Mirror Symmetry Breaking in Helical Polysilanes: Preference between Left and Right of Chemical and Physical Origin


Abstract
:1. Introduction
2. Results and Discussion
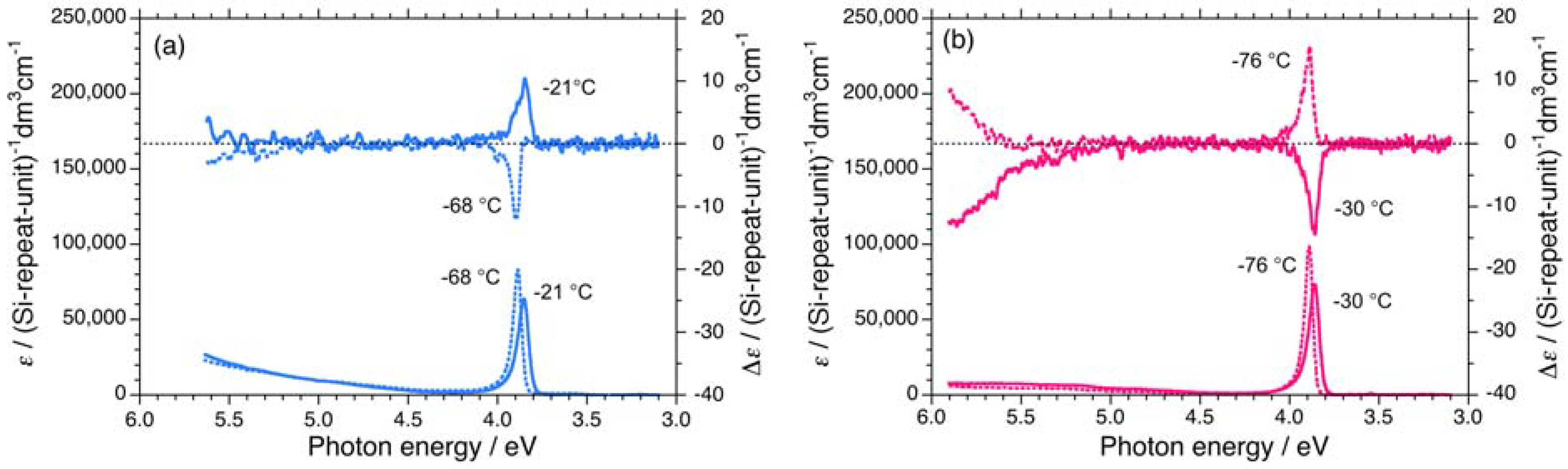
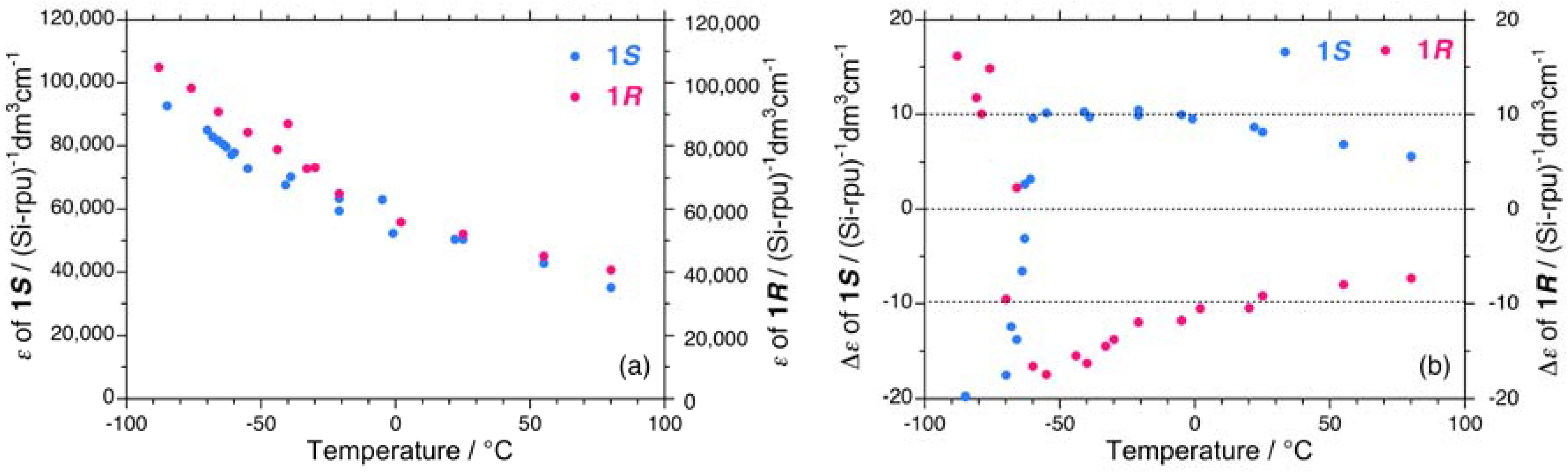
