbon dioxide [184,185]. To facilitate this process and to make optimum use of the incident light this process is split into two different light reactions. One, now called photosystem II (PS II), catalyzes the oxidation of water [186], while the other (PS I) serves as an acceptor for the electrons such generated via transfer from plastocyanin to ferredoxin. The two photosystems are linked via an electron transfer chain containing cytochromes. The overall arrangement is typically illustrated through the Z-scheme (Figure 26) [187]. The Z-scheme highlights the two light-dependent reactions in photosynthetic systems of higher plants and exemplifies that two photosystems function in sequence to convert solar energy into chemical energy.
4.3. Anoxygenic Photosynthesis
Anoxygenic photosynthesis is the evolutionary precursor to oxygenic photosynthesis and thus the photosynthetic apparatuses of both share many similarities [188]. The main difference are that the older systems utilize only one photosystem (either a PS I-type or PS II-type) and that, while performing CO2 fixation and generation of ATP, they do not utilize water and do not release oxygen. Instead the electron donor can be hydrogen or sulfur species. Chemically, another difference is the utilization of bacteriochlorophylls instead of chlorophylls, which allows the utilization of different light wavelengths. In microbiological terms the most important, and best understood, bacterial groups are the purple phototrophic bacteria, green sulfur bacteria, green filamentous bacteria, and the heliobacteria. While the situation has rapidly changed in the last decade, historically, most of our structural understanding was derived from the first class [189,190].
The overall photochemistry is rather similar in all systems. It involves absorption of light by pigments in the antenna complexes, resulting in photo-excitation of the accessory pigments (see Figure 24). This is followed by Förster resonance energy transfer (FRET) to the reaction center pigments and then charge-separation [9]. Subsequently, a varying series of electron transfer reactions is coupled with proton translocation to generate a transmembrane potential. In structural terms the bacterial photosystems consist of a reaction center complex (RC) and a light-harvesting complex (typically denoted LHC-I). Some species utilize additional antenna systems (LHC2, 3, …) for light harvesting. The RC-LHCI complex is associated with a cytochrome bc complex which transfers electrons between a hydroquinone (QH, which differs from species to species) and a soluble electron transfer protein carrier [191]. This allows for a cyclic electron flow in the photosystems (Figure 27).
In the following we outline the involvement of the (bacterio)chlorophylls in these processes using selected examples from contemporary crystallographic analyses to illustrate the supramolecular character of the arrangements. The “picture” of photosynthesis is continuously showing more detail and has reached the state where complete arrangements of the whole photosynthetic apparatus and its dynamics in the membrane can be visualized [192,193].
5. Reaction Center Systems
The reaction centers are the primary sites of electron transfer reactions in photosynthesis. While different organisms have different reaction center components their overall structure and the arrangement of the photosynthetic pigments is strikingly similar. Likewise the sequence of electron transfer events differs more in the details of the cofactors than in overall strategy (Figure 27). This is related to the evolutionary development of photosynthesis, which must have started with one proto-photosystem [178]. Today we distinguish two reaction center systems, RC I and RC II. Both utilize a primary donor, a structurally closely packed dimer of two (bacterio)chlorophylls, the so-called “special pair” [194,195]. Electrons then pass through accessory tetrapyrroles to two different types of electron acceptors, an iron-sulfur type (RC I) and a pheophytin-quinone Type (RC II). Photosynthetic bacteria have only one of these, adapted to environmental conditions and optimized for different electron donors, while oxygenic photosynthetic organism use both reaction centers in sequence (see Z-scheme in Figure 26).
5.1. Reaction Center I and Photosystem I
Most of the initial structural detail on PS I was derived from the cyanobacterium Thermosynechococcus elongatus. The protein backbone is characterized by two main subunits (PsaA and PsaB), which are arranged in the photosystem core in an approximate two-fold symmetry. The core cofactors are chlorophyll and phylloquinone with two closely associated special pair pigments, one of which is believed to be Chl a′. Here the redox active [Fe4S4] clusters are involved in the electron transfer (Figure 28). Overall, the photosystem is more complex than, e.g., the reaction center of heliobacteria or green sulfur bacteria, with more than 90 chlorophylls and more than 20 carotenoids being part of the core structure in the former [196].
Significant advances have been made with the elucidation of the PS I structure from pea [197]. Like in other systems the core of PS I is highly similar to that of bacterial species. However, the PS I system consists of the core complex and a peripheral antenna complex (LHC I). The latter is more flexible in terms of function and composition and can vary from organism to organism and depends on the environmental conditions. The whole eukaryotic PS I system is complex and consists of 15 protein subunits, over 170 chlorophylls, 15 carotenes and two phylloquinones and three [Fe4S4] clusters.
5.2. Reaction Center II
5.2.1. Bacterial RC II
Historically most of our structural knowledge on the photosynthetic apparatus in general was derived from the first crystal structure of the reaction center from Rhodopseudomonas viridis, which is a RC II type [190]. At present the best high resolution structures are available for two species of purple photosynthetic bacteria Rhodobacter sphaeroides and Blastochloris viridis [198]. The bacteriochlorin cofactors of the electron transfer chain are bound by one of two reaction center proteins (L or M; synonymous with A and B, respectively), via numerous interactions including axial ligation by histidine residues in the case of the four bacteriochlorophylls, and are arranged in pairs of approximate C2 symmetry [190,199,200]. Each branch contains one of the primary donor BChls, a bacteriopheophytin, a BChl monomer and a quinone. There are a few different nomenclatures for the individual pigments in the electron transport chain and here we will distinguish the components of the special-pair dimer (P) by the addition of a subscript indicating the protein to which it is attached (i.e., DL and DM). This formality is continued for both the monomeric “accessory” BChls (BA and BB) and the BPheos (HA and HB) noting the distinction that in bacterial reaction centers it is conventional to describe the monomers of P using the L/M nomenclature and the accessories using the equivalent A/B labels. A close-up view of the chromophore arrangement in the Blastochloris reaction center is given in Figure 29.
Once P is excited to P*, an electron is transferred to the primary acceptor bacteriopheophytin (BPheo) (HA) in ∼2 ps and ∼3–5 ps, for B. viridis and R. sphaeroides, respectively, the beginning of the formation of the cross-membrane electron gradient that drives photosynthesis [9,202]. This takes place through a scarcely detectable P+BL− intermediate that is formed rapidly after photo-excitation. Subsequent reduction of QA by HA− is followed by electron transfer from QA− to QB. After re-reduction of P+ either directly by cytochrome c2 in R. sphaeroides or by the RC-cyt in B. viridis a second ET cycle takes place culminating in the reduction of QB− to QB2−. At this point, the fully reduced and protonated QH2 dissociates from its binding site in the reaction center and is replaced by another oxidized quinone from the cytoplasmic pool [203]. A crucial feature of the RC is that electron transfer occurs only along the l-branch despite the apparent C2 symmetry [204].
There are two striking differences between the reaction centers from R. sphaeroides and B. viridis (Figure 30). Specifically, the RC from B. viridis utilizes BChl b and is in possession of a bound tetraheme cytochrome (see Figure 31) whilst that from R. sphaeroides contains BChl a and does not have a bound cytochrome [9]. Other differences include the identity of the carotenoid close to the ET inactive accessory BChl (BB) and of the quinones (1,2-dihydroneurosporene, menaquinone-9 (QA) and ubiquinone-9 (QB) in B. viridis and spheroidene and ubiquinone-10 in R. sphaeroides) as well as the detailed binding interactions of the pigments with the chromophores [9]. There are also significant differences in the static physical properties of the reaction centers (e.g., P960 in B. viridis vs. P865 in R. sphaeroides) as well as the kinetic parameters with respect to photo-induced charge-separation between the two species [9,202]. By now many effects have been identified where the subtle interplay of protein and cofactor determines the physicochemical properties of the latter in these systems [205].
5.2.2. Photosystem II in Oxygenic Systems
The coupling of two separate reaction centers with specifically poised redox potentials so that one is capable of oxidizing water to oxygen (PS II) and the other of generating a strong reductant to produce NADPH (PS I) is the characteristic feature of oxygenic photosynthesis [206]. The Z-scheme shown above already outlined the energetic relationships between the electron transfer events in the light-reactions of photosynthesis that result overall in the reduction of NADP+ by H2O [207]. Immediately following photo-excitation of the primary donors, the initial charge-separated states are formed in both photosystems (P680+/Pheo− and P700+/A0−) producing the trapped energetic electrons that participate in the “downhill” reduction steps and, concurrently, the strong oxidant P680+ [208,209]. The relatively weak reductant produced by PS II is used to re-reduce P700+ through the interconnecting electron transport chain via the membrane mobile quinone that, after double protonation, affects electron transfer to the [2Fe–2S] cluster in cytochrome b6f. A subsequent electron transfer step within the cytochrome b6f complex sees the reduced [2Fe–2S] cluster pass its electron to oxidized plastocyanin (PC) via cytochrome f, which consequently reduces P700+, whilst the alternative electron transfer pathway mediated by the cytochrome b6 component is coupled to cyclic phosphorylation. After the accumulation of four oxidizing equivalents in the Mn-containing oxygen evolving complex (i.e., the S4-state is reached), water oxidation takes place and the fully reduced state (S0) of the oxygen evolving complex is recovered. Meanwhile, the electron transfer events downstream of PS II involve the reduction of a soluble ferredoxin that in-turn reduces the flavoprotein ferredoxin-NADP+ reductase [207].
Structural similarity of both PS I and II to the reaction centers is maintained in terms of the overall arrangement and number of cofactors present in the electron transfer chain although in both RCs BChls are substituted for Chl a and derivatives. Specifically, in PS II there are four Chl a pigments, the PD1/D2 dimer and the pair of accessories ChlD1/D2, and two pheophytin a (Figure 32) [210,211]. The symmetry of their arrangement and similarity to the bacterial center is evident in the crystal structure of the Thermosynechococcus vulcanus oxygen-evolving PS II (Figure 33). The symmetry of the cofactor arrangement is also maintained, as is the unidirectionality of the electron transfer [212]. Likewise the main protein subunits which are involved in the cofactor binding, D1 and D2, are symmetrically related around an approximate two-fold axis of symmetry similar to the bacterial species [196]. A detailed structural and biophysical comparison of RC II and PS II has been given by Allen and Williams [213].
In PS I the special-pair appears to be a heterodimer composed of Chl a and its C132 epimer Chl a′ and the four accessories and primary acceptors are all Chl a [214]. Further critical differences in the biophysical dynamics of these two reaction centers are apparent. Firstly, that the primary donors are believed to be “accessory” chlorophylls, specifically ChlD1 in PS II [215] and either Chl AA or -AB in PS I [216], contrary to the situation in reaction centers, even so, the photo-generated cation radicals are localized to the both P680 and P700 after initial charge-separation [217]. Additionally, as hinted by these identifications, electron transfer is not as asymmetric in PS I when compared to the reactions centers from the purple bacteria, although this feature is retained by PS II [217].
6. Light-Harvesting (Bacterio)chlorophyll Proteins
In order to make efficient use of the available light all photosynthetic organisms use light harvesting systems as “antennae” to optimize the supply of photons to the reaction center. This assures that requirement that P is photo-excited before it may serve as the primary donor in the electron transport chain so that it does not become a severely rate limiting step of charge-separation impinging on overall photosynthetic efficiency. The task of these dedicated light-harvesting complexes (LHCs) that typically consist of densely packed arrays of chromophores is the conversion of light into electronic excitations and their direction to the RCs. In contrast to RCs that bear comparatively close resemblances regardless of their origin, there is considerable architectural flexibility with respect to the structures of LHCs. Thus, as shown above, complete photosynthetic units consist of RC and LHC units. Depending on their location with regard to the RC “core” and “peripheral” antenna are distinguished. True to their light harvesting function often many antenna complexes are associated with one RC, with the exact ratio depending on the light conditions; oligomerization of photosystems can also occur.
6.1. Bacterial Antennae Systems
6.1.1. FMO Protein
The Fenna–Matthews–Olson (FMO) protein provided the first crystal structure of a chlorophyll-containing protein (Figure 34) [218]. The FMO complex serves as the excitation energy transfer (EET) intermediate between the chlorosome baseplate and the reaction center of the photosynthetic green sulfur bacteria [219]. The complex consists of seven BChl a pigments encapsulated between an oval-shaped β-sheet and a few α-helix stitches, in what has been referred to as the “taco shell protein”, and crystallizes as a trimer with (in some species) an eighth BChl situated at the interface of each monomeric unit. Specific pathways for the excitation energy transfer through the individual pigments have been suggested [220] and this system is probably the most widely investigated LHC in terms of single chromophore contributions.
There are two spectral classes of FMO protein that are differentiated by the relative intensities of their absorption maxima [222]. Type I complexes show a greater intensity of the second lowest-energy excitation and include the species Prosthecochloris aestuarii and Pelodictyon phaeum, whereas in type II species (e.g., Chlorobaculum tepidium) the intensity of this excitation is suppressed such that it is lower than the third lowest-energy optical transition. Speculation as to the factors that lead to the spectral classes has included citations of the differences in the macrocycle conformations of the pigments (including nonplanarity and C3-acetyl orientation) and alternatively differences in the binding of BChl 8, specifically its ligation state [223]. This latter suggestion has since been superseded on the basis that it has been recently suggested that the FMO from P. phaeum does not include BChl 8 after all [224]; although it is possible that this is a preparative issue as crystal structure determinations usually ascribe this cofactor a low-occupancy and it is considered to be the most labile cofactor.
The FMO protein is considered by many to be a useful model complex for studying photosynthetic energy transfer, Olson himself referred to the crystal structures as, “… a godsend for physicists and physical chemists interested in spectral properties, excited states, and energy transfer”. As a result, numerous studies of the site-energy distributions and exciton dynamics have been reported [222], and have included explicit investigations of the influence of variations in BChl conformation [64,65,104,225]. So far, the best agreement between structure-based ab initio calculations of the site-energies with those empirically determined by parametric fitting to the experimental spectra, have been obtained by consideration of the effect of the electrostatic environments of the BChl binding-sites on the relative stabilities of their ground- and excited-states [226,227].
6.1.2. LHC1 and 2 from Purple Bacteria
The antenna complexes of purple bacteria consist of the RC-associated light-harvesting complex I (LHC1; Figure 35), or B875, and the peripheral antenna light-harvesting complex II (LHC2; Figure 36), or B800/850 [228]. Both proteins contain numerous BChl a pigments held by the apoprotein in strictly defined circular arrangements. Specifically, LHC2 consists of two BChl rings denoted B800 and B850 containing 9 monomeric BChls and 9 pairs of BChl pseudo dimers, respectively, with Mg–Mg distances of ∼21 and 9 Å, also respectively [229–231]. The RC-LHC1 complex contains the B875 ring of 30 BChls composed of 15 BChl a dimers with similar Mg–Mg separations to B850 [232,233]. The energetic hierarchy of the bacterial light-harvesting complexes suggests excitation energy transfer occurs in a stepwise sequence: (LHC3 →) LHC2 → LHC1 → RC [234,235]. Interestingly, differences in the planarity of the BChl macrocycle conformations have been noted [236] and further insight is gained from advances in NMR spectroscopic studies of such systems [237].
Aside from differences in excitonic coupling arising from the various BChl packing-densities present in the B850 and B800 rings that contributes to their spectral differences [238,239], the detailed pigment-protein interactions experienced by each BChl may affect unique site-energies (uncoupled excitation energies) that influence the exciton dynamics. For instance, computational studies utilizing models based on crystallographic data have indicated that the protein environment red-shifts the exciton energies of the B800 ring to a greater extent than in B850 [240,241]. Moreover, direct calculation of the pigment site-energies has shown that differences caused by the pigment conformations alone echo those that are found when more of the protein environment is included. In detail, the isolated pigment models of the LHC2 chromophores yielded Qy energies of 1.59, 1.57 and 1.61 eV for the αB850, βB850 and B800 BChls, respectively, and the relative difference between the B850 BChls was maintained when the effects of neighboring BChls, axial-ligands or H-bonding residues were explicitly included [242,243] An earlier study found even greater variation, suggesting the nonplanar βB850 BChl’s site-energy to be ∼0.04 eV lower (i.e., red-shifted) than its planar αB850 counterpart [244].
In order to provide environmental adaptability, a modified form of LHC2 may be expressed by some species of purple bacteria under low-light conditions known-as LHC3 in which the absorbance maximum of the B850 ring is blue-shifted to 820 nm and hence is termed B820 [228]. Structural studies of LHC3 [246] have led to the suggestion that altered H-bonding to the C3-acetyl of BChl a in the B820 ring was responsible for the spectroscopic variation, an idea that has recently been extended to include the effect that this has on the excitonic coupling in the complex [247].
The highly ordered (and symmetric) arrangement of the pigments in LHCI and II illustrates one of several basic concepts used for light harvesting in nature. Here, a circular arrangement of chromophores results in rapid delocalization of the excitation energy which is then transferred between the various spectral species and funneled to P870. Such optimized geometrical arrangements not only allows an efficient functioning of photosynthesis but also prevents the formation of chlorophyll triplet states, which, through formation of singlet oxygen, could result in damage to the photosynthetic apparatus.
6.2. Antenna Systems in Oxygenic Photosynthesis
Alternative strategies for the pigment arrangements are used in the light harvesting systems of plants. Here, the arrangement of the chlorophylls is less “symmetric” and often different spectroscopic classes of pigments can be distinguished. The best known system is LHCII from pea [248]. Each of the subunits contains four xanthophylls (i.e., oxygen containing carotenoids), 8 Chl a, 6 Chl b and lipids in addition to the protein backbone [249]. More recently the structure of light harvesting complex b4 (LHCb4) revealed overall similar structural features to LHCII but different numbers, types and arrangements of the accessory pigment binding sites therein [250].
The light harvesting chlorophyll a/b complex of PS II, LHCII, is also an example for “global” chiral effects to play a regulatory role in photosynthesis. It has been implicated in controlling the assembly of chirally organized PSII macrodomains, which might help to separate the two photosystems. While the terms “global chirality” or “long-range chiral order” are normally used in physics for the description of spin distributions it is used here to describe macroscopic areas with distinct CD signals. Crystals of LHCII trimer also form aggregates with long range chiral order and this can be further modulated by individual components in the LHCII [251,252]. Such changes could also be induced in grana thylakoids by light [253]. Changes at the molecular level in PS II crystals were also described as the result of different light conditions [254] and this has given rise to the thermo-optical mechanism for the reorganization of the complex [255]. For the chiral macroaggregates left- and right-handed forms have been identified which differ in their thermal response [256].
LHCII is also an example for a photosynthetic system, where a clear structural requirement for specific carotenoid end groups, configurations and chiral centers for a correct assembly of the complex has been shown [257]. It should also not be forgotten, that, despite all the focus on structure and structure function relationships in the literature and in our work, the whole photosynthetic machinery, esp. in the thylakoids is flexible and adaptable [258]. In functional terms PS II is typically associated with two antenna systems, the major trimer-forming LHCII protein and a monomeric, minor LHCII protein. The latter comprises systems such as CP29, CP26, and CP24 [259]. In PS I the situation is even more complex and the composition of the associated LHC varies significantly from organism to organism [259].
7. Chlorosomes—the Case of Chlorophyll Self-Organization
7.1. Chlorosome Structure and Function
The chlorosomes are the main photosynthetic antenna complexes in green bacteria. In contrast to the protein-organized complexes described previously, chlorosomes contain very little protein and instead are comprised almost entirely of self-assembled chlorophyll aggregates; a small BChl a binding-protein is associated with the chlorosome may serve as an intermediate between the chlorosome and the RC apparatuses (including the FMO protein described above) and is referred to as the “baseplate” [260]. Depending on the particular species, Chlorobiaceae (green sulfur bacteria) contain one of either BChls c, d or e (the Chlorobium chlorophylls) and their corresponding homologues, which differ in the length of the alkyl substituents attached to C8 and C12 as well as the identity of the esterifying alcohol, whereas all Chloroflexaceae (green nonsulfur bacteria) usually contain the BChl c derivatives [260].
Chlorosomes represent the most efficient antenna in nature and their photosynthetic light-harvesting structures. Their efficiency can be largely attributed to the large size of the structure and large amount of pigment molecules inside. It remains the last known light-harvesting complex for which no high-resolution structural information is available. Chlorosomes from different species can be ellipsoidal, conical or irregular in shape—depending on the bacterial strain involved—and are attached to the inner side of the cytoplasmic membrane. Within the chlorosome, the light-harvesting BChls c, d and e, typically in the order of magnitude of 105, form aggregates, which arrange into tubular constructs and lamellar sheets that fill the interior, along with carotenoids, quinones, galactolipids and wax esters. The entire structure is surrounded by a monolipid-protein casing. Also within the chlorosome are small amounts of BChl a, and it is thought that these are located in paracrystalline layer or “baseplate” to which the chlorosomes are attached and provides their link to the plasma membrane. BChl a in the layer binds to the FMO protein (trimeric in nature and binds 7–8 BChl a molecules through H-bonding, hydrophobic interactions and ligand binding with Mg2+). This interaction mediates the excitation energy from the chlorosome to the reaction center embedded in the plasma membrane. Figure 37 shows a cartoon representation of the chlorosome structure [220,261].
The particular BChl substitution pattern (i.e., specific homologue) of the compounds isolated from chlorosomes in some species is governed by the light-conditions maintained during culture, with reduced light leading to increased methylation [109]. This is considered to affect altered spectral properties by modulating the in vitro aggregation properties of the chromophores. However, structural studies of the demetallated derivatives of the homologues [104] from Chlorobium vibrioforme indicate substantial alterations to the macrocycle conformation, which may contribute to the red-shift of the low-light chlorosomes, although analysis of the spectral properties of the isolated chromophores [262] did not show any systematic variation corresponding to the extent of methylation. Additionally, model compounds have indicated that the C20-methyl substituent induces local nonplanarity at this position, which may contribute to the 10 nm red shift of BChl c relative to BChl d (see Chapter 2.3) [46,96].
7.2. Chlorin Aggregation and Biomimetic Studies
Several models for the organization of BChl aggregates within chlorosomes have been proposed and the exact supramolecular organization of such has been the subject of debate for many decades. The absence of high-resolution structural information for the chlorosome is due to its heterogeneous nature restricting analysis via crystallographic means. A significant proportion of these BChl c, d and e pigments self-arrange without any assistance from peptides (contrary to the light-harvesting proteins in green plants) and it is this organization of pigments that modulates the overall optical properties of the BChls, resulting in extremely efficient energy transfer. It is largely accepted that within natural chlorosomes three structural features are required for self-assembly: (a) the central magnesium; (b) the peripheral 3′–OH group and (c) the 13–C=O group. Typically J-aggregation occurs due to coordination between the Mg and 3′–OH, H-bonding between the 3′–OH and the 13–C=O and π–π interactions between the macrocycles, in addition to electrostatic interactions (e.g., the polarized keto group and the Mg atom) and the partitioning of the long chain farnesyl groups into the hydrophobic parts of the construct. Through various spectroscopic and microscopic techniques, it has been shown that these aggregates form both rod-like elements and lamellar sheets within the chlorosome [263–266].