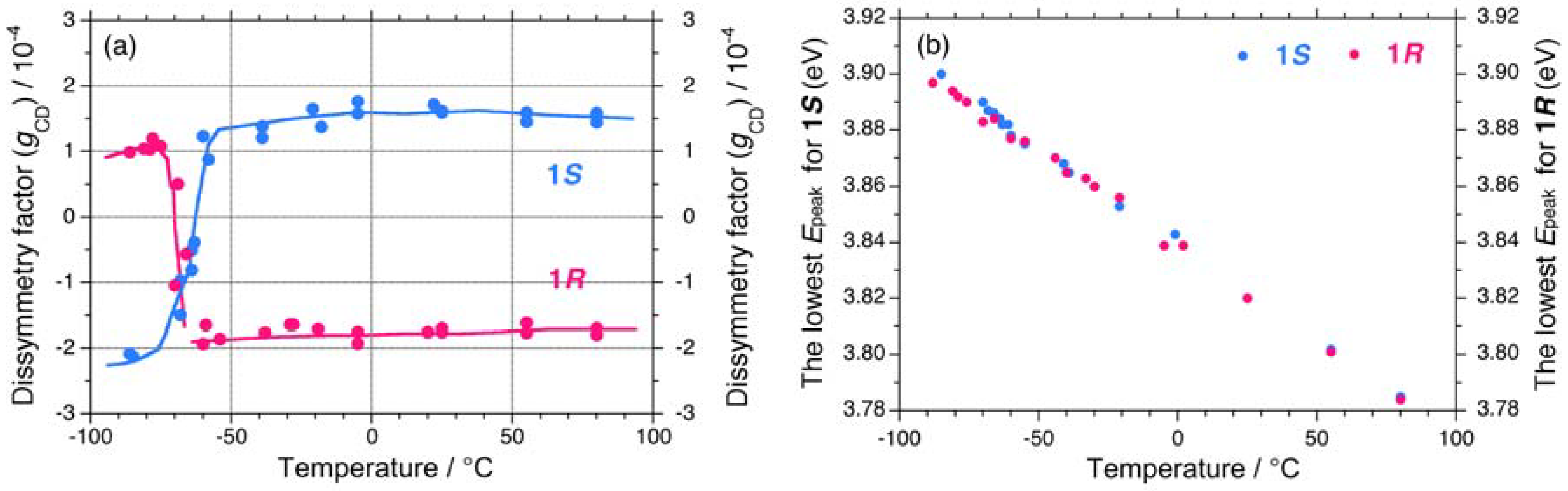
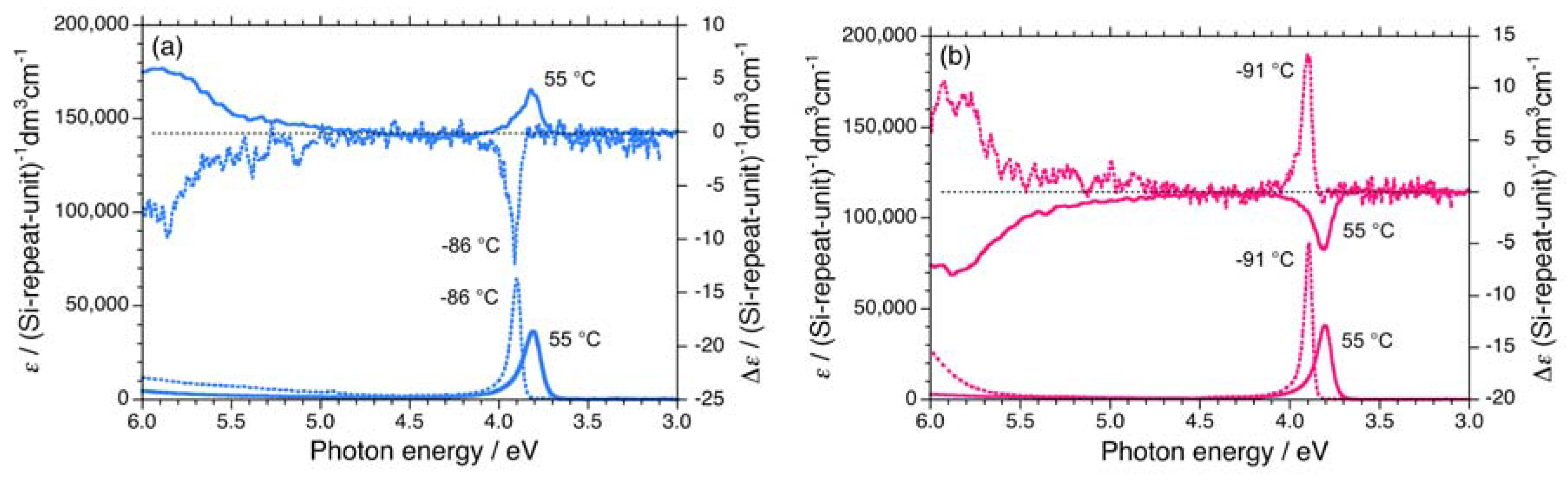
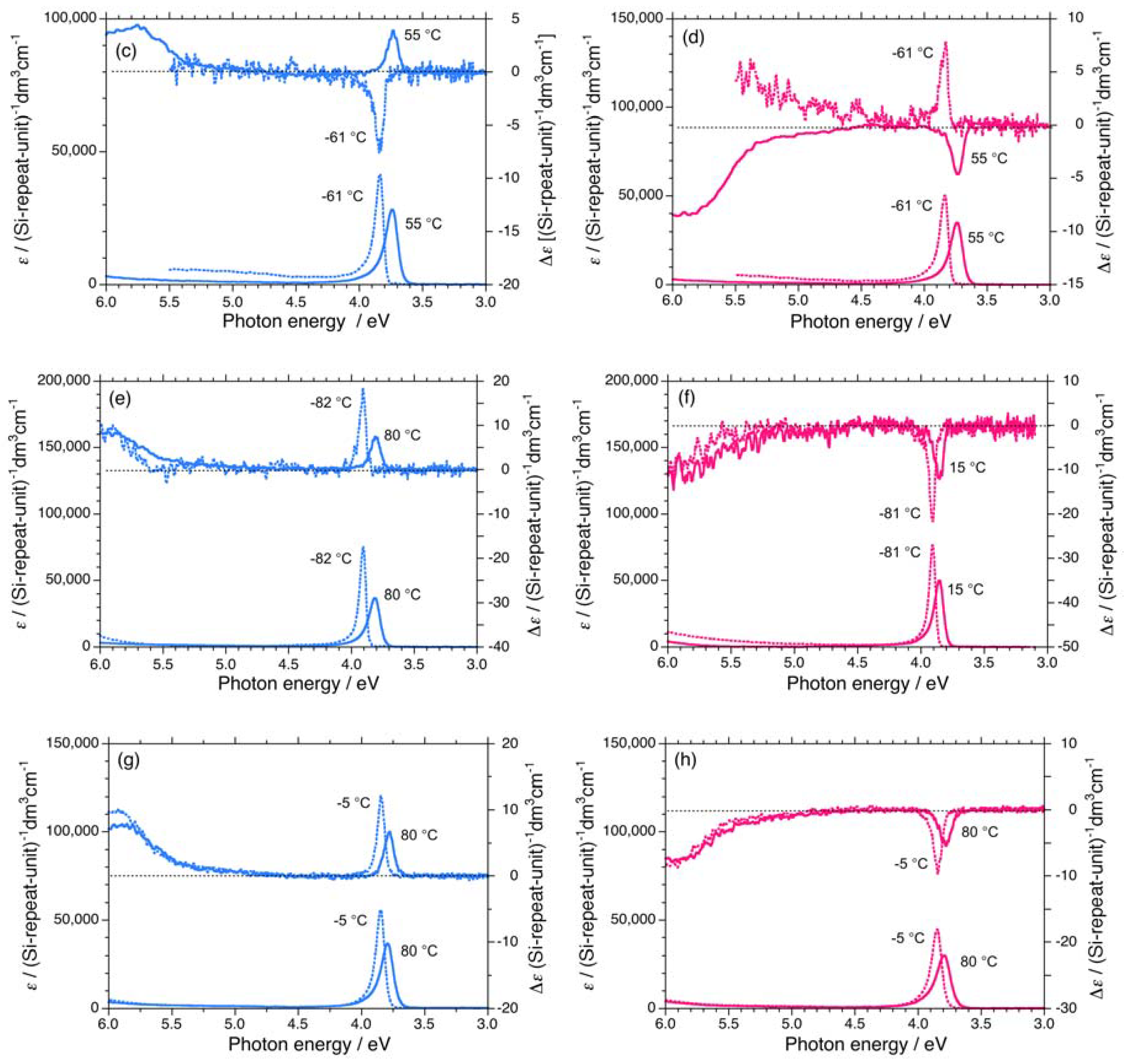