In the structures of BChl c, d and e, there is a chiral secondary alcohol at the 31 position. This alcohol plays an important role in the self-aggregation of these pigments. Both the 31R- and 31S-stereoisomers are found in natural chlorosomes and these stereoisomers are diastereotopic as both the 17- and 18-chiral centers have S-configuration. Self-aggregates of biomimetic compounds show that depending on the stereochemistry at this position, different supramolecular structures form, which in turn affects the optical properties of the aggregate. Although most chlorosomes are usually more populated with the R-stereoisomers of BChls, the exact ratio depending on the bacterial strain involved and its living conditions, the S-stereoisomer plays a critical role in the self-assembly process and co-aggregation of both stereoisomers is likely to exist in natural systems [263,264].
Many biomimetic aggregates of natural BChls have been prepared and provide extensive in vitro insight into the supramolecular structures of chlorosomal antennae systems. Isolated BChls c, d, e have been shown to readily self-assembly into J-aggregates, whereby their interaction was investigated using spectroscopic studies. Many conflicting models have been proposed based for these aggregates and up until recently these models could be generalized into two categories, based on the asymmetric repeating unit: (a) parallel chain model—where all the BChl molecules align in the same direction starting from a BChl monomer and (b) anti-parallel double-chain model, where the molecules have altering direction alignment and begin with a BChl dimer, i.e., the “piggy-back” arrangement as shown in Figure 37b or a “face-to-face” configuration [267]. Depending on the arrangement, the intermediate aggregates can have varying configurations leading to differences in the overall aggregation. The stepwise arrangement of these molecules is highly influenced by the 3′ stereochemistry and this has been demonstrated by Tamiaki and co-workers, amongst others, in vitro using epimerically-pure BChls. The 3′-R epimer gave primarily anti-parallel arrangements whilst the 3′-S epimer yielded the parallel derivative. In the presence of small amounts of the 3′-S epimer, the 3′-R aggregates changed from an anti-parallel arrangement and resulted in an overall bilayer tubular supramolecular structure, consistent with those observed in natural chlorosomes. The self-assembly of equal mixtures of the BChl epimers was hence shown to proceed more readily than with the isolated derivatives, further proving the hypothesis that a heterochiral environment for self-assembly is thermodynamically more favored than that of a homochiral one [263].
The above models are based on the rod-like arrangement of BChl aggregates within chlorosomes which have long been proposed using electron microscopy studies. Another proposed organization is that of lamellar sheets [265], whereby the BChl a molecules and proteins of the “baseplate” may serve as the nucleation site for lamellar assembly. Here it is proposed that linear chains of BChls (parallel or anti-parallel aggregates) can be organized into planar structures instead of rod-like elements, whereby the farnesyl tails protrude from the plane as a hydrophobic surface for this formation. It is now widely accepted that chlorosomes most likely contain both elements, i.e., lamellar and cylindrical structures. This hypothesis was further enhanced by the recent work by Ganapathy et al., although the aggregation model conflicts those described above [266,268]. Here, four aggregate models, namely the parallel monomer stack, “piggy-back dimer model, anti-parallel monomer model and syn-anti monomer stack (see Figure 37b for a schematic representation of all 4 models), were compared using 1H NMR ring current shift calculations. It was concluded that the syn-anti monomer stack was the only model which was consistent with their NMR calculations and observations. This syn-anti model consists of alternating molecular conformations of the 3′ side chain, with syn-anti referring to the orientation of the OH ligation with the Mg atom of neighboring BChl molecules (syn ligation on the same side as farnesyl chain, anti ligation on the opposite side). These syn-anti aggregates can self-assemble into co-axial cylinders to form tubular-shaped elements, in addition to sheet-like elements, both of which are likely coexisting in natural chlorosomes.
8. Outlook
A discussion such as this can only scratch the surface of our knowledge on the photosynthetic pigments. A focus of chlorophylls omits the photo-protective, structural and accessory role of carotenoids in photosynthesis [269,270] and the beautiful structures of the phycobilisomes [271] with their constituent allophycocyanins, phycocyanins, phycoerythrins or phycoerythrocyanins [272,273]. The latter contain phycobilins, linear tetrapyrroles (e.g., phycocyanobilin, phycoerythrobilin, and others) with photophysical and stereochemical properties which are highly susceptible to the protein environment [274]. For example, their highly efficient chromatic adaptation [275] warrants a closer look at the pigment-protein interplay. Similarly, phytochrome [276], while not photosynthetic, a photo-responsive system utilizing the porphyrin-derived phytochromobilin chromophore must be mentioned here [277].
The (bacterio)chlorophylls are clearly a versatile class of photosynthetic pigments. The question of “Why Chlorophyll?” has been the subject of two titled reviews [217,278]. In Mauzerall’s 1973 discussion the transition from the requirements of a prototypical photooxidizing chromophore toward those of a photoreductant are compared for uroporphyrin and the chlorophylls [278]. For Chls, the general properties conferred by the macrocycle’s excited states are said to be augmented by the inclusion of Mg, which substantially improves its abilities as a photoreductant, and the absorption characteristics enhanced by reduction to the chlorin and the presence of the cyclopentanone ring. The phytol chain is noted to improve lipid solubility whilst the additional peripheral substituents are considered a mixture of remnants from the biosynthetic pathway and to impart stability to potentially reactive β-positions. Bjorn et al.’s consideration is more specific, and considers why Chl a has been selected to perform the dual roles of light-harvesting and function as the photochemical cofactors in oxygenic photosynthesis, capable of acting as a powerful oxidant and reductant, over all other Chl derivatives that serve only as excitation energy capture and transfer agents [217]. The argument is largely phenomenological and is based on the fact that there are no RCs in oxygenic phototrophs that contain any other type of Chl (with the rare exception of Chl d) and the conclusion is similarly so in the suggestion that its dominance is the result of an earlier appearance than chlorophyll b. However, another well-known point raised is that despite the many chlorophylls present in photosynthetic proteins, only the few in the RCs are involved in the oxidation-reduction photochemistry. Thus, Bjorn emphasizes the prevailing view that the details of the architecture of the protein environment is responsible for determining the precise role of the individual chlorophylls and says that, “the major tasks of a protein may be to exclude or position functionally compatible electron donors or acceptors, depending upon whether the complex will act as an antenna or a reaction center” [217].
Likewise the evolutionary relationship of the various PS is understood in general terms, both in terms of structural analogy of the protein and cofactor arrangements and the genetics of the coding sequences. Less well known are regulatory and dynamic aspects of the adaptation of the complexes to environmental conditions in structural terms, despite the often very detailed photophysical and spectroscopic studies thereof. Thus, many areas for further studies remain.
More difficult, if not impossible to answer is the question suggested by a reviewer, “suppose chlorophyll was achiral—would photosynthesis actually be any different”? Photosynthetic systems as we know them today are remarkably tolerant to the exchange of individual pigments, albeit with a loss in efficiency. Likewise, the central metal of some pigments in the RC can be replaced with attendant impact on the axial ligation. Similarly, many artificial antenna systems have been prepared without chiral control. All this could be taken as an indication that “chiral” chlorophylls are not required for the construction of an efficient photosynthetic system. On the other hand, chlorophylls, as (bacterio)chlorin-type porphyrins, appear to have evolved in terms of efficient spectral use and reduction of the achiral protoporphyrin (as an example of a “achiral Chl”) would automatically generate chiral centers. For antenna systems “chirality” is not required. The C132 chiral center is clearly not necessary, as exemplified by the BChls c–e. Likewise, the porphyrin-type Chl c lack the chiral centers at C17 and C18. Thus, “achiral photosynthesis” should still work, but be very limited with regard to their photobiological ecological niche (in terms of using the sun spectrum).
Some suggestions—purely along the lines of personal preference of an organic chemist—focus more on the individual chromophores. For example, while the biosynthesis of the chlorophylls is now well understood chemically and most of the enzymes have been characterized much less is known about the role the chlorophyll cofactors play in the biosynthesis and correct folding of the apoproteins [279]. Crucial points for understanding the molecular properties of the cofactors in vivo are their ligation and protein interactions. More attention should be paid to the cofactor-protein interactions at an atomic level and to the macrocycle conformation of the tetrapyrrole cofactors. With the low resolution of protein crystal structures compared to small molecule ones, this requires a more statistical approach and the comparative analysis of many protein structures which might ultimately yield a unified picture of 3D-tetrapyrrole functions. The basic analytical tools, protein crystallography [196], normal-structural decomposition for chromophore conformational analysis [107], and multivariate statistical cluster analysis of different structures [280] are in hand. For the bacterial RC II this has already resulted in the unambiguous identification of “conserved” chlorophyll conformations for the different functional classes of cofactors [281] and a similar analysis of the bacterial LHC is warranted [282]. Another example is the diastereoselectivity of the axial ligand binding in chlorophylls, which has now been confirmed using data from PS II and LHCII [283].
In terms of “small molecule” chemistry, the area of chlorophyll chemistry is well established. Synthesis and functionalization methods are in hand or under development and their use in areas such as photodynamic cancer therapy (PDT) is quite advanced. On the other hand, due to their low stability they have not been used much in ET models or in solar energy conversion materials except for chlorophyllin [284]. This might be overcome with appropriate chemical manipulations taking into account “construction” principles from nature. For example, nonplanar chlorins with their significantly altered properties have not attracted much interest yet [285] and catalytic systems based on chlorophylls are in their infancy. Many synthetic examples of reaction center or light-harvesting mimics have been prepared over the years, but aspects of π–π interactions or chirality have often been ignored [286].
Acknowledgments
This work was supported by a grant from Science Foundation Ireland (P.I. 09/IN.1/B2650).
Author Contributions
All authors shared in the conception, literature search and analysis of the field, and writing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Milgrom, L.R. The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. Applications: Past, Present, and Future, the Porphyrin Handbook; CRC Press: Boca Raton, FL, USA, 2000; Volume 6. [Google Scholar]
- Chapman, S.K.; Daff, S.; Munro, A.W. Heme: The most versatile redox centre in biology. In Metal Sited in Proteins and Models, Iron Centres; Hill, H.A.O., Sadler, P.J., Thomson, A.J., Eds.; Springer: Berlin, Germany, 1997; Volume 88, pp. 39–70. [Google Scholar]
- Perutz, M.F.; Wilkinson, A.J.; Paoli, M.; Dodson, G.G. The Stereochemical mechanism of the cooperative effects in hemoglobin revisited. Ann. Rev. Biophys. Biomol. Struct 1998, 27, 1–34. [Google Scholar]
- Schneider, S.; Marles-Wright, J.; Sharp, K.H.; Paoli, M. Diversity and conservation of interactions for binding heme in b-type heme proteins. Nat. Prod. Rep 2007, 3, 621–630. [Google Scholar]
- Poulos, T.L. Heme Enzyme structure and function. Chem. Rev 2014, 114, 3919–3962. [Google Scholar]
- Poulos, T.L. Heme enzyme crystal structures. Adv. Inorg. Biochem 1988, 7, 1–36. [Google Scholar]
- Isin, E.M.; Guengerich, P. Complex reactions catalyzed by cytochrome P450 enzyme. Biochim. Biophys. Acta 2007, 1770, 314–329. [Google Scholar]
- Hoff, A.J.; Deisenhofer, J. Photophysics of photosynthesis. Structure and spectroscopy of the reaction centers of purple bacteria. Phys. Rep 1997, 287, 1–247. [Google Scholar]
- Mochizuki, N.; Tanaka, R.; Grimm, B.; Nasuda, T.; Moulin, M.; Smith, A.G.; Tanaka, A.; Terry, M.J. The cell biology of tetrapyrroles: A life and death struggle. Trends Plant Sci 2010, 15, 488–498. [Google Scholar]
- Scheer, H. Chlorophylls; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique metalorganic compounds and the most complex vitamins. Molecules 2010, 15, 3328–3259. [Google Scholar]
- Gruber, K.; Puffer, B.; Kräutler, B. Vitamin B12-derivatives-enzyme cofactors and ligands of proteins and nucleic acids. Chem. Soc. Rev 2011, 40, 4346–4363. [Google Scholar]
- Pfaltz, A.; Jaun, B.; Flassler, A.; Eschenmoser, A.; Jaenchen, R.; Gilles, H.H.; Diekert, G.; Thauer, R.K. Factor-F430 from methanogenic bacteria structure of the porphinoid ligand system. Helv. Chim. Acta 1982, 65, 828–865. [Google Scholar]
- Crane, B.R.; Siegel, L.M.; Getzoff, E.D. Sulfite reductase structure at 1.6 angstrom-evolution and catalysis for reduction of inorganic anions. Science 1995, 270, 59–67. [Google Scholar]
- Battersby, A.R. Tetrapyrroles: The pigments of life. Nat. Prod. Rep 2000, 17, 507–526. [Google Scholar]
- Quail, P.H.; Boylan, M.T.; Parks, B.M.; Short, T.W.; Xu, Y.; Wagner, D. Phytochromes: Photosensory perception and signal-transduction. Science 1995, 268, 675–680. [Google Scholar]
- Wasielewski, M.R. Photoinduced electron-transfer in supramolecular systems for artificial photosynthesis. Chem. Rev 1992, 92, 435–461. [Google Scholar]
- Gust, D.; Moore, T.A.; Moore, A.L. Mimicking photosynthetic solar energy transduction. Acc. Chem. Res 2001, 34, 40–48. [Google Scholar]
- Fukuzumi, S.; Honda, T.; Kojima, T. Structures and photoinduced electron transfer of protonated complexes of porphyrins and metallophthalocyanines. Coord. Chem. Rev 2012, 256, 2488–2502. [Google Scholar]
- Bringmann, G.; Rüdenauer, S.; Götz, D.C.G.; Gulder, T.A.M.; Reichert, M. Axially chiral directly beta,beta-linked bisporphyrins: Synthesis and stereostructure. Org. Lett 2006, 8, 4743–4746. [Google Scholar]
- Senge, M.O.; Rössler, B.; von Gersdorff, J.; Schäfer, A.; Kurreck, H. The meso-beta-linkage as structural motif in porphyrin-based donor-acceptor compounds. Tetrahedron Lett 2004, 45, 3363–3367. [Google Scholar]
- Götz, D.C.G.; Bruhn, T.; Senge, M.O.; Bringmann, G. Synthesis and stereochemistry of highly unsymmetric beta,meso-linked porphyrin arrays. J. Org. Chem 2009, 74, 8005–8020. [Google Scholar]
- Gaudemer, A.; Gaudemer, F.; Merienne, C. Structural studies of metalloporphyrins. VIII. NMR evidence for planar chirality in natural porphyrins. Org. Magn. Res 1983, 21, 83–85. [Google Scholar]
- Borovkov, V.V. Supramolecular chirality in porphyrin chemistry. Symmetry 2014, 6, 256–294. [Google Scholar]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev 2005, 105, 2253–2277. [Google Scholar]
- A similar case could be made for the biological function of linear tetrapyrroles where several cases of chiral templating or stereocontrol have been demonstrated in vitro and in vivo in seminal works by Lightner and McDonagh [28–30].
- McDonagh, A.F.; Lightner, D.A.; Reisinger, M.; Palma, L.A. Human-Serum albumin as a chiral template. Stereoselective photocyclization of bilirubin. J. Chem. Soc. Chem. Commun 1986, 249–250. [Google Scholar]
- Lightner, D.A.; Gawronski, J.K.; Wikekoon, W.M.D. Complementarity and chiral recognition: Enantioselective complexation of bilirubin. J. Am. Chem. Soc 1987, 109, 6354–6362. [Google Scholar]
- Krois, D.; Lehner, H. Helically fixed chiral bilirubins and biliverdins—A new insight into the conformational associative and dynamic features of linear tetrapyrroles. J. Chem. Soc. Perkin. Trans. 1993, 2, 1351–1360. [Google Scholar]
- Holliday, G.L.; Thornton, J.M.; Marquet, A.; Smith, A.G.; Rébeillé, F.; Mendel, R.; Schubert, H.L.; Lawrence, A.D.; Warren, M.J. Evolution of enzymes and pathways for the biosynthesis of cofactors. Nat. Prod. Rep 2007, 24, 972–987. [Google Scholar]
- Fleming, I. Absolute configuration and the structure of chlorophyll. Nature 1967, 216, 151–152. [Google Scholar]
- Miyashita, H.; Ikemoto, H.; Kurano, N.; Adachi, K.; Chihara, M.; Miyachi, S. Chlorophyll d as a major pigment. Nature 1996, 383, 402. [Google Scholar]
- Granick, S. The pheoporphyrin nature of chlorophyll-c. J. Biol. Chem 1949, 179, 505. [Google Scholar]
- Mizoguchi, T.; Kimura, Y.; Tamiaki, H. Exclusive observation of the (132R)-Enantiomer of chlorophyll-c from a diatom Chaetoseros calcitrans. Photochem. Photobiol 2010, 86, 311–315. [Google Scholar]
- Willstätter, R.; Stoll, A. Untersuchungen über Chlorophyll; Methoden und Ergebniss; Springer: Berlin, Germany, 1913. [Google Scholar]
- Fischer, H.; Wenderoth, H. Optisch aktives hämotricarbonsäureimid aus chlorophyll. Justus Liebigs. Ann. Chem 1940, 545, 140–147. [Google Scholar]
- Fischer, H.; Orth, H. Die Chemie des Pyrrols; Akademische Verlagsgesellschaft: Leipzig, Germany, 1937. [Google Scholar]
- It must also be noted that the addition of a single or two different axial ligands to the Mg central metal generates a sixth asymmetric center and this will be discussed in Section 1.3.2.
- Ficken, G.E.; Johns, R.B.; Linstead, R.P. Chlorophyll and related compounds. Part IV. The position of the extra hydrogens in chlorophyll. The oxidation of pyropheophorbide-a. J. Chem. Soc 1956, 2272–2280. [Google Scholar]
- Woodward, R.B.; Ayer, W.A.; Beaton, J.M.; Bickelhaupt, F.; Bonnett, R.; Buchschacher, P.; Closs, G.L.; Dutler, H.; Hannah, J.; Hauck, F.P.; et al. The total synthesis of chlorophyll. J. Am. Chem. Soc 1960, 82, 3800–3802. [Google Scholar]
- Woodward, R.B.; Ayer, W.A.; Beaton, J.M.; Bickelhaupt, F.; Bonnett, R.; Buchschacher, P.; Closs, G.L.; Dutler, H.; Hannah, J.; Hauck, F.P.; et al. The total synthesis of chlorophyll a. Tetrahedron 1990, 46, 7599–7659. [Google Scholar]
- Wolf, H.; Brockmann, H., Jr.; Biere, H.; Inhoffen, H.H. Zur weiteren kenntnis des chlorophylls und des hämins, XIII. Darstellung der diastereomeren 10-Methoxy-(pyro)-methylphäophorbide a und bestimmung der relativen konfiguration am C-atom 10. Liebigs Ann. Chem 1967, 704, 208–225. [Google Scholar]
- Seybold, A.; Hirsch, G. Bakteriochlorophyll a-Das grüne pigment gelbgrüner bakterien. Naturwissenschaften 1954, 41, 258. [Google Scholar]
- Burrell, J.W.K.; Garwood, R.F.; Jackman, L.M.; Oskay, E.; Weedon, B.C.L. Carotenoids and related compounds. Part XIV. Stereochemistry and synthesis of geraniol, nerol, farnesol, and phytol. J. Chem. Soc. C 1966, 2144–2154. [Google Scholar]
- For a comprehensive discussion of all relevant “Chl” crystal structures see:; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific/Imperial College Press: Singapore, 2010; Volume 13, pp. 253–297.
- Fleming, I. The absolute configuration and the structures of chlorophyll and bacteriochlorophyll. J. Chem. Soc. C 1968, 2765–2770. [Google Scholar]
- International Union of Pure and Applied Chemistry and International Union of Biochemistry, Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature of tetrapyrroles (Recommendations 1986). Pure Appl. Chem 1987, 59, 779–832.
- Kemmer, T.; Brockmann, H.; Risch, N. Die einzelkomponenten der bacteriophäophorbidmethylester von bacteriochlorophyll c und d. Z. Naturforsch 1979, 34b, 633–637. [Google Scholar]
- Kobayashi, M.; Oh-oka, H.; Akutsu, S.; Akiyama, M.; Tominaga, K.; Kise, H.; Nishida, F.; Watanabe, T.; Amesz, J.; Koizumi, M.; et al. The primary electron acceptor of green sulfur bacteria, bacteriochlorophyll 663, is chlorophyll a esterified with Δ2,6-phytadienol. Photosynth. Res 2000, 63, 269–280. [Google Scholar]
- Caple, M.B.; Chow, H.; Strouse, C.E. Photosynthetic pigments of green sulfur bacterial. The esterifying alcohols of bacteriochlorophylls c from Chlorobium limicola. J. Biol. Chem 1978, 253, 6730–6737. [Google Scholar]
- Nishimori, R.; Mizoguchi, T.; Tamiaki, H.; Kashimura, S.; Saga, Y. Biosynthesis of unnatural bacteriochlorophyll c derivatives esterified with α,ω-diols in the green sulfur photosynthetic bacterium Chlorobaculum Tepidum. Biochem 2011, 50, 7756–7764. [Google Scholar]
- Brockmann, H., Jr.; Knobloch, G.; Schweer, I.; Trowitsch, W. Die alkoholkomponente des bakteriochlorophyll a aus Rhodospirillum rubrum. Arch. Mikrobiol 1973, 90, 161–164. [Google Scholar]
- Various other esterified alcohols have been found in smaller amounts. Especially the esterase of the bacteriochlorophylls appears to tolerate a range of alcohol substrates. See:; Frigaard, N.U.; Chew, A.G.M.; Maresca, J.A.; Bryant, D.A. Bacteriochlorophyll Biosynthesis in Green Bacteria. In Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, Netherlands, Advances in Photosynthesis and Respiration; 2006; Volume 25, pp. 201–221. [Google Scholar]
- Stevens, J.M.; Daltrop, O.; Allen, J.W.A.; Ferguson, S.J. C-Type cytochrome formation: Chemical and biological enigmas. Acc. Chem. Res 2004, 37, 999–1007. [Google Scholar]
- Balaban, T.S.; Fromme, P.; Holzwarth, A.R.; Krauß, N.; Prokhorenko, V.I. Relevance of the diastereotopic ligation of magnesium atoms of chlorophylls in Photosystem I. Biochim. Biophys. Acta 2002, 1556, 197–207. [Google Scholar]
- Balaban, T.S. Are syn-ligated (bacterio)chlorophyll dimers energetic traps in light-harvesting systems? FEBS Lett 2003, 545, 97–102. [Google Scholar]
- Balaban, T.S. Relevance of the diastereotopic ligation of magnesium atoms of chlorophylls in the major light-harvesting complex II (LHC II) of green plants. Photosynth. Res 2005, 86, 251–262. [Google Scholar]
- Oba, T.; Tamiaki, H. Which side of the π-macrocycle plane of (bacterio)chlorophylls is favored for binding of the fifth ligand? Photosynth. Res 2002, 74, 1–10. [Google Scholar]
- Taniguchi, M.; Mass, O.; Boyle, P.D.; Tang, Q.; Diers, J.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Structural studies of sparsely substituted synthetic chlorins and phorbines establish benchmarks for changes in the ligand core and framework of chlorophyll macrocycles. J. Mol. Struct 2010, 979, 27–45. [Google Scholar]
- Fleischer, E.B.; Miller, C.K.; Webb, L.E. Crystal and molecular structures of some metal tetraphenylporphines. J. Am. Chem. Soc 1964, 86, 2342–2347. [Google Scholar]
- Senge, M.O. Highly substituted porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; CRC Press: Boca Raton, FL, USA, 2000; Volume 1, pp. 239–347. [Google Scholar]
- Senge, M.O. Exercises in molecular gymnastics—Bending, stretching and twisting porphyrins. Chem. Commun 2006, 243–256. [Google Scholar]
- Forman, A.; Renner, M.W.; Fujita, E.; Barkigia, K.M.; Evans, M.C.W.; Smith, K.M.; Fajer, J. ESR and ENDOR probes of skeletal conformations implications for conformations and orientations of chlorophylls in vivo. Isr. J. Chem 1989, 29, 57–64. [Google Scholar]
- Senge, M.O. The conformational flexibility of tetrapyrroles—Current model studies and photobiological relevance. J. Photochem. Photobiol. B Biol 1992, 16, 3–36. [Google Scholar]
- Shelnutt, J.A.; Song, X.Z.; Ma, J.G.; Jia, S.L.; Jentzen, W.; Medforth, C.J. Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev 1998, 27, 31–41. [Google Scholar]
- Senge, M.O.; Wiehe, A.; Ryppa, C. Synthesis, reactivity and structure of chlorophylls. In Chlorophylls and Bacteriochlorophylls: Biochemistry Biophysics, Functions and Applications; Grimm, B., Porra, R., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 25, pp. 27–37. [Google Scholar]
- Nyman, E.S.; Hynninen, P.H. Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B Biol 2004, 73, 1–28. [Google Scholar]
- Johnston, L.G.; Watson, W.F. The allomerization of chlorophyll. J. Chem. Soc 1956, 1203–1212. [Google Scholar]
- Schaber, P.M.; Hunt, J.E.; Fries, R.; Katz, J.J. High-Performance liquid-chromatographic study of the chlorophyll allomerization reaction. J. Chromatogr 1984, 316, 25–41. [Google Scholar]
- Hynninen, P.H.; Assandri, S. Chlorophylls. II. Allomerization of chlorophylls a and b. Acta Chem. Scand 1973, 27, 1478–1486. [Google Scholar]
- Senge, M.; Struck, A.; Dörnemann, D.; Scheer, H.; Senger, H. Hydroxylation of chlorinated and unchlorinated chlorophylls in vitro. Z. Naturforsch 1988, 43c, 515–518. [Google Scholar]
- Walker, J.S.; Jie, C.; Keely, B.J. Identification of diastereomeric chlorophyll allomers by atmospheric pressure chemical ionisation liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom 2003, 17, 1125–1131. [Google Scholar]
- Grese, R.P.; Cerny, R.L.; Gross, M.L.; Senge, M. Determination of structure and properties of modified chlorophylls by using fast atom bombardment combined with tandem mass spectrometry. J. Am. Soc. Mass Spectrom 1990, 1, 72–84. [Google Scholar]
- Maeda, H.; Watanabe, T.; Kobayashi, M.; Ikegami, I. Presence of 2 chlorophyll a′ molecules at the core of Photosystem I. Biochim. Biophys. Acta 1992, 1099, 74–80. [Google Scholar]
- Kobayashi, M.; Vandemeent, E.J.; Erkelens, C.; Amesz, J.; Ikegami, I.; Watanabe, T. Bacteriochlorophyll g epimer as possible reaction center component of heliobacteria. Biochim. Biophys. Acta 1991, 1057, 89–96. [Google Scholar]
- Helfrich, M.; Schoch, S.; Lempert, U.; Cmiel, E.; Rüdiger, W. Chlorophyll synthetase cannot synthesize chlorophyll a′. Eur. J. Biochem 1994, 219, 267–275. [Google Scholar]
- Scheer, H. An overview of chlorophylls and bacterial chlorophylls: Biochemistry, biophysics, functions, and applications. In Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 25, pp. 1–26. [Google Scholar]
- Pavlov, V.Y.; Ponomarev, G.V. Modification of the peripheral substituents in chlorophylls a and b and their derivatives. Chem. Heterocycl. Compd 2004, 40, 393–425. [Google Scholar]
- Ethirajan, M.; Chen, Y.H.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev 2011, 40, 340–362. [Google Scholar]
- Chen, Y.H.; Li, G.L.; Pandey, R.K. Synthesis of bacteriochlorins and their potential utility in photodynamic therapy (PDT). Curr. Org. Chem 2004, 8, 1105–1134. [Google Scholar]
- Grin, M.; Mironov, A.F.; Shtil, A.A. Bacteriochlorophyll a and its derivatives: Chemistry and perspectives for cancer therapy. Anti-Cancer Agents Med. Chem 2008, 8, 683–697. [Google Scholar]
- Balaban, T.S.; Tamiaki, H.; Holzwarth, A.R. Chlorins programmed for self-assembly. Top. Curr. Chem 2005, 258, 1–38. [Google Scholar]
- Soret, J.L. Analyse spectrale: Sur le spectre d’absorption du sang dans la partie violette et ultra-violette. Compt. Rend 1883, 97, 1269–1270. [Google Scholar]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc 1961, 6, 138–163. [Google Scholar]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins: Part II. Four orbital model. J. Mol. Spectrosc 1963, 11, 108–127. [Google Scholar]
- Wang, X.-F.; Tamiaki, H. Cyclic tetrapyrrole based molecules for dye-sensitized solar cells. Energy Environ. Sci 2010, 3, 94–106. [Google Scholar]
- Brückner, C.; McCarthy, J.R.; Daniell, H.W.; Pendon, Z.D.; Ilagan, R.P.; Francis, T.M.; Ren, L.; Birge, R.R.; Frank, H.A. A spectroscopic and computational study of the singlet and triplet excited states of synthetic β-functionalized chlorins. Chem. Phys 2003, 294, 285–303. [Google Scholar]
- Kratky, C.; Dunitz, J.D. Comparison of the results of two independent analyses of the ethyl chlorophyllide a dihydrate crystal structure. Acta Crystallogr. B 1975, 31, 1586–1589. [Google Scholar]
- Chow, H.-C.; Serlin, R.; Strouse, C.E. Crystal and molecular structure and absolute configuration of ethyl chlorophyllide a–dihydrate. Model for the different spectral forms of chlorophyll a. J. Am. Chem. Soc 1975, 97, 7230–7237. [Google Scholar]
- Kratky, C.; Isenring, H.P.; Dunitz, J.D. Methyl pyrochlorophyllide a monohydrate monoetherate. Acta Crystallogr 1977, B33, 547–549. [Google Scholar]
- De Boer, I.; Matysik, J.; Amakawa, M.; Yagai, S.; Tamiaki, H.; Holzwarth, A.R.; de Groot, H.J.M. MAS NMR structure of a microcrystalline Cd-Bacteriochlorophyll d analogue. J. Am. Chem. Soc 2003, 125, 13374–13375. [Google Scholar]
- Senge, M.O.; Ruhlandt-Senge, K.; Smith, K.M. Structure and conformation of photosynthetic pigments and related compounds. VIII. Molecular structure of an iron(III) chlorophyll derivative, chloro(phytochlorinato methyl ester)iron(III). Z. Naturforsch 1995, 50b, 139–146. [Google Scholar]
- Knapp, S.; Huang, B.; Emge, T.J.; Sheng, S.; Krogh-Jespersen, K.; Potenza, J.A.; Schugar, H.J. A pyropheophorbide dimer with single pyrrole π overlap and a low-energy Q absorption. J. Am. Chem. Soc 1999, 121, 7977–7978. [Google Scholar]
- Senge, M.O.; Smith, K.M. Structure and conformation of photosynthetic pigments and related compounds. II. Nickel(II) methyl pyropheophorbide a: A severely distorted chlorophyll derivative. Photochem. Photobiol 1991, 54, 841–846. [Google Scholar]
- Senge, M.O.; Smith, K.M. Structure and conformation of photosynthetic pigments and related compounds. VII. On the conformation of the methyl ester of (20-methyl-phytochlorinato)nickel(II): A bacteriochlorophyll c model compound. Photochem. Photobiol 1994, 60, 139–142. [Google Scholar]
- Fischer, M.S.; Templeton, D.H.; Zalkin, A.; Calvin, M. Crystal and molecular structure of methyl pheophorbide with applications to the chlorophyll arrangement in photosynthetic lamellae. J. Am. Chem. Soc 1972, 94, 3613–3619. [Google Scholar]
- Senge, M.O.; Smith, K.M. Structure and conformation of photosynthetic pigments and related compounds. I. Methyl mesopyropheophorbide a. Z. Kristallogr 1992, 199, 239–248. [Google Scholar]
- Chee, C.F.; Rahman, N.A.; Zain, S.M.; Ng, S.W. Pheophorbide b ethyl ester from a Chlorella vulgaris dietary supplement. Acta Crystallogr. E 2008, 64, o1986. [Google Scholar]
- Kim, H.J.; Lindsey, J.S. De novo synthesis of stable tetrahydroporphyrinic macrocycles: Bacteriochlorins and a tetradehydrocorrin. J. Org. Chem 2005, 70, 5474–5486. [Google Scholar]
- Barkigia, K.M.; Gottfried, D.S.; Boxer, S.G.; Fajer, J. A high-precision structure of a bacteriochlorophyll derivative, methyl bacteriopheophorbide a. J. Am. Chem. Soc 1989, 111, 6444–6446. [Google Scholar]
- Barkigia, K.M.; Gottfried, D.S. A new crystal form of methyl bacteriopheophorbide a. Acta Crystallogr. C 1994, 50, 2069–2072. [Google Scholar]
- Smith, K.M.; Kehres, L.A.; Tabba, H.D. Structures of the bacteriochlorophyll c homologs: Solution to a longstanding problem. J. Am. Chem. Soc 1980, 102, 7149–7151. [Google Scholar]
- Barkigia, K.M.; Chantranupong, L.; Smith, K.M.; Fajer, J. Structural and theoretical models of photosynthetic chromophores. Implications for redox, light-absorption properties and vectorial electron flow. J. Am. Chem. Soc 1988, 110, 7566–7567. [Google Scholar]
- Eschenmoser, A. Chemistry of corphinoids. Ann. N. Y. Acad. Sci 1986, 471, 108–129. [Google Scholar]
- Scheidt, W.R.; Lee, Y.-J. Recent advances in the stereochemistry of metallotetrapyrroles. Struct. Bonding (Berlin) 1987, 64, 1–70. [Google Scholar]
- Jentzen, W.; Song, X.Z.; Shelnutt, J.A. Structural characterization of synthetic and protein-bound porphyrins in terms of the lowest-frequency normal coordinates of the macrocycle. J. Phys. Chem. B 1997, 101, 1684–1699. [Google Scholar]
- Jentzen, W.; Ma, J.-G.; Shelnutt, J.A. Conservation of the conformation of the porphyrin macrocycle in hemoproteins. Biophys. J 1998, 74, 753–763. [Google Scholar]
- Smith, K.M.; Bobe, F.W. Light adaptation of bacteriochlorophyll-d producing bacteria by enzymatic methylation of their antenna pigments. J. Chem. Soc. Chem. Commun 1987, 276–277. [Google Scholar]
- Senge, M.O. Recent advances in the biosynthesis and chemistry of the chlorophylls. Photochem. Photobiol 1993, 57, 189–206. [Google Scholar]
- Hörtensteiner, S.; Kräutler, B. Chlorophyll breakdown in higher plants. Biochim. Biophys. Acta 2011, 1807, 977–988. [Google Scholar]
- Kräutler, B.; Matile, P. Solving the riddle of chlorophyll breakdown. Acc. Chem. Res 1999, 32, 35–43. [Google Scholar]
- Grimm, B.; Porra, R.; Rüdiger, W.; Scheer, H. Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Springer: Dordrecht, The Netherlands, 2006; Advances in Photosynthesis and Respiration, Volume 25. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. Handbook of Porphyrin Science; World Scientific: Singapore, 2012; Volume 20. [Google Scholar]
- Shemin, D.; Russell, C.S. Delta-Aminolevulinic acid, its role in the biosynthesis of porphyrins and purines. J. Am. Chem. Soc 1953, 75, 4873–4874. [Google Scholar]
- Von Wettstein, D.; Gough, S.; Kannangara, C.G. Chlorophyll biosynthesis. Plant Cell 1995, 7, 1039–1057. [Google Scholar]
- Jaffe, E.K. Porphobilinogen synthase, the first source of heme symmetry. J. Bioenerg. Biomembr 1995, 27, 169–179. [Google Scholar]
- Raronskaya, E.; Grimm, B. The pathway from 5-aminolevulinic acid to protochlorophyllide and protoheme. In Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 25, pp. 173–188. [Google Scholar]
- Senge, M.O. Struktur und Biosynthese der Bakteriochlorophylle. Chem. Unserer Zeit 1992, 26, 86–93. [Google Scholar]
- Shoolingin-Jordan, P.M. Porphobilinogen deaminase and uroporphyrinogen III synthase: Structure, molecular-biology, and mechanism. J. Bioenerg. Biomembr 1995, 27, 181–195. [Google Scholar]
- Hart, G.J.; Miller, A.D.; Battersby, A.R. Evidence that pyrromethane cofactor of hydroxymethylbilane synthase (porphobilinogen deaminase) is bound through the sulphur atom of a cysteine residue. Biochem. J 1987, 252, 909–912. [Google Scholar]
- Jordan, P.M.; Warren, M.J. Evidence for a dipyrromethane cofactor at the catalytic site of E. coli porphobilinogen deaminase. FEBS Lett 1987, 225, 87–92. [Google Scholar]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci 2010, 19, 1137–1161. [Google Scholar]
- Battersby, A.R.; Fookes, C.J.R.; Gustafson-Potter, K.E.; Matcham, G.W.J.; McDonald, E. Proof by synthesis that unarranged hydroxymethylbilane is the product from deaminase and the substrate for cosynthetase in the biosynthesis of uro’gen-III. J. Chem. Soc. Chem. Commun 1979, 1155–1158. [Google Scholar]
- Stark, W.M.; Hart, G.J.; Battersby, A.R. Synthetic studies on the proposed spiro intermediate for biosynthesis of the natural porphyrins: Inhibition of cosynthetase. J. Chem. Soc. Chem. Commun 1986, 465–467. [Google Scholar]
- Beale, S.I. Enzymes of chlorophyll biosynthesis. Photosynth. Res 1999, 60, 43–73. [Google Scholar]
- Layer, G.; Moser, J.; Heinz, D.W.; Jahn, D.; Schubert, W.D. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J 2003, 22, 6214–6224. [Google Scholar]
- Koch, M.; Breithaupt, C.; Kiefersauer, R.; Freigang, J.; Huber, R.; Messerschmidt, A. Crystal structure of protoporphyrinogen IX oxidase: A key enzyme in haem and chlorophyll biosynthesis. EMBO J 2004, 23, 1720–1728. [Google Scholar]
- Walker, C.J.; Willows, R.D. Mechanism and regulation of Mg-chetalase. Biochem. J 1997, 327, 321–333. [Google Scholar]
- Minamizaki, K.; Mizoguchi, T.; Goto, T.; Tamiaki, H.; Fujita, Y. Identification of two homologous genes, chlA I and chlA II, that are differentially involved in isocyclic ring formation of chlorophyll a in the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem 2008, 283, 2684–2692. [Google Scholar]
- Bollivar, D.W.; Beale, S.I. The chlorophyll biosynthetic enzyme Mg-protoporphyrin IX monomethyl ester (oxidative) cyclase (characterization and partial purification from Chlamydomonas reinhardtii and Synechocystis sp. PCC 6803). Plant Physiol 1996, 112, 105–114. [Google Scholar]
- Granick, S. Magnesium vinyl pheoporphyrin A-5, another intermediate in the biological synthesis of chlorophyll. J. Biol. Chem 1950, 183, 713–730. [Google Scholar]
- Gough, S.P.; Petersen, B.O.; Jens, O.D. Anaerobic chlorophyll isocyclic ring formation in Rhodobacter capsulatus requires a cobalamin cofactor. Proc. Natl. Acad. Sci. USA 2000, 97, 6908–6913. [Google Scholar]
- Bröcker, M.J.; Jahn, D.; Moser, J. Key enzymes of chlorophyll biosynthesis. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2012; Volume 20, pp. 2–43. [Google Scholar]
- Wang, P.; Gao, J.; Wan, C.; Zhang, F.; Xu, Z.; Huang, X.; Sun, X.; Deng, X. Divinyl chlorophyll(ide) a can be converted to monovinyl chlorophyll(ide) a by a divinyl reductase in rice. Plant Physiol 2010, 153, 994–1003. [Google Scholar]
- Apel, K.; Santel, H.J.; Redlinger, T.E.; Falk, H. The protochlorophyllide holochrome of barley (Hordeum vulgare L.). Isolation and characterization of the NADPH:protochlorophyllide oxidoreductase. Eur. J. Biochem 1980, 111, 251–258. [Google Scholar]
- Sundqvist, C.; Dahlin, C. With chlorophyll pigments from prolamellar bodies to light-harvesting complexes. Physiol. Plant 1997, 100, 748–759. [Google Scholar]
- Reinbothe, C.; Buhr, F.; Bartsch, S.; Desvignes, C.; Quigley, F.; Pesey, H.; Reinbothe, S. In vitro-mutagenesis of NADPH:protochlorophyllide oxidoreductase B: Two distinctive protochlorophyllide binding sites participate in enzyme catalysis and assembly. Mol. Genet. Genom 2006, 275, 540–552. [Google Scholar]
- Heyes, D.J.; Ruban, A.V.; Wilks, H.M.; Hunter, C.N. Enzymology below 200 K: The kinetics and thermodynamics of the photochemistry catalyzed by protochlorophyllide oxidoreductase. Proc. Natl. Acad. Sci. USA 2002, 99, 11145–11150. [Google Scholar]
- Valera, V.; Fung, M.; Wessler, A.N.; Richards, W.R. Synthesis of 4R- and 4S-tritium labeled NADPH for the determination of the coenzyme stereospecificity of NADPH:protochlorophyllide oxidoreductase. Biochem. Biophys. Res. Commun 1987, 148, 515–520. [Google Scholar]
- Heyes, D.J.; Ruban, A.V.; Hunter, C.N. Protochlorophyllide oxidoreductase: “dark” reactions of a light-driven enzyme. Biochemistry 2002, 42, 523–528. [Google Scholar]
- Helfrich, M.; Schoch, S.; Schäfer, W.; Ryberg, M.; Rüdiger, W. Absolute configuration of protochlorophyllide a and substrate specificity of NADPH-protochlorophyllide oxidoreductase. Plant Biochem 1996, 118, 2606–2611. [Google Scholar]
- Adapted from Figure 8 in reference [134].
- Burke, D.H.; Alberti, M.; Hearst, J.E. bchFNBH bacteriochlorophyll synthesis genes of Rhodobacter capsulatus and identification of the third subunit of light-independent protochlorophyllide reductase in bacteria and plants. J. Bacteriol 1993, 175, 2414–2422. [Google Scholar]
- Sarma, R.; Barney, B.M.; Hamilton, T.L.; Jones, A.; Seefeldt, L.C.; Peters, J.W. Crystal structure of the L protein of Rhodobacter sphaeroides light-independent protochlorophyllide reductase with MgADP bound: A homologue of the nitrogenase Fe protein. Biochemistry 2008, 47, 13004–13015. [Google Scholar]
- Bröcker, M.J.; Virus, S.; Ganskow, S.; Heathcote, P.; Heinz, D.W.; Schubert, W.-D.; Jahn, D.; Moser, J. ATP-Driven reduction by dark-operative protochlorophyllide oxidoreductase from Chlorobium tepidium mechanistically resembles nitrogenase catalysis. J. Biol. Chem 2008, 283, 10559–10567. [Google Scholar]
- Bröcker, M.J.; Schomburg, S.; Heinz, D.W.; Jahn, D.; Schubert, W.D.; Moser, J. Crystal structure of the nitrogenase-like dark operative protochlorophyllide oxidoreductase catalytic complex (ChlN/ChlB)2. J. Biol. Chem 2010, 285, 27336–27345. [Google Scholar]
- Muraki, N.; Nomata, J.; Ebata, K.; Mizoguchi, T.; Shiba, T.; Tamiaki, H.; Kurisu, G.; Fujita, Y. X-ray crystal structure of the light independent protochlorophyllide reductase. Nature 2010, 465, 110–114. [Google Scholar]
- Rüdiger, W.; Böhm, S.; Helfrich, M.; Schulz, S.; Schoch, S. Enzymes of the last steps of chlorophyll biosynthesis: Modification of the substrate structure helps to understand the topology of the active centers. Biochemistry 2005, 44, 10864–10872. [Google Scholar]
- Oster, U.; Bauer, C.E.; Rüdiger, W. Characterization of chlorophyll a and bacteriochlorophyll a synthases by heterologous expression in Escherichia coli. J. Biol. Chem 1997, 272, 9671–9676. [Google Scholar]
- Schneegurt, M.A.; Beale, S.I. Origin of the chlorophyll b formyl oxygen in Chlorella vulgaris. Biochemistry 1992, 31, 11677–11683. [Google Scholar]
- Oster, U.; Tanaka, R.; Tanaka, A.; Rüdiger, W. Cloning and functional expression of the gene encoding the key enzyme for chlorophyll b biosynthesis (CAO) from Arabidopsis thaliana. Plant J 2000, 21, 305–210. [Google Scholar]
- Scheumann, V.; Helfrich, M.; Schoch, S.; Rüdiger, W. Reduction of the formyl group of zinc pheophorbide b in vitro and in vivo: A model for the chlorophyll b to a transformation. Z. Naturforsch 1996, 51c, 185–194. [Google Scholar]
- Scheumann, V.; Schoch, S.; Rüdiger, W. Chlorophyll a formation in the chlorophyll b reductase reaction requires reduced ferrodoxin. J. Biol. Chem 1998, 273, 35102–35108. [Google Scholar]
- Ito, H.; Ohtsuka, T.; Tanaka, A. Conversion of chlorophyll b to chlorophyll a via 7-hydroxymethyl chlorophyll. J. Biol. Chem 1996, 271, 1475–1479. [Google Scholar]
- Burke, D.H.; Alberti, M.; Hearst, J.E. The Rhodobacter capsulatus chlorin reductase-encoding locus, bchA, consists of three genes, bchX, bchY, bchZ. J. Bacteriol 1993, 175, 2407–2413. [Google Scholar]
- Nomata, J.; Mizoguchi, T.; Tamiaki, H.; Fujita, Y. A second nitrogenase-like enzyme for bacteriochlorophyll biosynthesis: Reconstitution of chlorophyllide a reductase with purified x-protein (BchX) and yz-protein (BchY-BchZ) from Rhodobacter capsulatus. J. Biol. Chem 2006, 281, 15021–15028. [Google Scholar]
- McGlynn, P.; Hunter, C.N. Genetic analysis of the bchC and bchA genes of Rhodobacter sphaeroides. Mol. Gen. Genet 1993, 236, 227–234. [Google Scholar]
- Addlesee, H.A.; Hunter, C.N. Rhodospirillum rubrum possesses a variant of the bchP gene, encoding geranylgeranyl-bacteriophytin reductase. J. Bacteriol 2002, 184, 1578–1586. [Google Scholar]
- Tsukatani, Y.; Yamamoto, H.; Harada, J.; Yoshitomi, T.; Nomata, J.; Kasahara, M.; Mizoguchi, T.; Fujita, Y.; Tamiaki, H. An unexpectedly branched biosynthetic pathway for bacteriochlorophyll b capable of absorbing near-infrared light. Sci. Rep 2013, 3. [Google Scholar] [CrossRef]
- In the older literature they are often referred to as the Chlorobium. chlorophylls as they were first isolated from Chlorobium strains.