2.1. Chiroptical Spectral Analysis
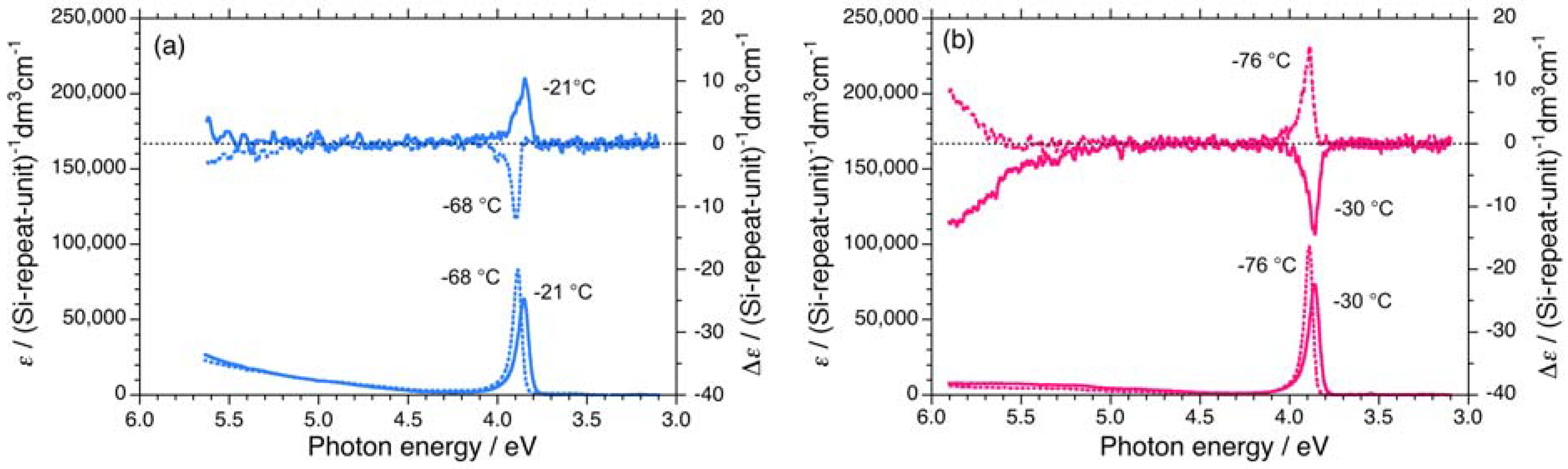
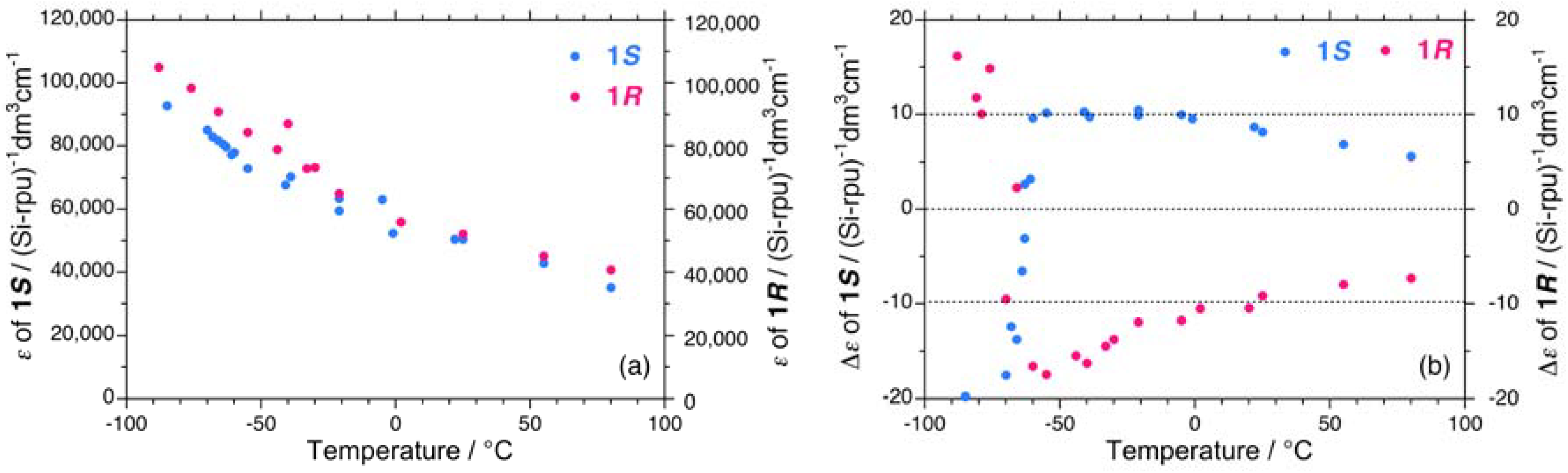
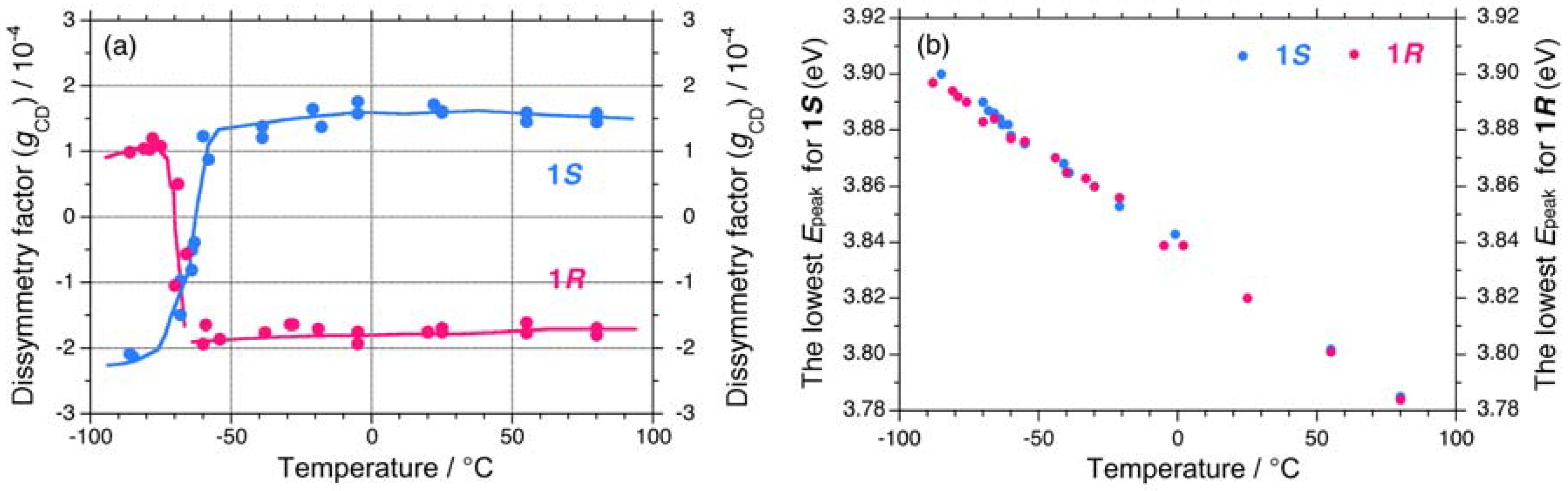