- Risch, N.; Brockmann, H. Zur absoluten Konfiguration der Chlorophylle, VII. Bacteriochlorophyll e, ein neues Chlorophyll aus braunen Arten von Chlorobiaceae. Liebigs Ann. Chem 1976, 578–583. [Google Scholar]
- Mizoguchi, T.; Harada, J.; Tsukatani, Y.; Tamiaki, H. Isolation and characterization of a new bacteriochlorophyll c bearing a neopentyl substituent at the 8-position from the bciD-deletion mutant of the brown-coloured green sulfur bacterium Chlorobaculum limnaeum. Photosyn. Res 2014, 121, 3–12. [Google Scholar]
- Risch, N.; Koster, B.; Schormann, A.; Siemens, T.; Brockmann, H. Bacteriochlorophyll f—Partialsynthese und eigenschaften einiger derivate. Liebigs Ann. Chem 1988, 343–347. [Google Scholar]
- Vogl, K.; Tank, M.; Orf, G.S.; Blankenship, R.E.; Bryant, D.A. Bacteriochlorophyll f: Properties of chlorosomes containing the “forbidden chlorophyll”. Front. Microbiol 2012, 3. [Google Scholar] [CrossRef]
- Earlier suggestions included a direct cyclization of Mg(II) protoporphyrin IX accompanied by decarboxylation to yield [3,8-divinyl]-protochlorophyllide a, or passing through an intermediate such as [13-vinyl]-protoporphyrin IX [54].
- Suzuki, Y.; Doi, M.; Shioi, Y. Two enzymatic reaction pathways in the formation of pyropheophorbide a. Photosynth. Res 2002, 74, 225–233. [Google Scholar]
- Huster, M.S.; Smith, K.M. Biosynthetic studies of substituent homologation in bacteriochlorophylls c and d. Biochemistry 1990, 29, 4348–4355. [Google Scholar]
- Chew, A.-G.-M.; Frigaard, N.-U.; Bryant, D.A. Bacteriochlorophyllide c C-82 and C-121 methyltransferases are essential for adaptation to low light in Chlorobaculum tepidum. J. Bacteriol 2007, 189, 6176–6184. [Google Scholar]
- Liu, Z.; Bryant, D.A. Bacteriochlorophyll biosynthesis and assembly in green chlorophototrophic bacteria: Theme and variations. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R, Eds.; World Scientific: Singapore, 2012; Volume 20, pp. 107–140. [Google Scholar]
- Maresca, J.A.; Gomez Maqueo Chew, A.; Ros Ponsatí, M.; Frigaard, N.-U.; Ormerod, J.G.; Bryant, D.A. The bchU gene of Chlorobium tepidum encodes the C-20 methyltransferase in bacteriochlorophyll c biosynthesis. J. Bacteriol 2004, 186, 2558–2566. [Google Scholar]
- Frigaard, N.-U.; Voigt, G.D.; Bryant, D.A. Chlorobium tepidum mutant lacking bacteriochlorophyll c made by inactivation of the bchK gene, encoding bacteriochlorophyll c synthase. J. Bacteriol 2002, 184, 3368–3376. [Google Scholar]
- Harada, J.; Mizoguchi, T.; Satoh, S.; Tsukatani, Y.; Yokono, M.; Noguchi, M.; Tanaka, A.; Tamiaki, H. Specific gene bciD for C7-methyl oxidation in bacteriochlorophyll e biosynthesis of brown-coloured green sulfur bacteria. PLoS One 2013, 8, e60026. [Google Scholar] [CrossRef]
- Smith, K.M.; Craig, G.W.; Kehres, L.A.; Pfennig, N. Reversed-Phase high-performance liquid-chromatography and structural assignment of the bacteriochlorophylls-c. J. Chromatogr 1983, 281, 209–223. [Google Scholar]
- Smith, K.M.; Goff, D.A.; Fajer, J.; Barkigia, K.M. Chirality and structures of bacteriochlorophylls d. J. Am. Chem. Soc 1982, 104, 3747–3749. [Google Scholar]
- Nelson, N.; Yocum, C.F. Structure and function of photosystems I and II. Ann. Rev. Plant Biol 2006, 57, 521–565. [Google Scholar]
- James, B. Photosynthetic energy conversion: Natural and artificial. Chem. Soc. Rev 2009, 38, 185–196. [Google Scholar]
- Blankenship, R.E. Origin and early evolution of photosynthesis. Photosynth. Res 1992, 33, 91–111. [Google Scholar]
- Van Grondelle, R.; Novoderezhkin, V.I. Energy transfer in photosynthesis: Experimental insights and quantitative models. Phys. Chem. Chem. Phys 2006, 8, 793–807. [Google Scholar]
- Gust, D.; Moore, T.A. Mimicking photosynthesis. Science 1989, 244, 35–41. [Google Scholar]
- Wasielewski, M.R. Self-Assembly strategies for integrating light harvesting and charge separation in artificial photosynthetic systems. Acc. Chem. Res 2009, 42, 1910–1921. [Google Scholar]
- Sergeeva, N.N.; Senge, M.O. Photochemical transformations involving porphyrins and phthalocyanines. In CRC Handbook of Organic Photochemistry and Photobiology; Griesbeck, A., Oelgemöller, M., Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 831–876. [Google Scholar]
- Krause, G.H.; Weis, E. Chlorophyll fluorescence and photosynthesis: The basics. Ann. Rev. Plant Physiol. Plant Mol. Biol 1991, 42, 313–349. [Google Scholar]
- Olson, J.M.; Blankenship, R.E. Thinking about the evolution of photosynthesis. Photosynth. Res 2004, 80, 373–386. [Google Scholar]
- Orr, L.; Govindjee. Photosynthesis web resources. Photosynth. Res 2013, 115, 179–214. [Google Scholar]
- Rappaport, F.; Diner, B.A. Primary photochemistry and energetics leading to the oxidation of the (Mn)4Ca cluster and to the evolution of molecular oxygen in photosystem II. Coord. Chem. Rev 2008, 252, 259–272. [Google Scholar]
- Hill, R.; Bendall, F. Function of the two cytochrome components in chloroplasts: A working hypothesis. Nature 1960, 186, 136–137. [Google Scholar]
- Gupta, R.S. Evolutionary relationships among photosynthetic bacteria. Photosynth. Res 2003, 76, 173–183. [Google Scholar]
- Feher, G.; Allen, J.P.; Okamura, M.Y.; Rees, D.C. Structure and function of bacterial photosynthetic reaction centers. Nature 1989, 339, 111–116. [Google Scholar]
- Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. Structure of the protein subunits in the photosynthetic reaction center of Rhodopseudomonas viridis at 3 Å resolution. Nature 1985, 318, 618–624. [Google Scholar]
- Cape, J.L.; Bowman, M.K.; Kramer, D.M. Understanding the cytochrome bc complexes by what they don’t do. The Q-cycle at 30. Trends Plant Sci 2006, 11, 46–55. [Google Scholar]
- Liu, L.-N.; Scheuring, S. Investigation of photosynthetic membrane structure using atomic force microscopy. Trends Plant Sci 2013, 18, 277–286. [Google Scholar]
- Nevo, R.; Charuvi, D.; Tsabari, O.; Reich, Z. Composition, architecture and dynamics of the photosynthetic apparatus in higher plants. Plant J 2012, 70, 157–176. [Google Scholar]
- The primary donors in different photosynthetic systems are abbreviated as PXXX, where XXX is the absorption band where absorption of a photon causes bleaching of absorbance. Thus, it is indicative for the “special pair”.
- Ermler, U.; Fritzsch, G.; Buchanan, S.K.; Michel, H. Structure of the photosynthetic reaction-center from Rhodobacter sphaeroides at 2.65-Ångstrom resolution: Cofactors and protein-cofactor interactions. Structure 1994, 2, 925–936. [Google Scholar]
- Allen, J.P.; Seng, C.; Larson, C. Structures of proteins and cofactors: X-ray crystallography. Photosynth. Res 2009, 102, 231–240. [Google Scholar]
- Busch, A.; Hippler, M. The structure and function of eukaryotic photosystem I. Biochim. Biophys. Acta 2011, 1807, 864–877. [Google Scholar]
- Olson, T.L.; Williams, J.C.; Allen, J.P. The three-dimensional structures of bacterial reaction centers. Photosynth. Res 2014, 120, 87–98. [Google Scholar]
- Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. X-ray structure analysis of a membrane protein complex: Electron density map at 3 Å resolution and a model of the chromophores of the photosynthetic reaction center from Rhodopseudomonas viridis. J. Mol. Biol 1984, 180, 385–398. [Google Scholar]
- Deisenhofer, J.; Michel, H. The photosynthetic reaction center from the purple bacterium Rhodopseudomonas viridis. Science 1989, 245, 1463–1473. [Google Scholar]
- Deisenhofer, J.; Epp, O.; Sinning, I.; Michel, H. Crystallographic refinement at 2.3 Å resolution and refined model of the photosynthetic reaction centre from Rhodopseudomonas viridis. J. Mol. Biol 1995, 246, 429–457. [Google Scholar]
- Parson, W.W.; Warshel, A. Mechanism of charge separation in purple bacterial reaction centers. In The Purple Phototrophic Bacteria; Hunter, C.N., Daldal, F., Thurnauer, M.C., Beatty, J.T., Eds.; Springer: Dordrecht, The Netherlands, 2009; Volume 28, pp. 355–377. [Google Scholar]
- Lancaster, C.R.; Michel, H. The coupling of light-induced electron transfer and proton uptake as derived from crystal structures of reaction centres from Rhodopseudomonas viridis modified at the binding site of the secondary quinone, QB. Structure 1997, 5, 1339–1359. [Google Scholar]
- Michel-Beyerle, M.E.; Plato, M.; Deisenhofer, J.; Michel, H.; Bixon, M.; Jortner, J. Unidirectionality of charge separation in reaction centers of photosynthetic bacteria. Biochim. Biophys. Acta 1988, 932, 52–70. [Google Scholar]
- Olson, T.L.; Williams, J.C.; Allen, J.P. Influence of protein interactions on oxidation/reduction midpoint potentials of cofactors in natural and de novo metalloproteins. Biochim. Biophys. Acta 2013, 1827, 914–922. [Google Scholar]
- Ishikita, H.; Saenger, W.; Biesiadka, J.; Loll, B.; Knapp, E.W. How photosynthetic reaction centers control oxidation power in chlorophyll pairs P680, P700, and P870. Proc. Natl. Acad. Sci. USA 2006, 103, 9855–9860. [Google Scholar]
- Govindjee; Bjorn, L.O.; Nickelsen, K. Evolution of the Z-scheme of electron transport in oxygenic photosynthesis. In Photosynthesis Research for Food, Fuel and the Future; Kuang, T., Lu, C., Zhang, L., Eds.; Springer: Berlin, Germany, 2012; pp. 827–833. [Google Scholar]
- Golbeck, J.H. Structure and function of photosystem I. Ann. Rev. Plant Physiol. Plant Mol. Biol 1992, 43, 293–324. [Google Scholar]
- Barber, J; Photosystem, II. The engine of life. Quart. Rev. Biophys 2003, 36, 71–89. [Google Scholar]
- Umena, Y.; Kawakami, K.; Shen, J.R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar]
- Koua, F.H.; Umena, Y.; Kawakami, K.; Shen, J.-R. Structure of Sr-substituted photosystem II at 2.1 Å resolution and its implications in the mechanism of water oxidation. Proc. Natl. Acad. Sci. USA 2013, 110, 3889–3894. [Google Scholar]
- Xiong, L.; Seibert, M.; Gusev, A.V.; Wasielewski, M.R.; Hemann, C.; Hille, C.R.; Sayre, R.T. Substitution of a chlorophyll into the inactive branch pheophytin-binding site impairs charge separation in photosystem II. J. Phys. Chem. B 2004, 108, 16904–16911. [Google Scholar]
- Allen, J.P.; Williams, J.C. The evolutionary pathway from anoxygenic to oxygenic photosynthesis examined by comparison of the properties of photosystem II and bacterial reaction centers. Photosynth. Res 2011, 107, 59–69. [Google Scholar]
- Grotjohann, I.; Fromme, P. Structure of cyanobacterial photosystem I. Photosynth. Res 2005, 85, 51–72. [Google Scholar]
- Holzwarth, A.R.; Muller, M.G.; Reus, M.; Nowaczyk, M.; Sander, J.; Rogner, M. Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: Pheophytin is the primary electron acceptor. Proc. Natl. Acad. Sci. USA 2006, 103, 6895–6900. [Google Scholar]
- Holzwarth, A.R.; Muller, M.G.; Niklas, J.; Lubitz, W. Ultrafast transient absorption studies on photosystem I reaction centers from Chlamydomonas reinhardtii. II: Mutations near the P700 reaction center chlorophylls provide new insight into the nature of the primary electron donor. Biophys. J 2006, 90, 552–565. [Google Scholar]
- Bjorn, L.O.; Papageorgiou, G.C.; Blankenship, R.E. Govindjee. A viewpoint: Why chlorophyll a? Photosynth. Res 2009, 99, 85–98. [Google Scholar]
- Fenna, R.E.; Matthews, B.W. Chlorophyll arrangement in a bacteriochlorophyll protein from Chlorobium limicola. Nature 1975, 258, 573–577. [Google Scholar]
- Olson, J.M. The FMO protein. In Discoveries in Photosynthesis; Govindjee, Beatty, J.T., Gest, H., Allen, J.F., Eds.; Springer: Berlin, Germany, 2005; Volume 20, pp. 421–427. [Google Scholar]
- Schmidt am Busch, M.; Müh, F.; El-Amine Madjet, M.; Renger, T. The eighth bacteriochlorophyll completes the excitation energy funnel in the FML protein. J. Phys. Chem. Lett 2011, 2, 93–98. [Google Scholar]
- Tronrud, M.F.; Schmid, F.; Matthews, B.W. Structure and X-ray amino acid sequence of a bacteriochlorophyll a protein from Prosthecochloris aestuarii refined at 1.9 Å resolution. J. Mol. Biol 1986, 188, 443–454. [Google Scholar]
- Milder, M.T.; Bruggemann, B.; van Grondelle, R.; Herek, J.L. Revisiting the optical properties of the FMO protein. Photosynth. Res 2010, 104, 257–274. [Google Scholar]
- Tronrud, D.E.; Wen, J.; Gay, L.; Blankenship, R.E. The structural basis for the difference in absorbance spectra for the FMO antenna protein from various green sulphur bacteria. Photosynth. Res 2009, 100, 79–87. [Google Scholar]
- Tronrud, D.E.; Allen, J.P. Reinterpretation of the electron density at the site of the eighth bacteriochlorophyll in the FMO protein from Pelodictyon phaeum. Photosynth. Res 2012, 112, 71–74. [Google Scholar]
- Gudowska-Nowak, E.; Newton, M.D.; Fajer, J. Conformational and environmental effects on bacteriochlorophyll optical spectra: Correlations of calculated spectra with structural results. J. Phys. Chem 1990, 94, 5795–5801. [Google Scholar]
- Muh, F.; El-Amine Madjet, M.; Adolphs, J.; Abdurahman, A.; Rabenstein, B.; Ishikita, H.; Knapp, E.W.; Renger, T. α-Helices direct excitation energy flow in the Fenna-Matthews-Olson protein. Proc. Natl. Acad. Sci. USA 2007, 104, 16862–16867. [Google Scholar]
- Adolphs, J.; Muh, F.; El-Amine Madjet, M.; Renger, T. Calculation of pigment transition energies in the FMO protein. Photosynth. Res 2008, 95, 197–209. [Google Scholar]
- Zuber, H.; Cogdell, R.J. Structure and organization of purple bacterial antenna complexes. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer: Dordrecht, The Netherlands, 1995; Volume 2, pp. 315–348. [Google Scholar]
- McDermott, G.; Prince, S.M.; Freer, A.A.; Hawthornthwaite-Lawless, A.M.; Papiz, M.Z.; Cogdell, R.J.; Isaacs, N.W. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 1995, 374, 517–521. [Google Scholar]
- Papiz, M.Z.; Prince, S.M.; Howard, T.; Cogdell, R.J.; Isaacs, N.W. The structure and thermal motion of the B800–850 LH2 complex from Rps. Acidophila at 2.0 Å resolution and 100K: New structural features and functionally relevant motions. J. Mol. Biol 2003, 326, 1523–1538. [Google Scholar]
- Koepke, J.; Hu, X.; Muenke, C.; Schulten, K.; Michel, H. The crystal structure of the light-harvesting complex II (B800–850) from Rhodospirillum molischianum. Structure 1996, 4, 581–597. [Google Scholar]
- Roszak, A.W.; Howard, T.D.; Southall, J.; Gardiner, A.T.; Law, C.J.; Isaacs, N.W.; Cogdell, R.J. Crystal structure of the RC-LH1 core complex from Rhodopseudomonas palustris. Science 2003, 302, 1969–1972. [Google Scholar]
- Richter, M.F.; Baier, J.; Southall, J.; Cogdell, R.J.; Oellerich, S.; Kohler, J. Refinement of the X-ray structure of the RC-LH1 core complex from Rhodopseudomonas palustris by single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 20280–20284. [Google Scholar]
- Hu, X.; Ritz, T.; Damjanović, A.; Schulten, K. Pigment organization and transfer of electronic excitation in the photosynthetic unit of purple bacteria. J. Phys. Chem. B 1997, 101, 3854–3871. [Google Scholar]
- Hu, X.; Damjanović, A.; Ritz, T.; Schulten, K. Architecture and mechanism of the light-harvesting apparatus of purple bacteria. Proc. Natl. Acad. Sci. USA 1998, 95, 5935–5941. [Google Scholar]
- Freer, A.; Prince, S.; Sauer, K.; Papiz, M.; Lawless, A.H.; McDermott, G.; Cogdell, R.; Isaacs, N.W. Pigment-pigment interactions and energy transfer in the antenna complex of the photosynthetic bacterium Rhodopseudomonas acidophila. Structure 1996, 4, 449–462. [Google Scholar]
- Pandit, A.; de Groot, H.J.M. Solid-State NMR applied to photosynthetic light-harvesting complexes. Photosynth. Res 2012, 111, 219–226. [Google Scholar]
- Sener, M.; Strumpfer, J.; Hsin, J.; Chandler, D.; Scheuring, S.; Hunter, C.N.; Schulten, K. Forster energy transfer theory as reflected in the structures of photosynthetic light-harvesting systems. ChemPhysChem 2011, 12, 518–531. [Google Scholar]
- Alden, R.G.; Johnson, E.; Nagarajan, V.; Parson, W.W.; Law, C.J.; Cogdell, R.G. Calculations of spectroscopic properties of the LH2 bacteriochlorophyll-protein antenna complex from Rhodopseudomonas acidophila. J. Phys. Chem. B 1997, 101, 4667–4680. [Google Scholar]
- Linnanto, J.; Korppi-Tommola, J.E.I.; Helenius, V.M. Electronic states, absorption spectrum and circular dichroism spectrum of the photosynthetic bacterial LH2 antenna of Rhodopseudomonas acidophila as predicted by exciton theory and semiempirical calculations. J. Phys. Chem. B 1999, 103, 8739–8750. [Google Scholar]
- He, Z.; Sundström, V.; Pullerits, T. Influence of the protein binding site on the excited states of bacteriochlorophyll: DFT calculations of B800 in LH2. J. Phys. Chem. B 2002, 106, 11606–11612. [Google Scholar]
- Neugebauer, J. Photophysical properties of natural light-harvesting complexes studied by subsystem density functional theory. J. Phys. Chem. B 2008, 112, 2207–2217. [Google Scholar]
- Neugebauer, J. Chromophore-Specific theoretical spectroscopy: From subsystem density functional theory to mode-specific vibrational spectroscopy. Phys. Rep 2010, 489, 1–87. [Google Scholar]
- Scholes, G.D.; Gould, I.R.; Cogdell, R.J.; Fleming, G.R. Ab initio molecular orbital calculations of electronic couplings in the LH2 bacterial light-harvesting complex of Rps. Acidophila. J. Phys. Chem. B 1999, 103, 2543–2553. [Google Scholar]
- Prince, S.M.; Papiz, M.Z.; Freer, A.A.; McDermott, G.; Hawthornthwaite-Lawless, A.M.; Cogdell, R.J.; Isaacs, N.W. Apoprotein structure in the LH2 complex from Rhodopseudomonas acidophila strain 10050: Modular assembly and protein pigment interactions. J. Mol. Biol 1997, 268, 412–423. [Google Scholar]
- McLuskey, K.; Prince, S.M.; Cogdell, R.J.; Isaacs, N.W. The crystallographic structure of the B800–820 LH3 light-harvesting complex from the purple bacteria Rhodopseudomonas acidophila strain 7050. Biochemistry 2001, 40, 8783–8789. [Google Scholar]
- Chmeliov, J.; Songaila, E.; Rancova, O.; Gall, A.; Robert, B.; Abramavicius, D.; Valkunas, L. Excitons in the LH3 complexes from purple bacteria. J. Phys. Chem. B 2013, 117, 11058–11068. [Google Scholar]
- Liu, Z.; Yan, H.; Wang, K.; Kuang, T.; Zhang, J.; Gui, L.; An, X.; Chang, W. Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 2004, 428, 287–292. [Google Scholar]
- Ballottari, M.; Girardon, J.; Dall’Osto, L.; Bassi, R. Evolution and functional properties of Photosystem II light harvesting complexes in eukaryotes. Biochim. Biophys. Acta 2012, 1817, 143–157. [Google Scholar]
- Pan, X.; Li, M.; Wan, T.; Wang, L.; Jia, C.; Hou, Z.; Zhao, X.; Zhang, J.; Chang, W. Structural insights into energy regulation of light-harvesting complex CP29 from spinach. Nat. Struct. Mol. Biol 2011, 18, 309–315. [Google Scholar]
- Simidjiev, I.; Barzda, V.; Mustardy, L.; Garab, G. Isolation of lamellar aggregates of the light-harvesting chlorophyll a/b protein complex of photosystems II with long-range chiral order and structural flexibility. Anal. Biochem 1997, 250, 169–175. [Google Scholar]
- Barzda, V.; Mustardy, L.; Garab, G. Size dependency of circular-dichroism in macroaggregates of photosynthetic pigment-protein complexes. Biochemistry 1994, 33, 10837–10841. [Google Scholar]
- Barzda, V.; Istokovics, A.; Simidjiev, I.; Garab, G. Structural flexibility of chiral macroaggregates of light-harvesting chlorophyll a/b pigment-protein complexes. Light-Induced reversible structural changes associated with energy dissipation. Biochemistry 1996, 35, 8981–8985. [Google Scholar]
- Stoylova, S.; Flint, T.D.; Ford, R.C.; Holzenburg, A. Structural analysis of photosystem II in far-red-light-adapted thylakoid membranes. New crystal forms provide evidence for a dynamic reorganization of light-harvesting antennae subunits. Eur. J. Biochem 2000, 267, 207–215. [Google Scholar]
- Cseh, Z.; Vianelli, A.; Rajagopal, S.; Krumova, S.; Kovacs, L.; Papp, E.; Barzda, V.; Jennings, R.; Garab, G. Thermo-Optically induced reorganizations in the main light harvesting antenna of plants. I. Non-Arrhenius type of temperature dependence and linear light-intensity dependencies. Photosynth. Res 2005, 86, 263–273. [Google Scholar]
- Gussakovsky, E.E.; Ionov, M.V.; Giller, Y.E.; Ratner, K.; Aripov, T.F.; Shahak, Y. Left- and right-handed LHC II macroaggregates revealed by circularly polarized chlorophyll luminescence. Photosynth. Res 2006, 87, 253–265. [Google Scholar]
- Phillip, D.; Hobe, S.; Paulsen, H.; Molnar, P.; Hashimoto, H. The Binding of Xanthophylls to the Bulk Light-harvesting Complex of Photosystem II of Higher Plants. A specific requirement for carotenoids with a 3-hydroxy-end group. J. Biol. Chem 2002, 277, 25160–25169. [Google Scholar]
- Garab, G. Hierarchical organization and structural flexibility of thylakoid membranes. Biochim. Biophys. Acta 2014, 1837, 481–494. [Google Scholar]
- Neilson, J.A.D.; Dumford, D.G. Structural and functional diversification of the light-harvesting complexes in photosynthetic eukaryotes. Photosynth. Res 2010, 106, 57–71. [Google Scholar]
- Blankenship, R.E.; Olson, J.M.; Miller, M. Antenna complexes from green photosynthetic bacteria. In Anoxygenic Photosynthetic Bacteria; Blankenship, R.E., Madigan, M.T., Bauer, C.E., Eds.; Kluwer: Dordrecht, The Netherlands, 1995; Volume 2, pp. 399–435. [Google Scholar]
- Vassilieva, E.V.; Stirewalt, V.L.; Jakobs, C.U.; Frigaard, N.-U.; Inoue-Sakamoto, K.; Baker, M.A.; Sotak, A.; Bryant, D.A. Subcellular localization of chlorosome proteins in Chlorobium tepidum and characterization of three new chlorosome proteins: CsmF, CsmH, and CsmX. Biochemistry 2002, 41, 4358–4370. [Google Scholar]
- Otte, S.C.M.; van de Meent, E.J.; van Veelen, P.A.; Pundsnes, A.S.; Amesz, J. Identification of the major chlorosomal bacteriochlorophylls of the green sulphur bacteria Chlorobium vibrioforme and Chlorobium phaeovibroides; their function in lateral energy transfer. Photosynth. Res 1993, 35, 159–169. [Google Scholar]
- Miyatake, T.; Tamiaki, H. Self-Aggregates of bacteriochlorophylls-c, d, and e in a light-harvesting antenna system of green photosynthetic bacteria: Effect of stereochemistry at the chiral 3-(1-hydroxyethyl) group on the supramolecular arrangement of chlorophyllous pigments. J. Photochem. Photobiol. C Photochem. Rev 2005, 6, 89–107. [Google Scholar]
- Balaban, T.S. Tailoring porphyrins and chlorins for self-assembly in biomimetic artificial antenna systems. Acc. Chem. Res 2005, 38, 612–623. [Google Scholar]
- Pšenčík, J.; Ikonen, T.P.; Laurinmäki, P.; Merckel, M.C.; Butcher, S.J.; Serimaa, R.E.; Tuma, R. Lamellar organization of pigments in chlorosomes, the light harvesting complexes of green photosynthetic bacteria. Biophys. J 2004, 87, 1165–1172. [Google Scholar]
- Oostergetel, G.T.; van Amerongen, H.; Boekema, E.J. The chlorosome: A prototype for efficient light harvesting in photosynthesis. Photosynth. Res 2010, 104, 245–255. [Google Scholar]
- The terms parallel and anti-parallel refer to the orientation of the QY transition dipoles.
- Ganapathy, S.; Oostergetel, G.T.; Wawrzyniak, P.K.; Reus, M.; Gomez Maqueo Chew, A.; Buda, F.; Boekema, E.J.; Bryant, D.A.; Holzwarth, A.R.; de Groot, H.J.M. Alternating syn-anti bacteriochlorophylls form concentric helical nanotubes in chlorosomes. Proc. Natl. Acad. Sci. USA 2009, 106, 8525–8530. [Google Scholar]
- Frank, H.A.; Cogdell, R.J. Carotenoids in photosynthesis. Photochem. Photobiol 1996, 63, 257–264. [Google Scholar]
- Green, B.R.; Durnford, D.G. The chlorophyll-carotenoid proteins of oxygenic photosynthesis. Ann. Rev. Plant Physiol. Plant Mol. Biol 1996, 47, 685–714. [Google Scholar]
- Glazer, A.N. Phycobilisome: A macromolecular complex optimized for light energy transfer. Biochim. Biophys. Acta 1984, 768, 29–51. [Google Scholar]
- Grossman, A.R.; Schaefer, M.R.; Chiang, G.G.; Collier, J.L. The phycobilisome, a light-harvesting complex responsive to environmental conditions. Microbiol. Rev 1993, 57, 725–749. [Google Scholar]
- MacColl, R. Cyanobacterial phycobilisomes. J. Struct. Biol 1998, 124, 311–334. [Google Scholar]
- Scheer, H. Biliproteins. Angew. Chem. Int. Ed. Engl 1981, 20, 241–261. [Google Scholar]
- Bogorad, L. Phycobiliproteins and complimentary chromatic adaptation. Ann. Rev. Plant Physiol. Plant Mol. Biol 1975, 26, 369–401. [Google Scholar]