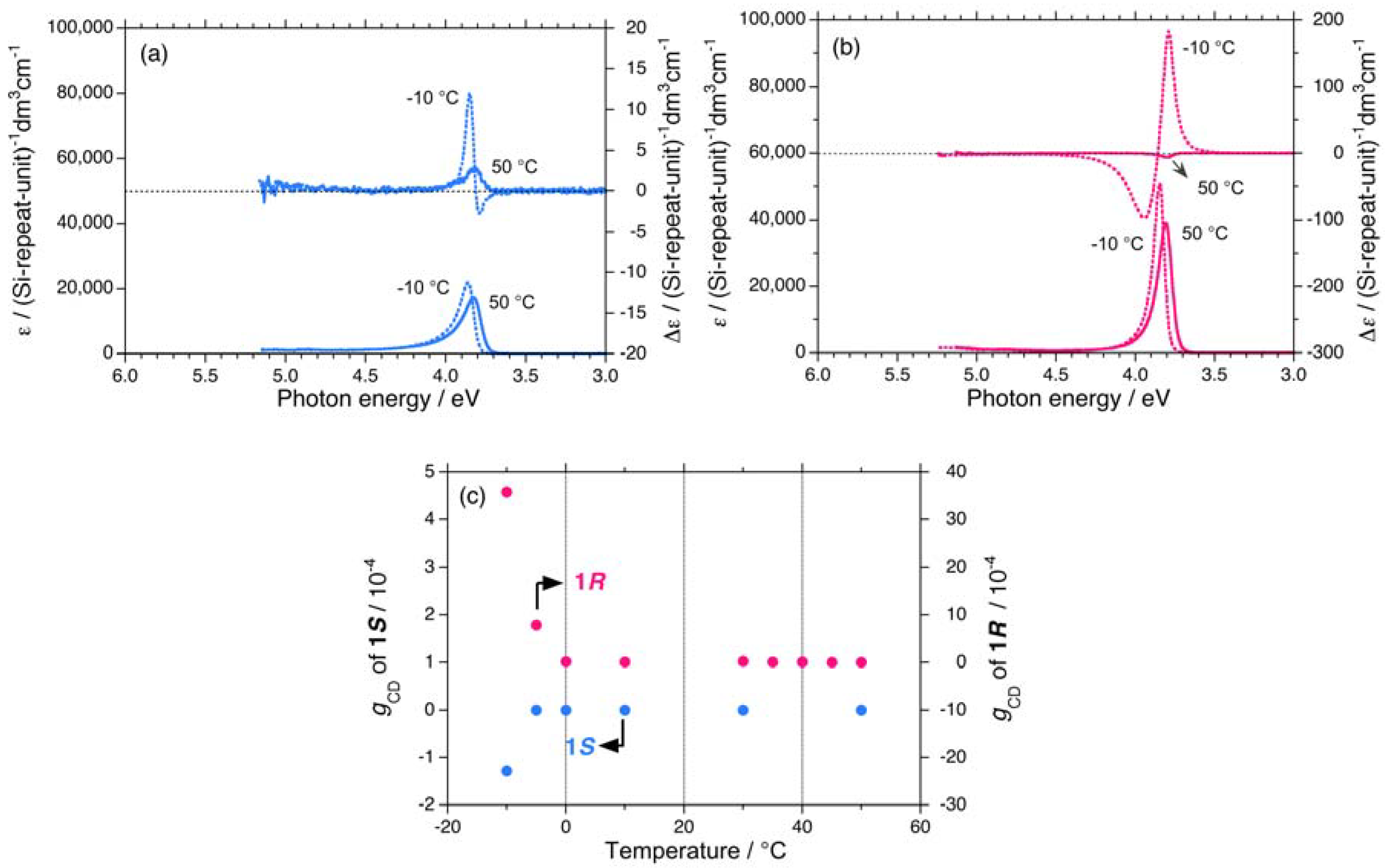
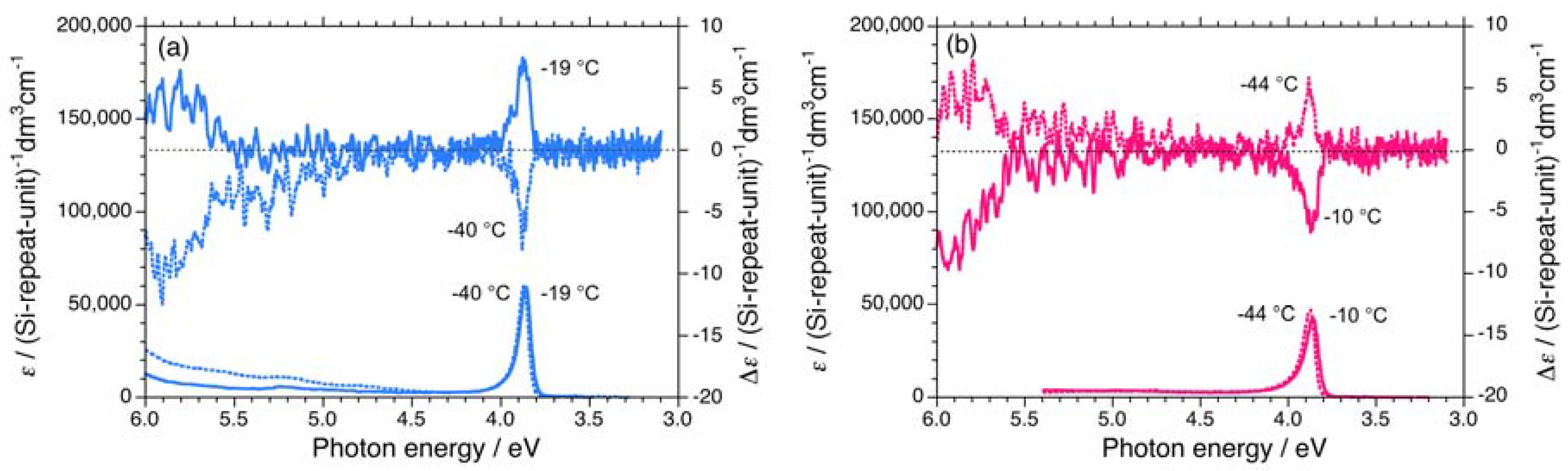
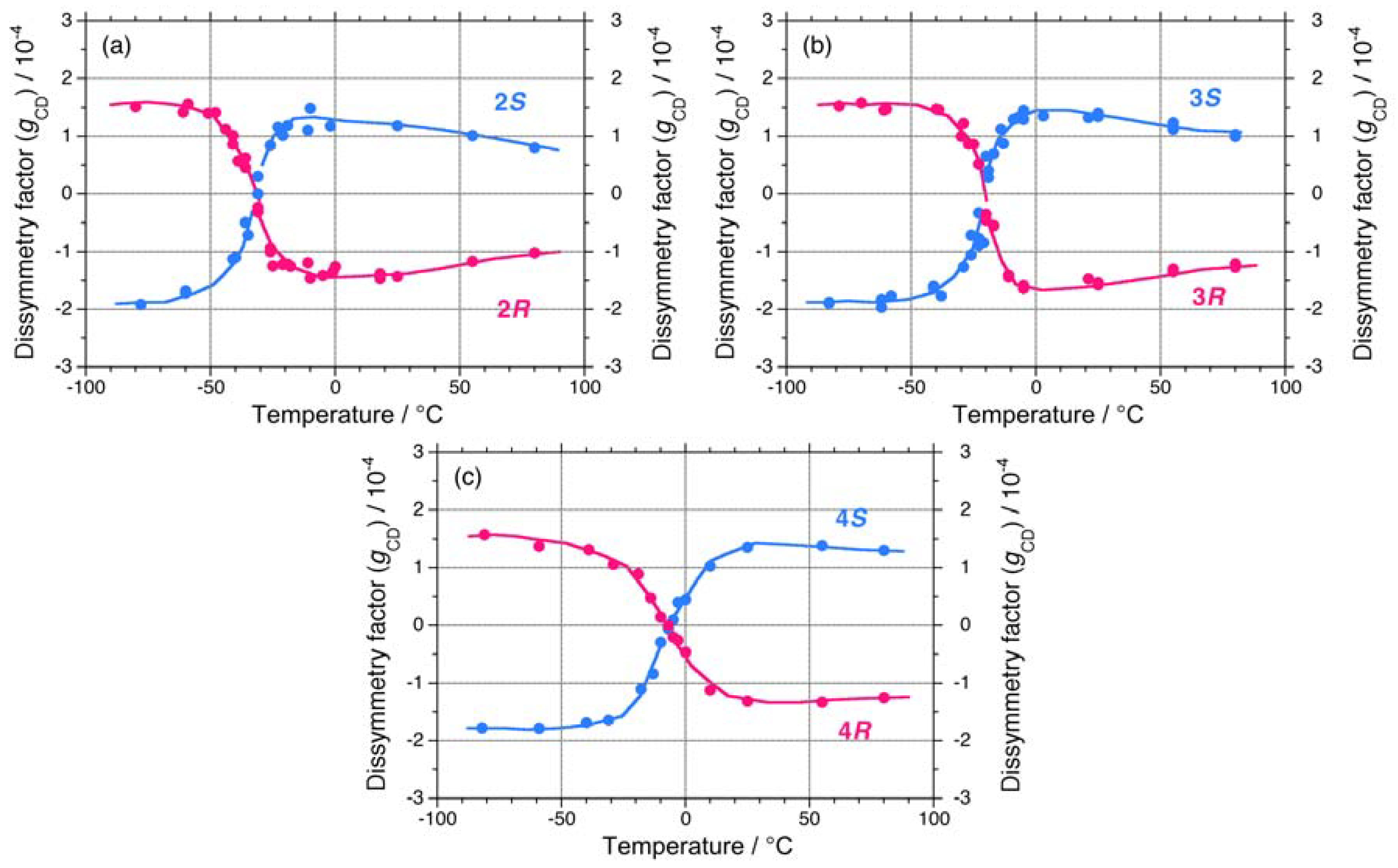