- Rockwell, N.C.; Su, Y.-S.; Lagarias, J.C. Phytochrome structure and signaling mechanisms. Ann. Rev. Plant Biol 2006, 57, 837–858. [Google Scholar]
- Takala, H.; Bjorling, A.; Berntsson, O.; Lehtivuori, H.; Niebling, S.; Hoernke, M.; Kosheleva, I.; Henning, R.; Menzel, A.; Ihalainen, J.A.; et al. Signal amplification and transduction in phytochrome photosensors. Nature 2014, 509, 245–248. [Google Scholar]
- Mauzerall, D. Why chlorophyll? Ann. N. Y. Acad. Sci 1973, 206, 483–494. [Google Scholar]
- Sobotka, R. Making proteins green; biosynthesis of chlorophyll-binding proteins in cyanobacteria. Photosynth. Res 2014, 119, 223–232. [Google Scholar]
- Fraley, C.; Raftery, A.E. Model-Based clustering, discriminant analysis, and density estimation. J. Am. Statist. Assoc 2002, 97, 611–631. [Google Scholar]
- MacGowan, S.A.; Senge, M.O. Conformational control of cofactors in nature–functional tetrapyrrole conformations in the photosynthetic reaction centers of purple bacteria. Chem. Commun 2011, 47, 11621–11623. [Google Scholar]
- At present the low number of available high resolution data for PS I and PS II and related LHC limits the application of this approach.
- Oba, T.; Tamiaki, H. Effects of peripheral substituents on diastereoselectivity of the fifth ligand-binding to chlorophylls, and nomenclature of the asymmetric axial coordination sites. Bioorg. Med. Chem 2005, 13, 5733–5739. [Google Scholar]
- Kay, A.; Grätzel, M. Artificial photosynthesis 1. Photosensitization of TiO2 solar-cells with chlorophyll derivatives and related natural porphyrins. J. Phys. Chem 1993, 97, 6272–6277. [Google Scholar]
- Senge, M.O.; Kalisch, W.W.; Runge, S. Conformationally distorted chlorins via diimide reduction of nonplanar porphyrins. Tetrahedron 1998, 54, 3781–3798. [Google Scholar]
- Kalisch, W.W.; Senge, M.O.; Ruhlandt-Senge, K. Synthesis and crystal structures of cofacial bisoctaethylchlorins as structural models for the special pair. Photochem. Photobiol 1998, 67, 312–323. [Google Scholar]
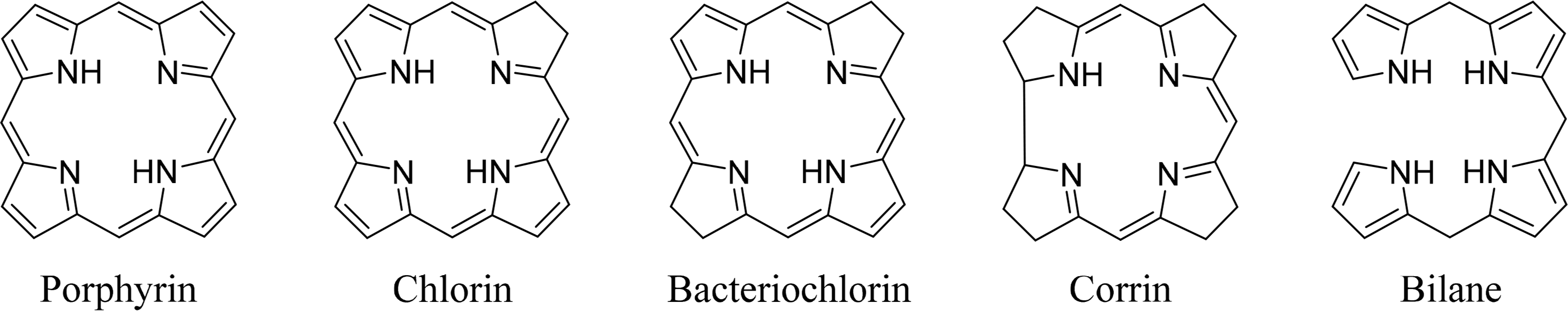
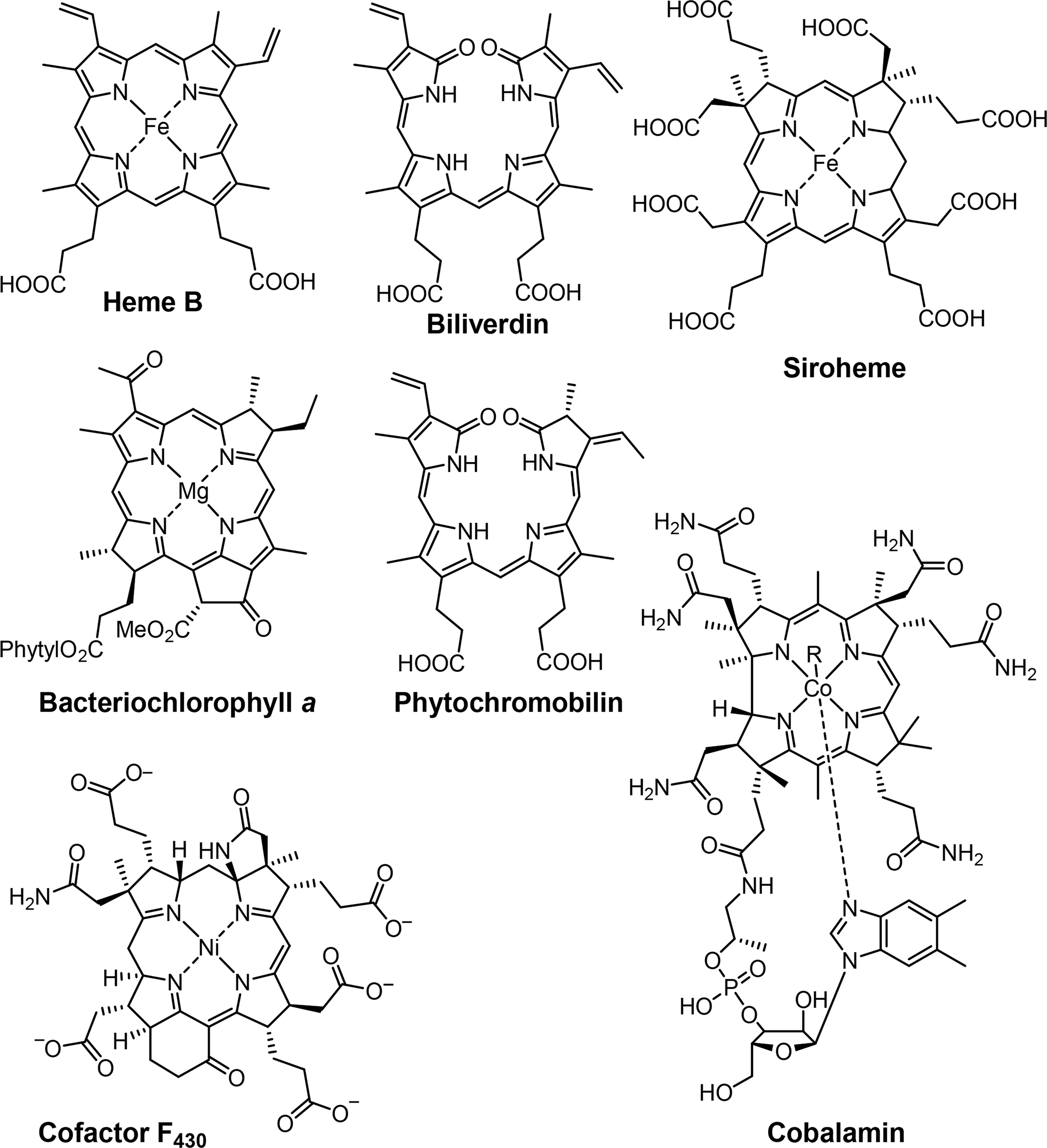

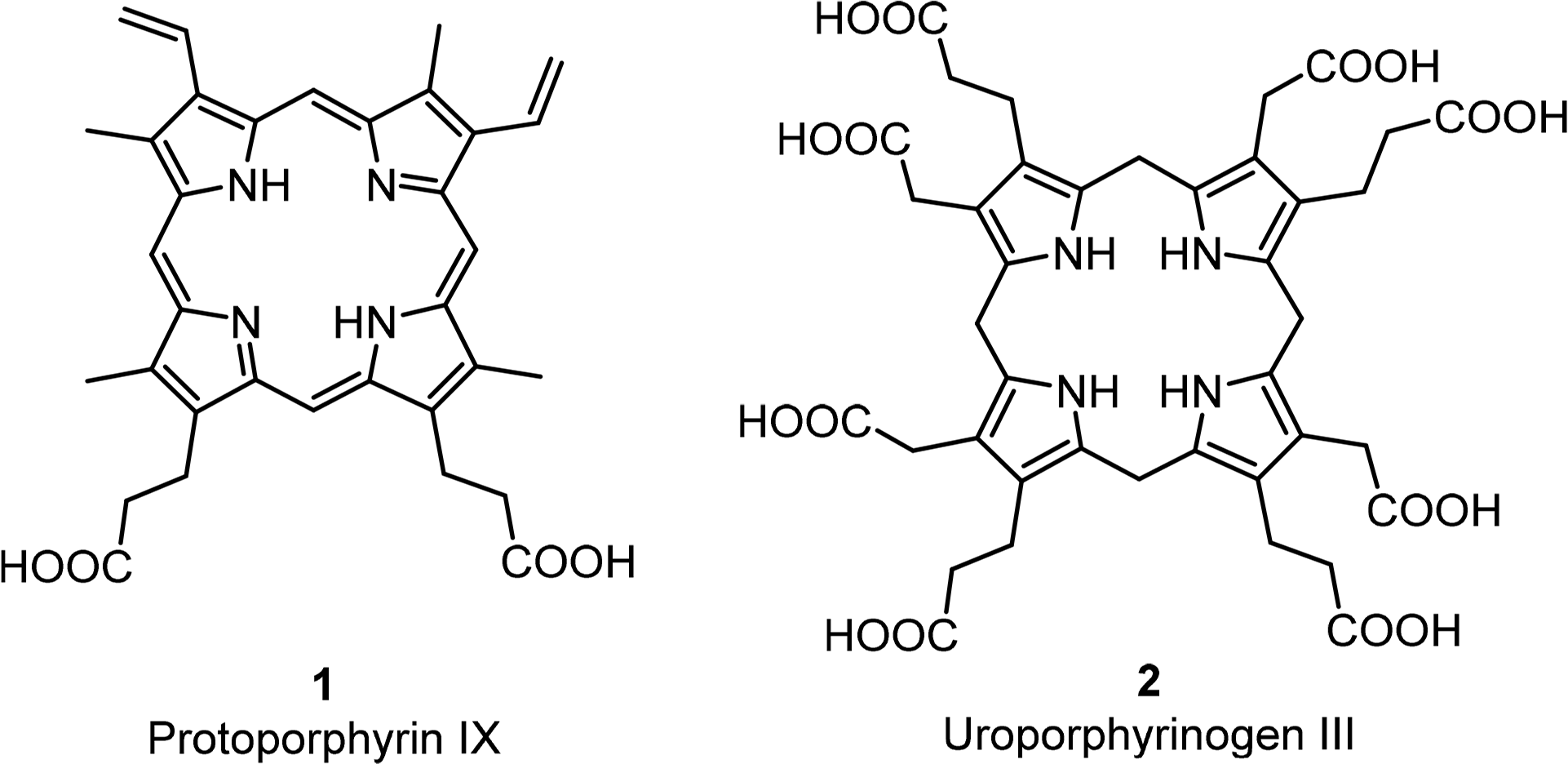
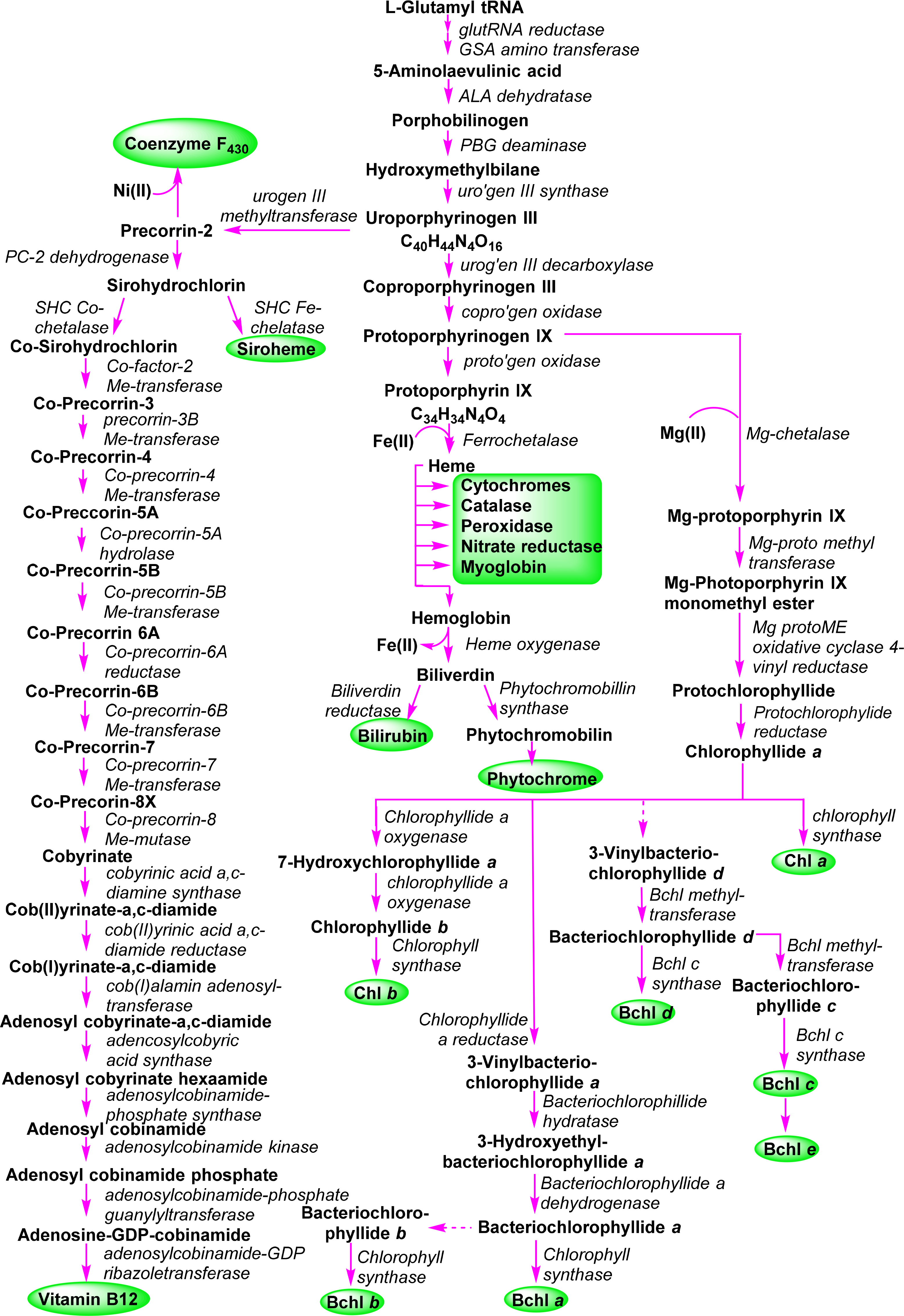
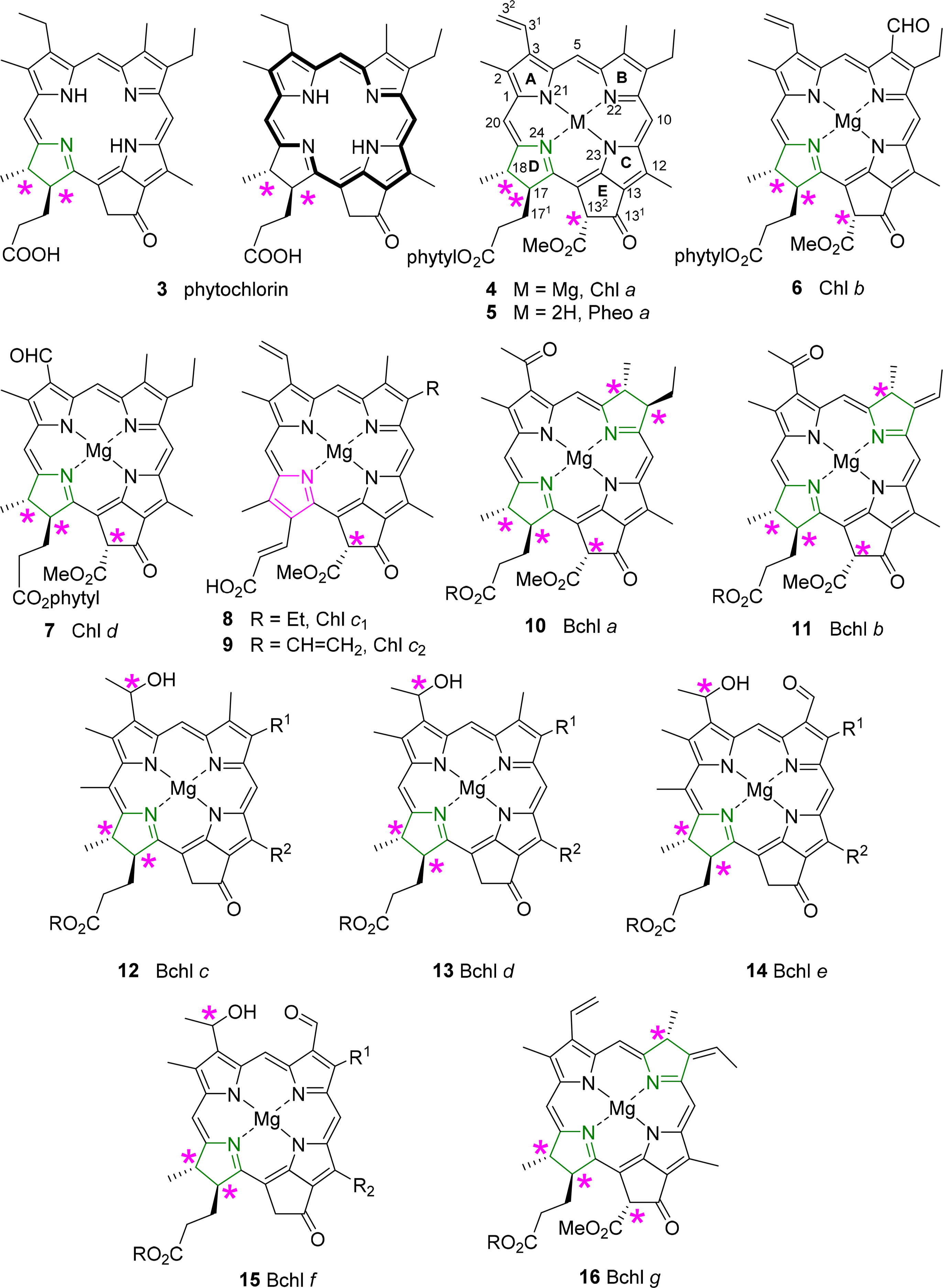
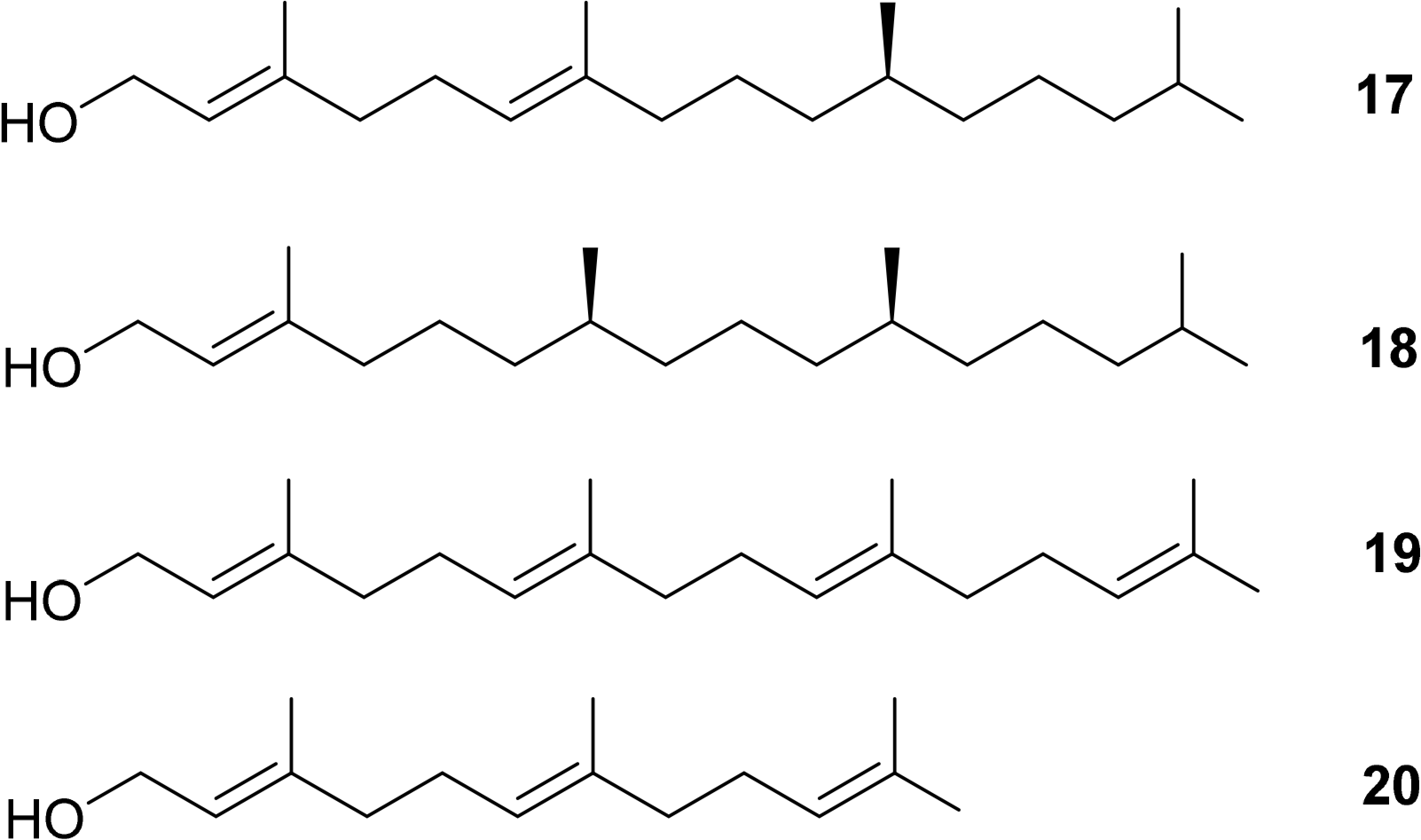
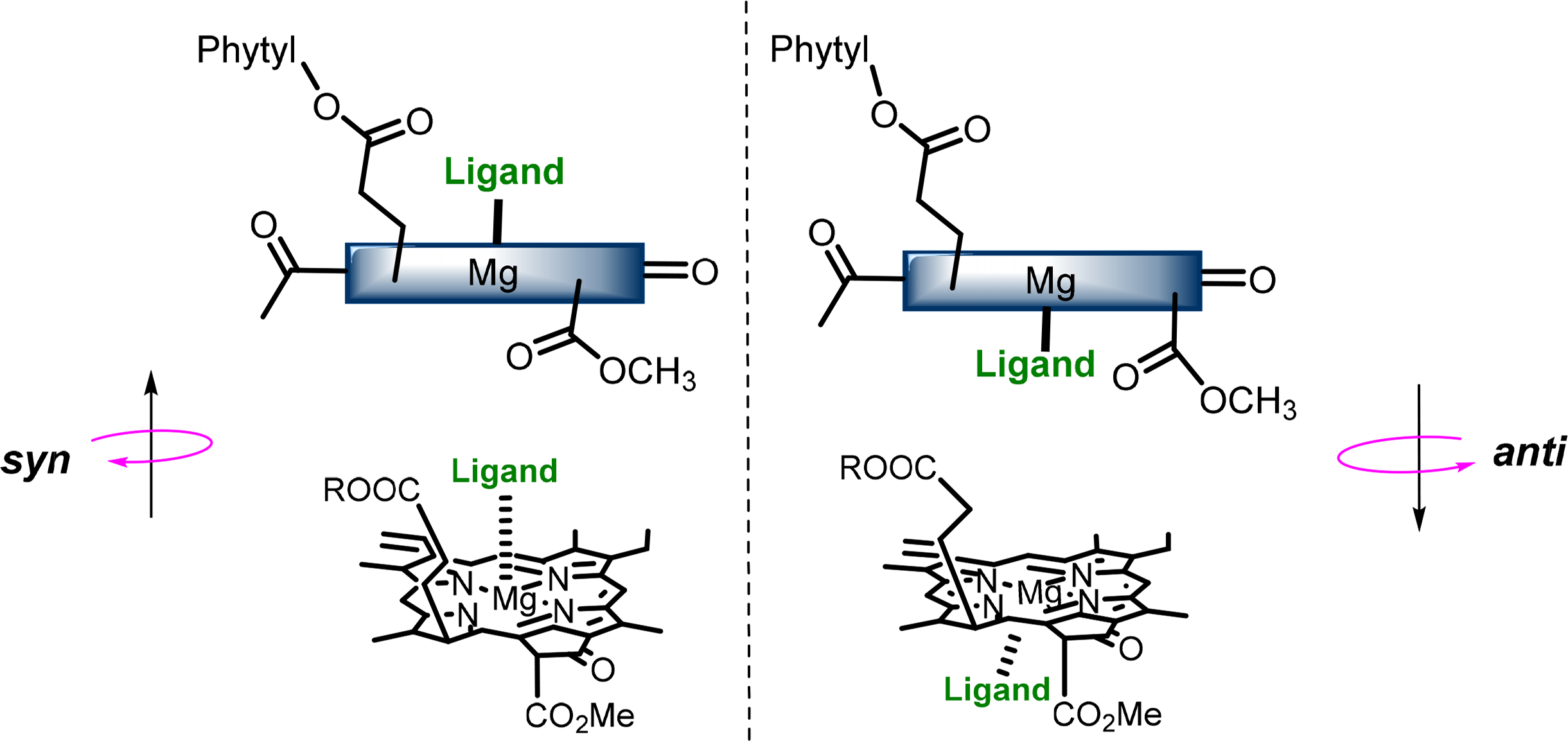

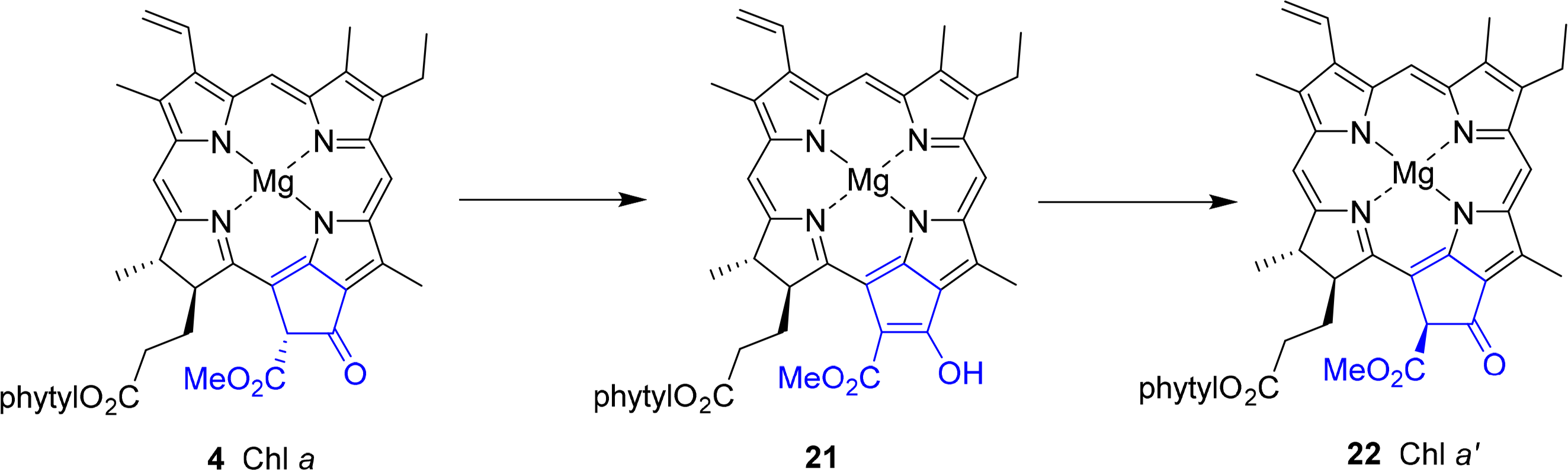

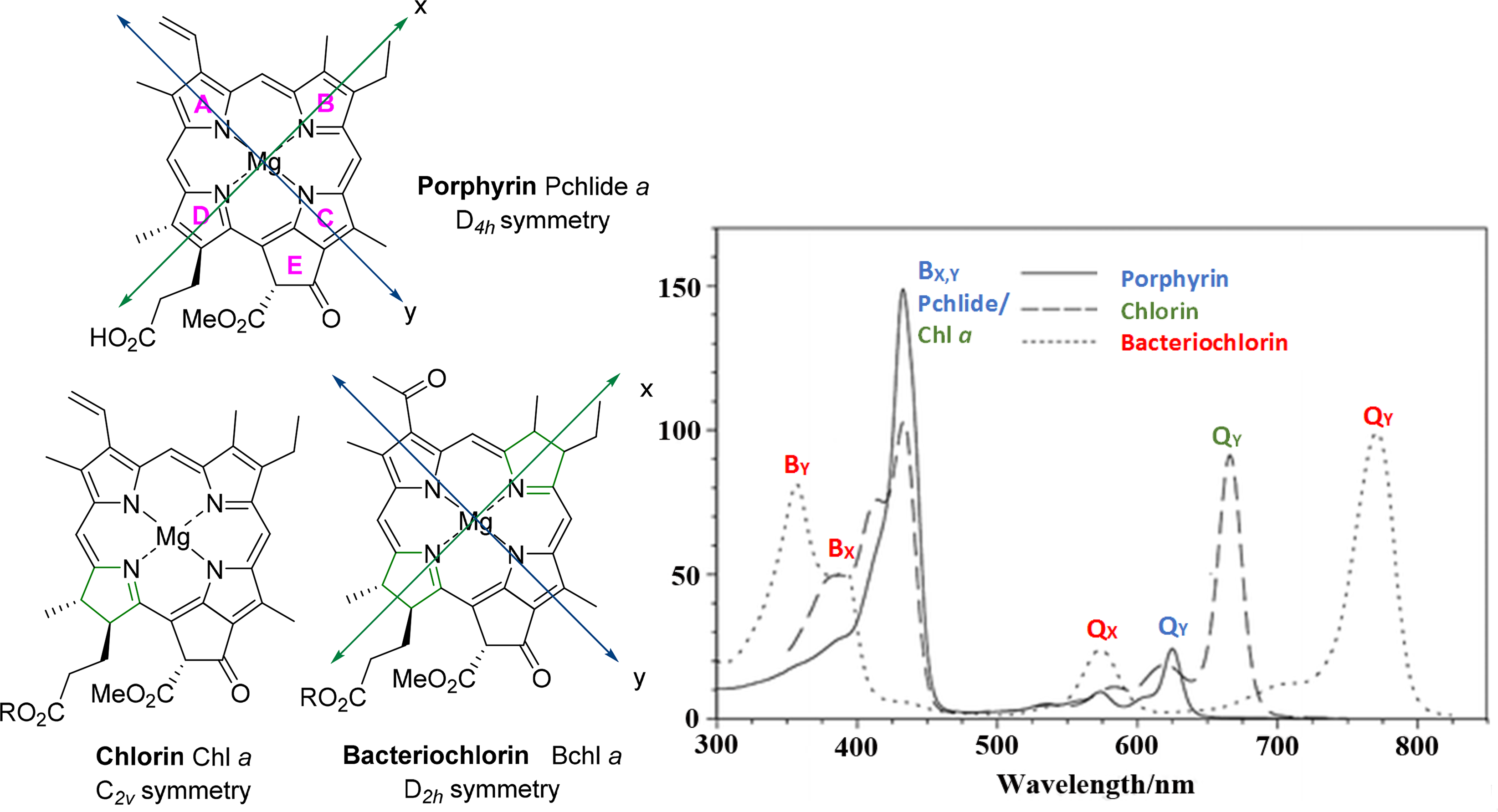
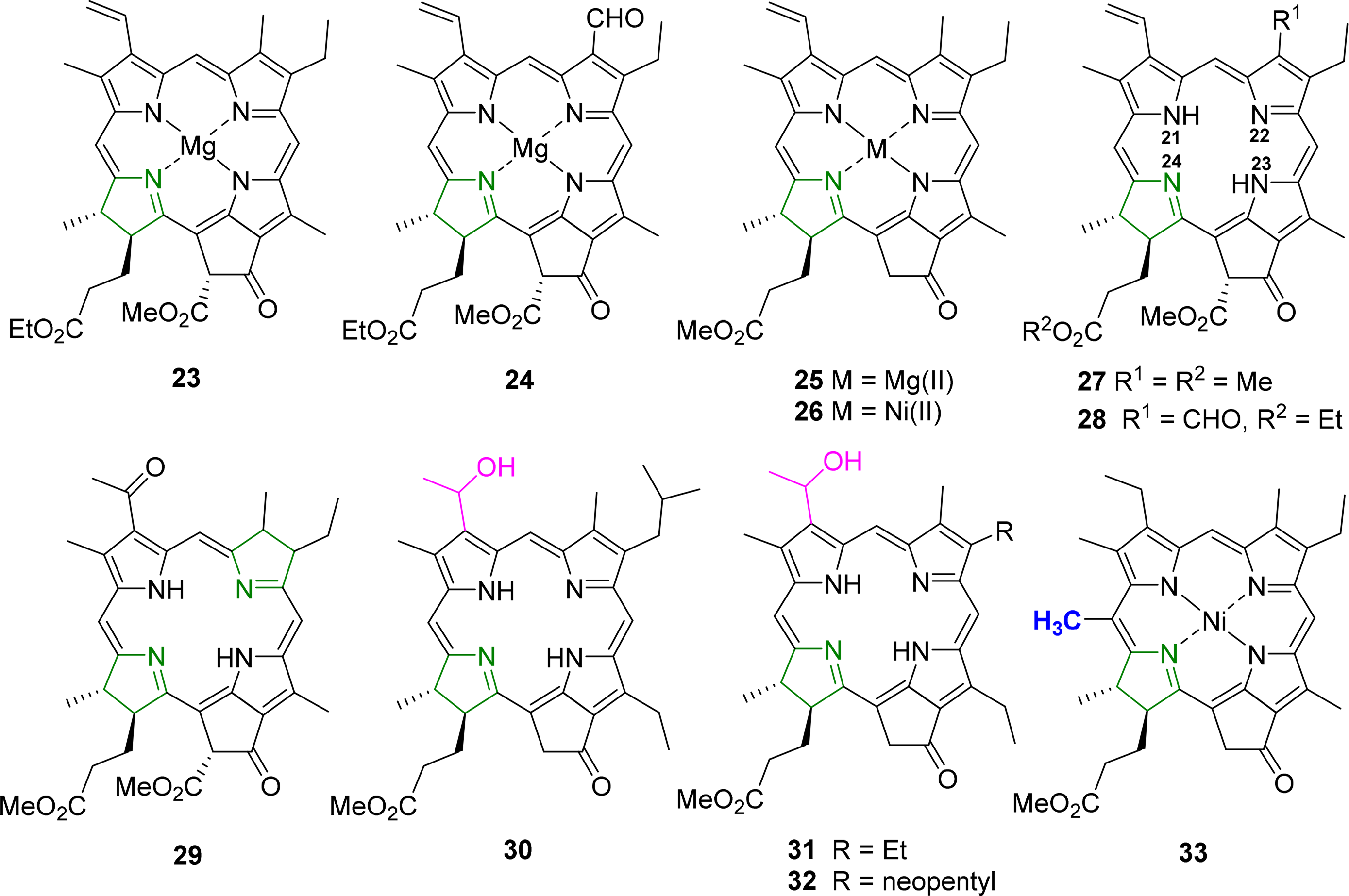

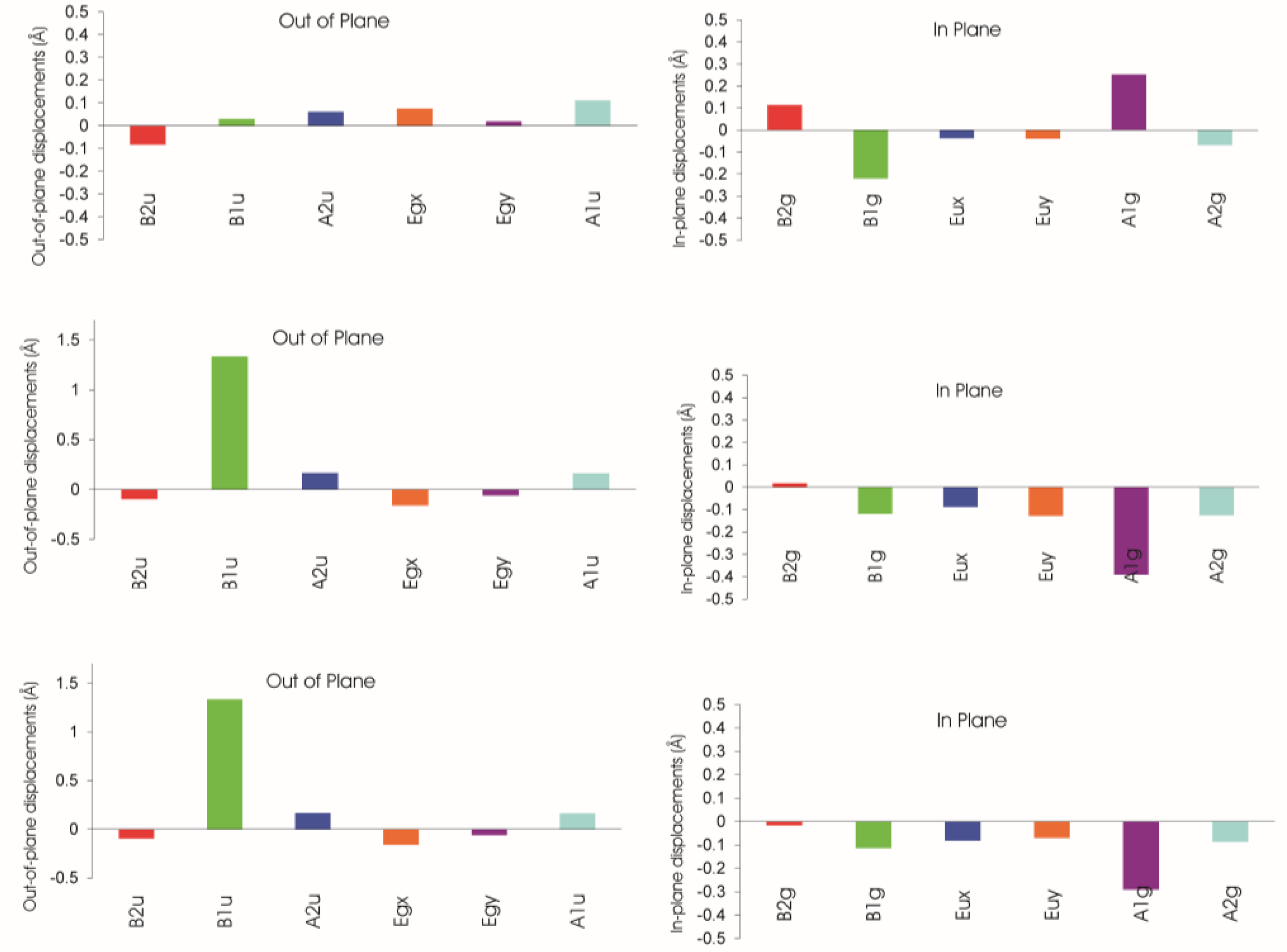
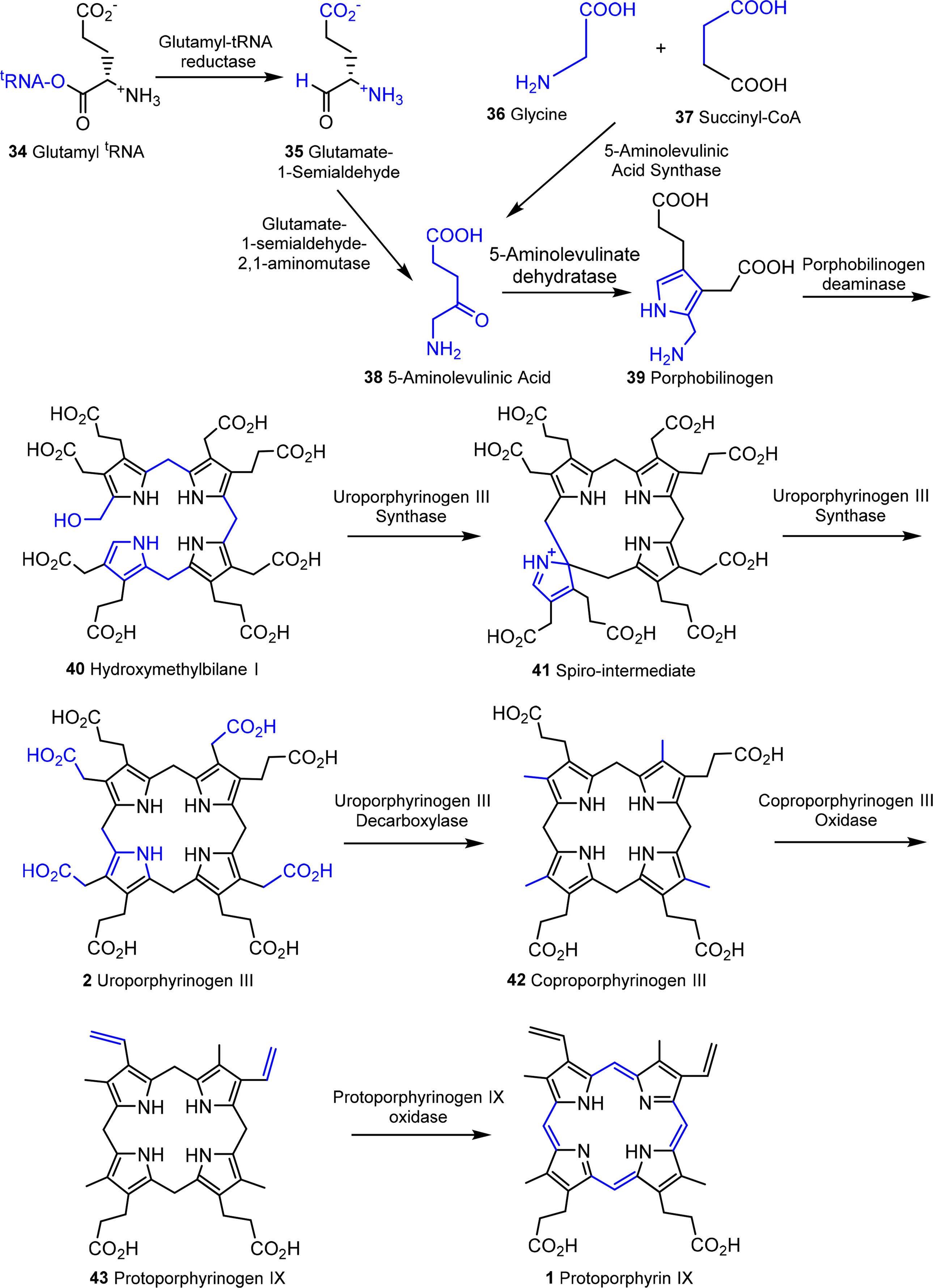
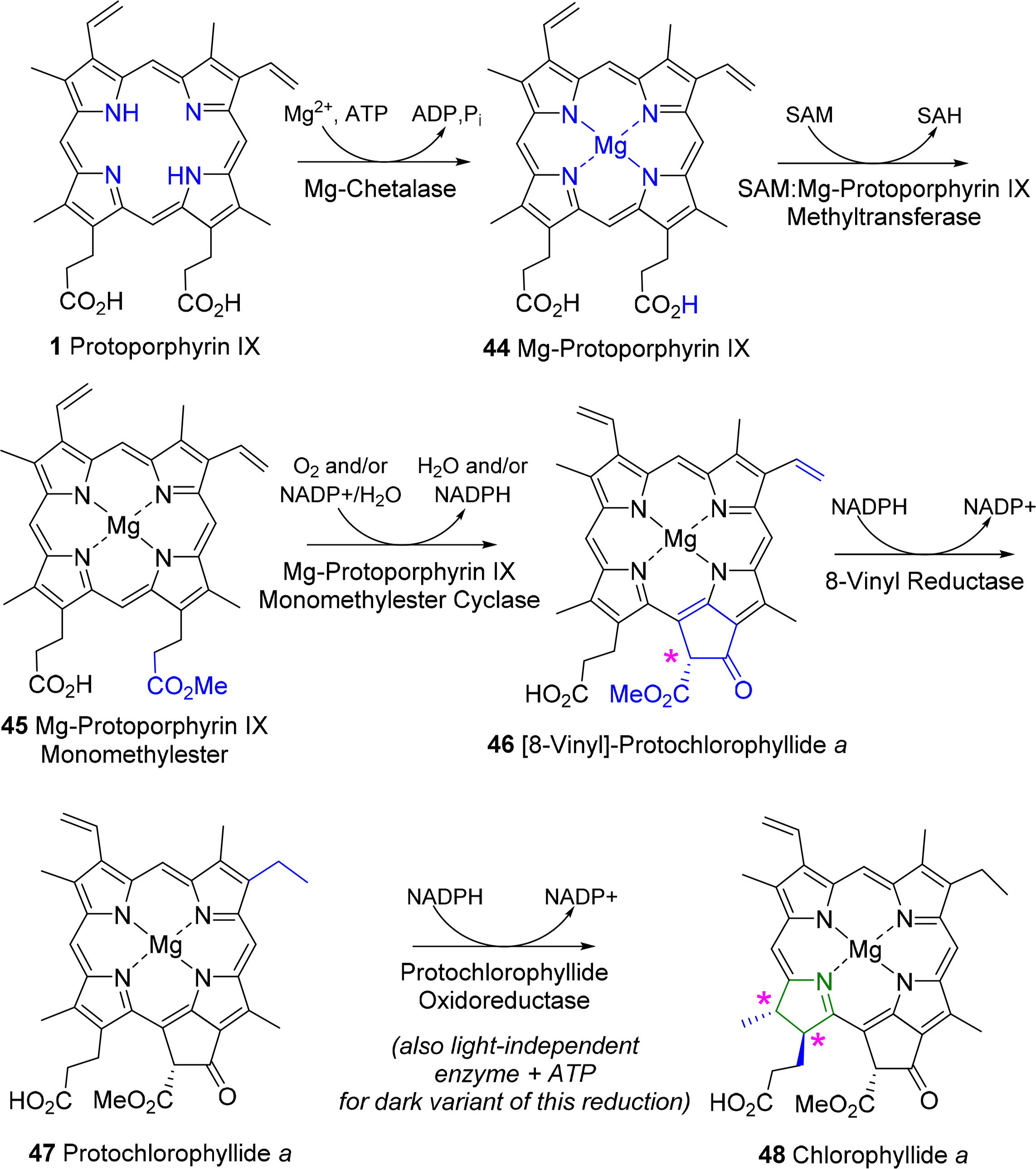