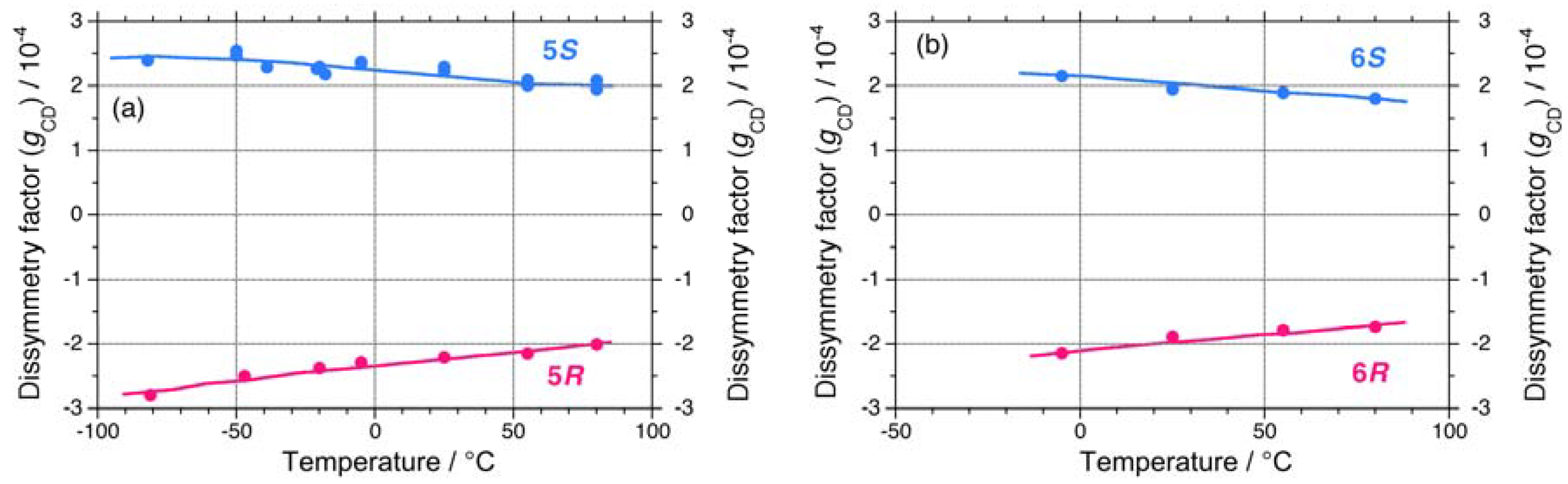
2.1.1. Helical Polysilane Symmetrically Substituted with Chiral Side Groups in Isotropic Solution