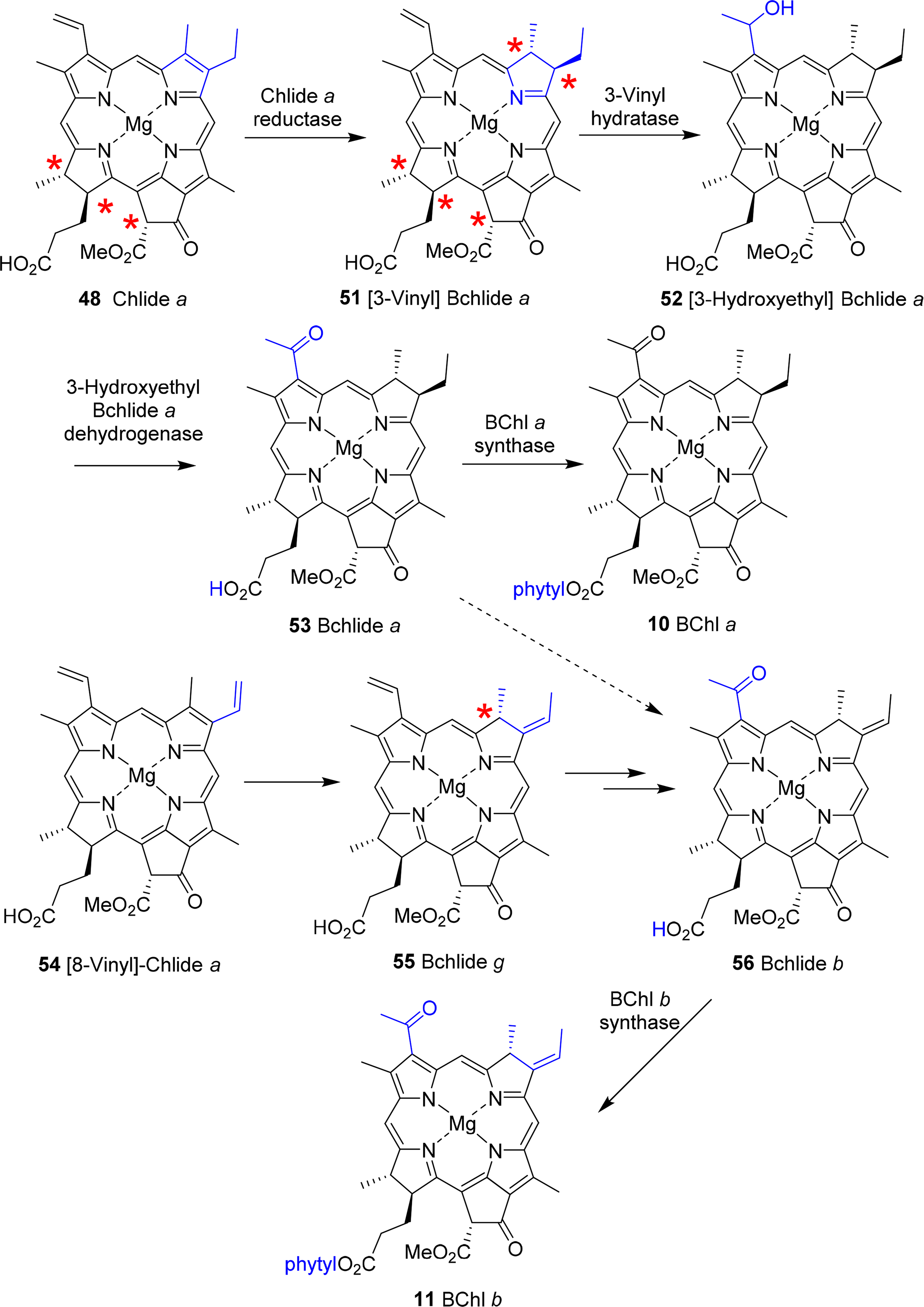
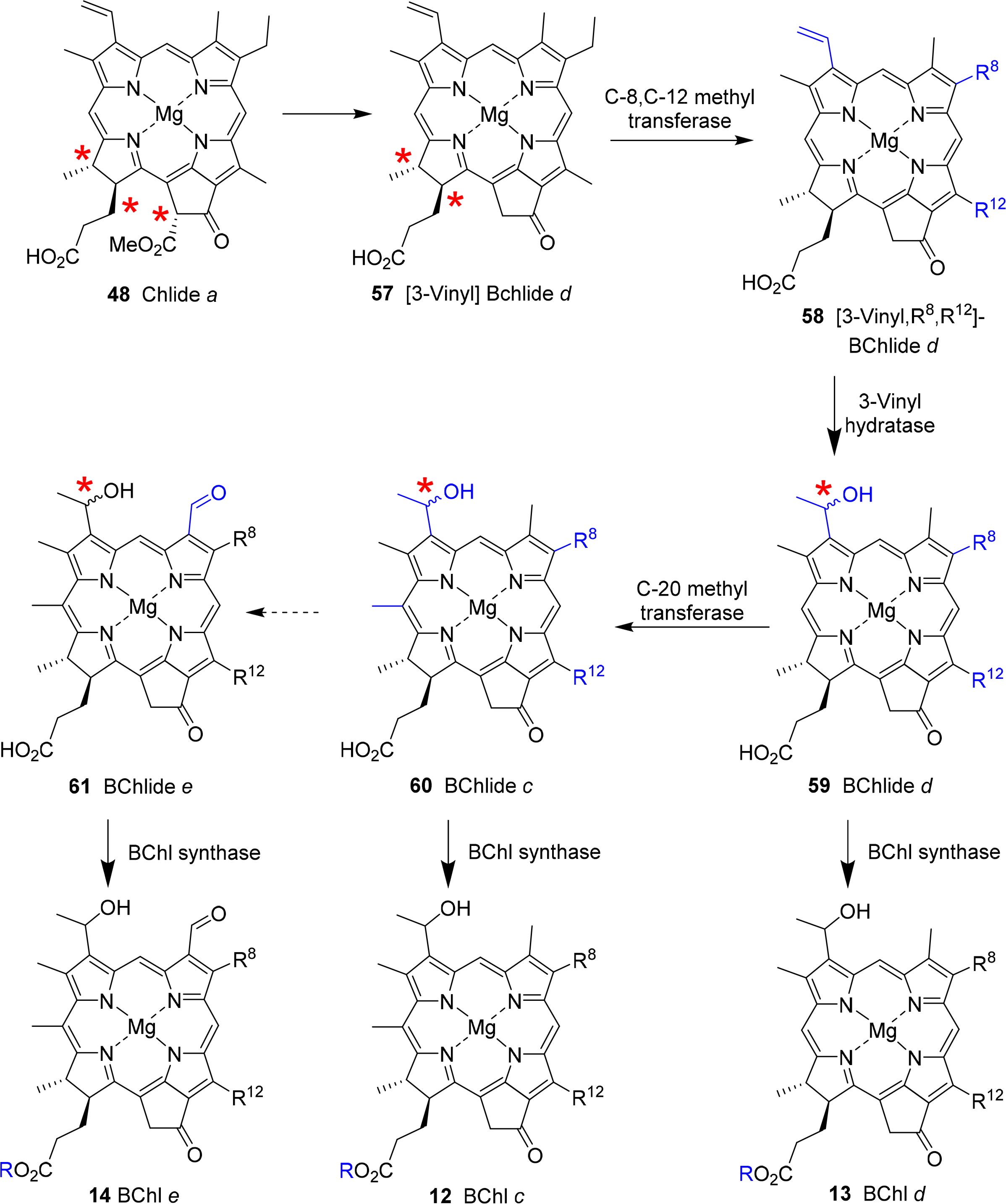
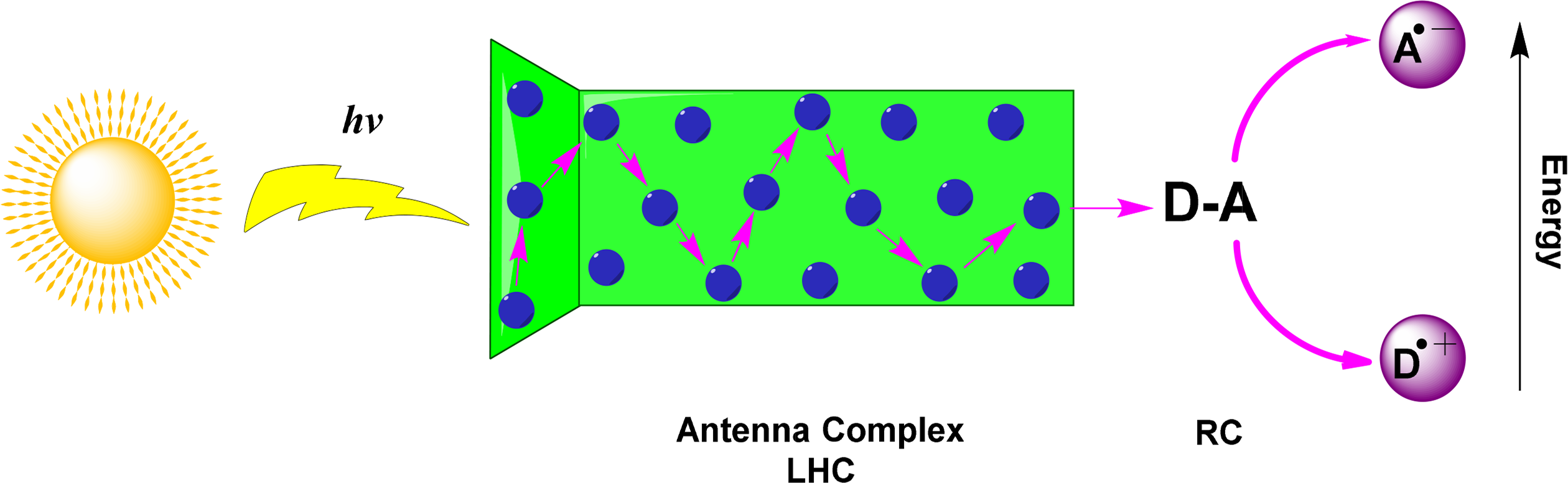


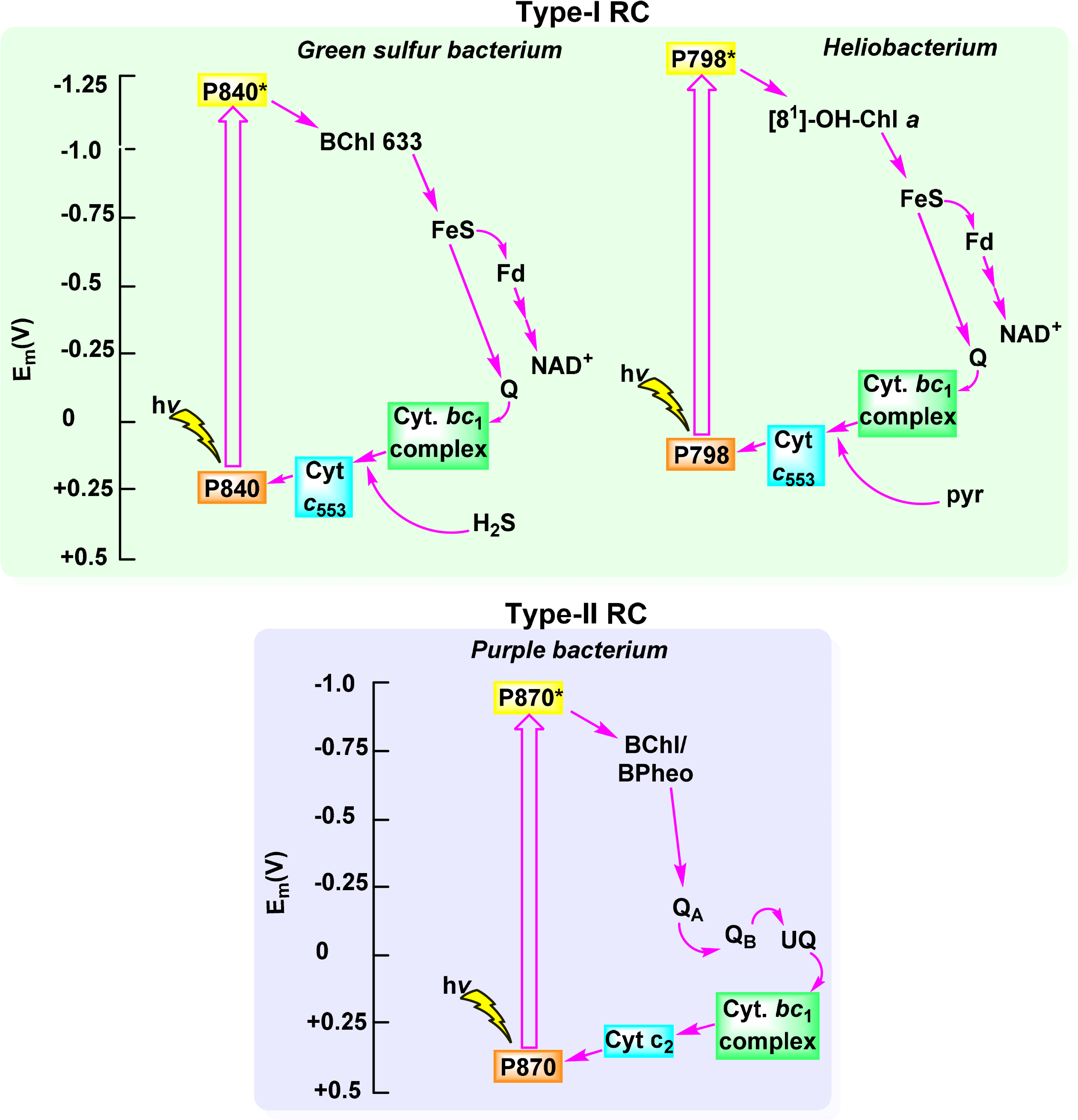
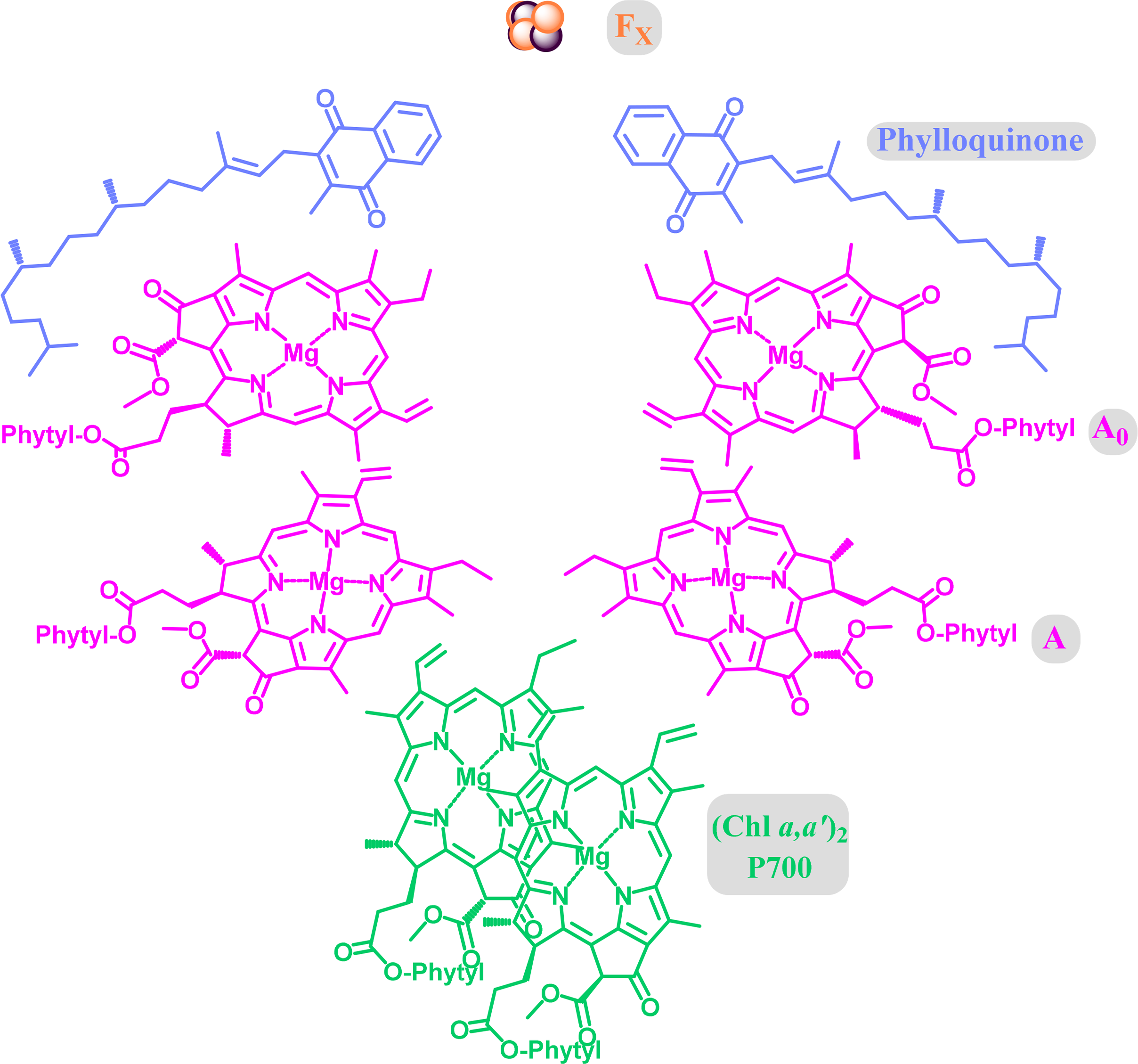


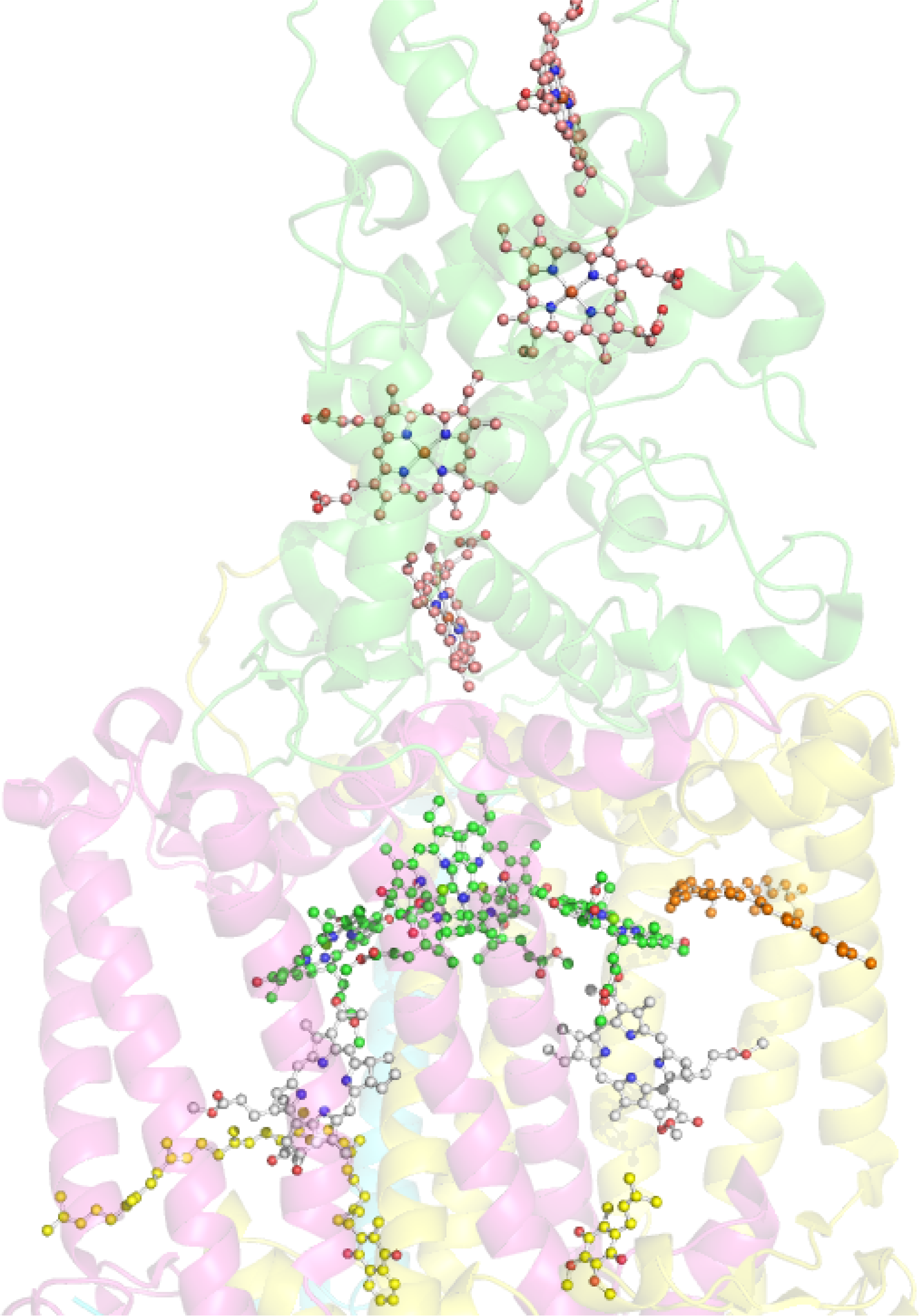



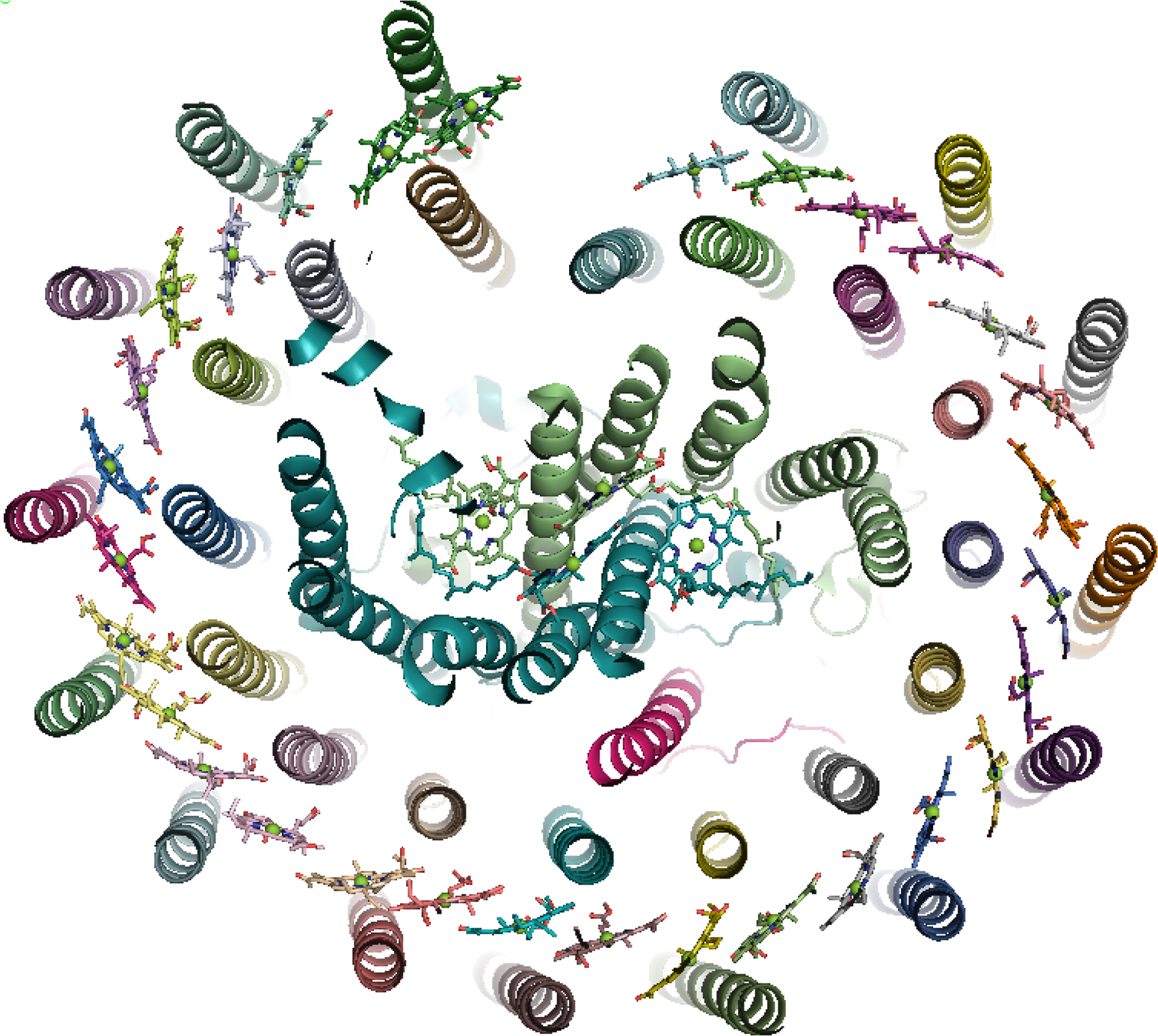
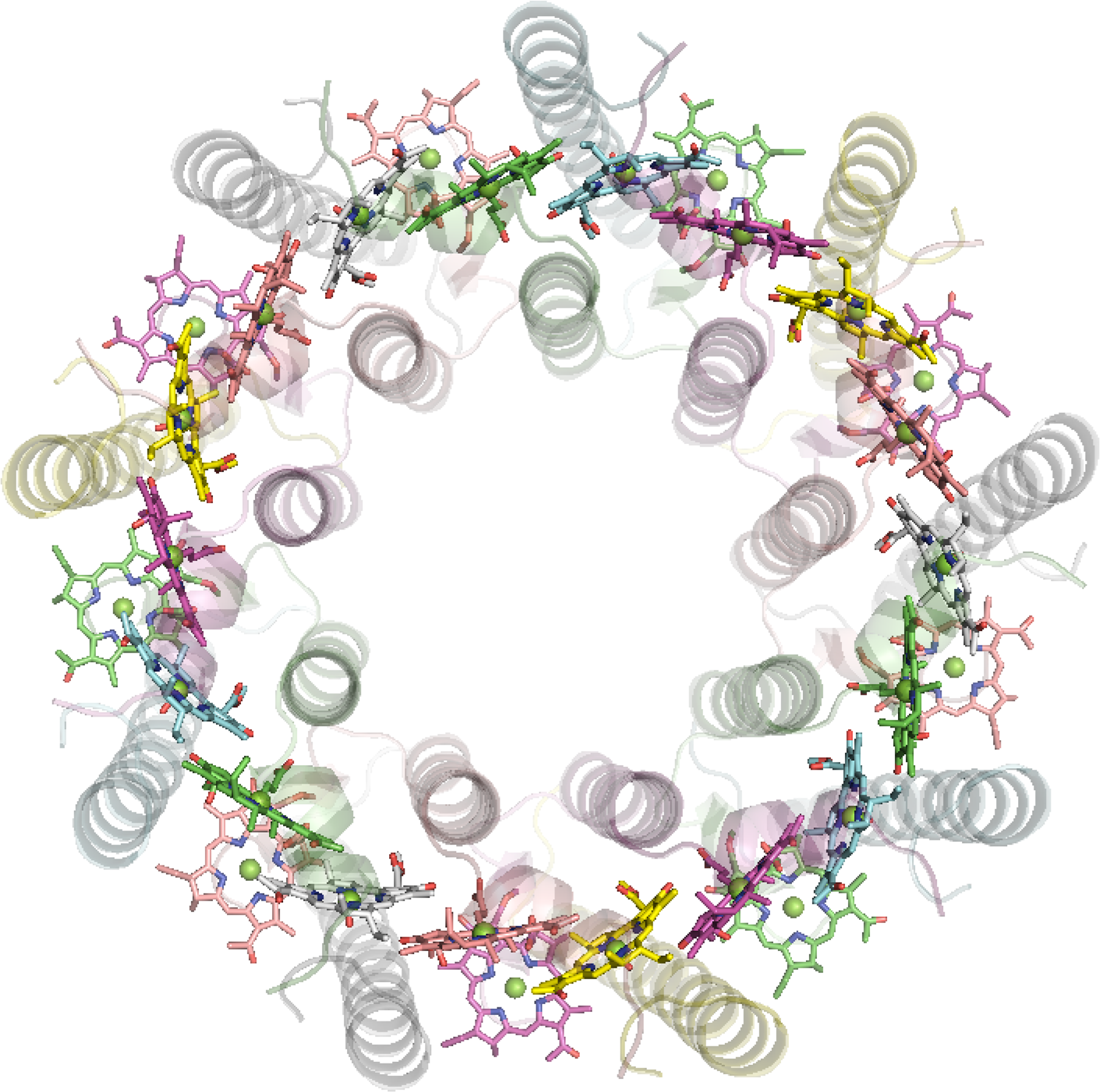
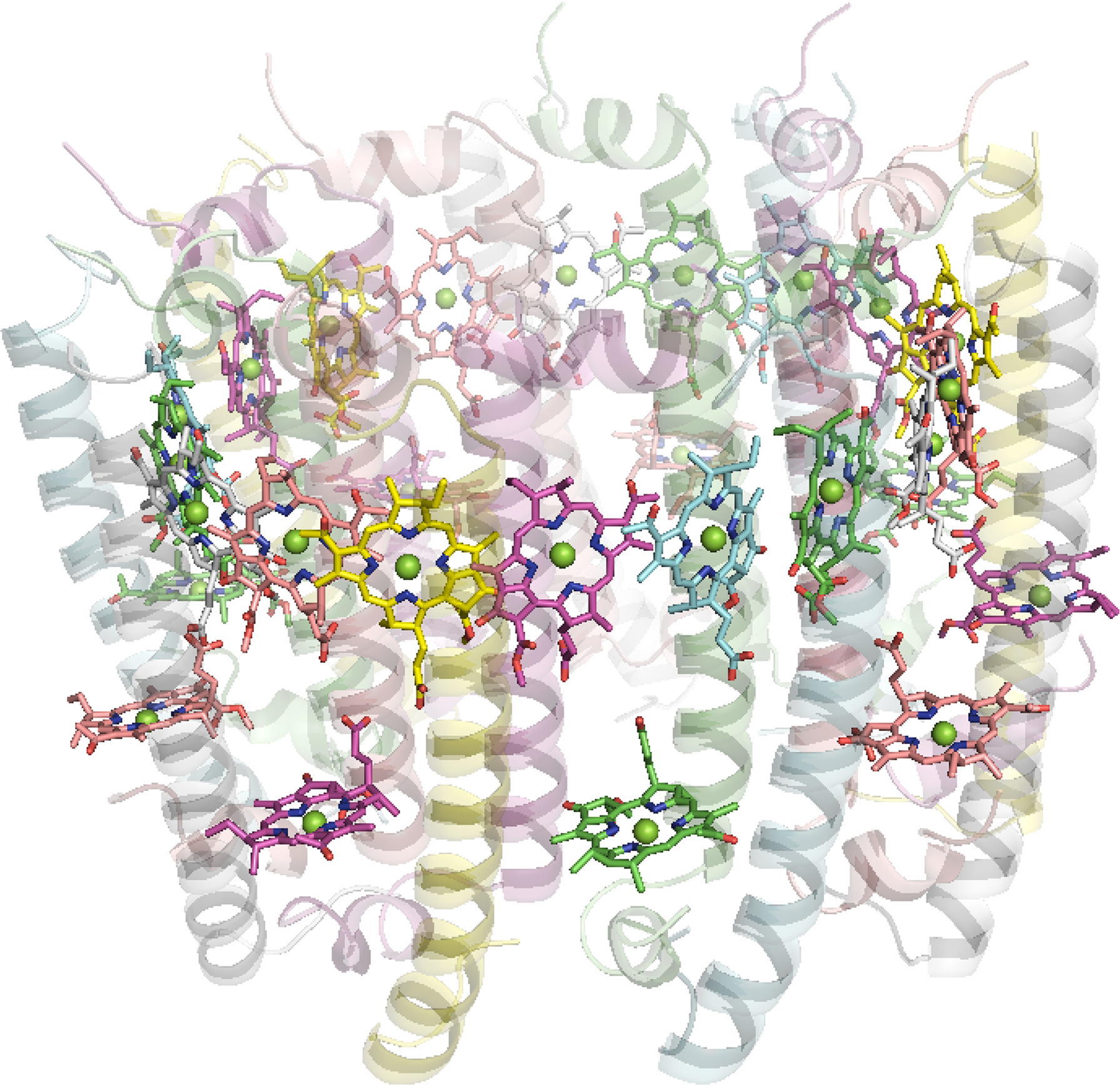
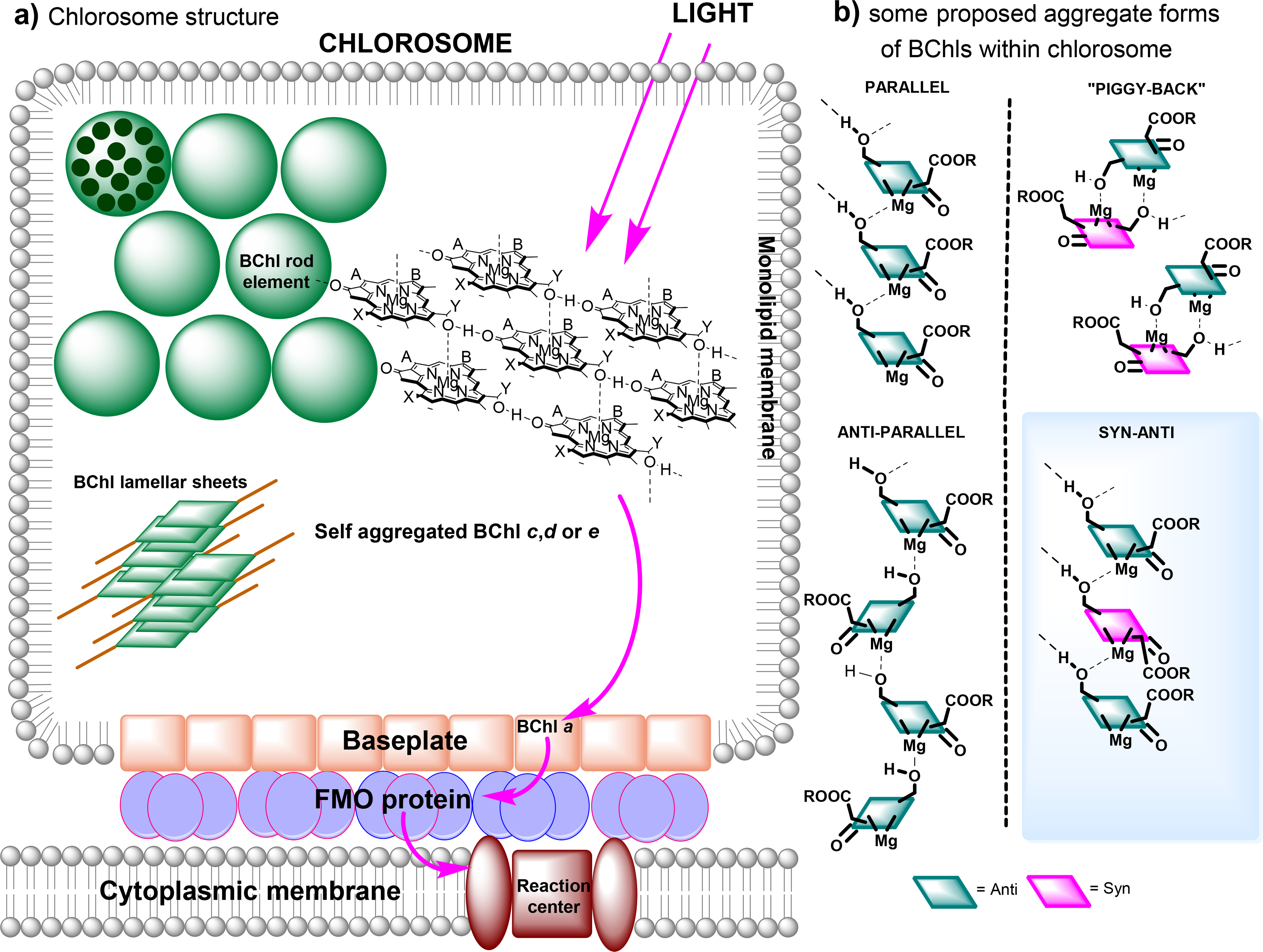
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Senge, M.O.; Ryan, A.A.; Letchford, K.A.; MacGowan, S.A.; Mielke, T. Chlorophylls, Symmetry, Chirality, and Photosynthesis. Symmetry 2014, 6, 781-843. https://doi.org/10.3390/sym6030781
Senge MO, Ryan AA, Letchford KA, MacGowan SA, Mielke T. Chlorophylls, Symmetry, Chirality, and Photosynthesis. Symmetry. 2014; 6(3):781-843. https://doi.org/10.3390/sym6030781
Chicago/Turabian StyleSenge, Mathias O., Aoife A. Ryan, Kristie A. Letchford, Stuart A. MacGowan, and Tamara Mielke. 2014. "Chlorophylls, Symmetry, Chirality, and Photosynthesis" Symmetry 6, no. 3: 781-843. https://doi.org/10.3390/sym6030781
APA StyleSenge, M. O., Ryan, A. A., Letchford, K. A., MacGowan, S. A., & Mielke, T. (2014). Chlorophylls, Symmetry, Chirality, and Photosynthesis. Symmetry, 6(3), 781-843. https://doi.org/10.3390/sym6030781