2.1.2. Helical Polysilane Aggregates Dispersed in Solution
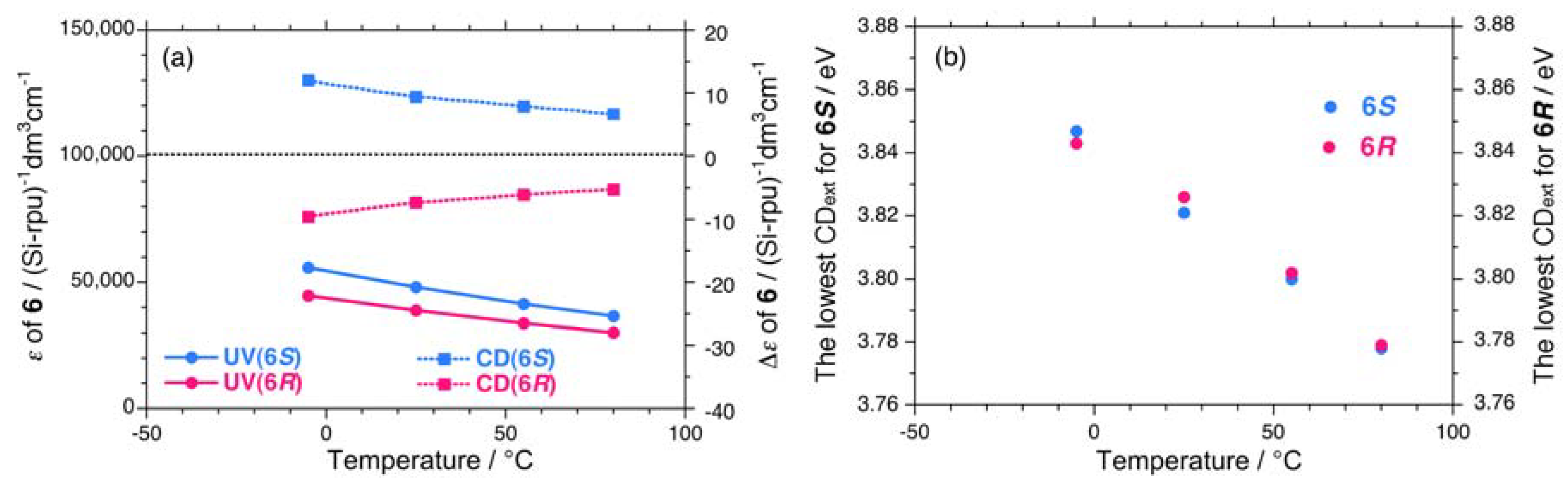
2.1.3. Helical Polysilanes Asymmetrically Substituted with Chiral and Achiral Side Groups in Isotropic Solution
2.2. NMR Spectral Analysis
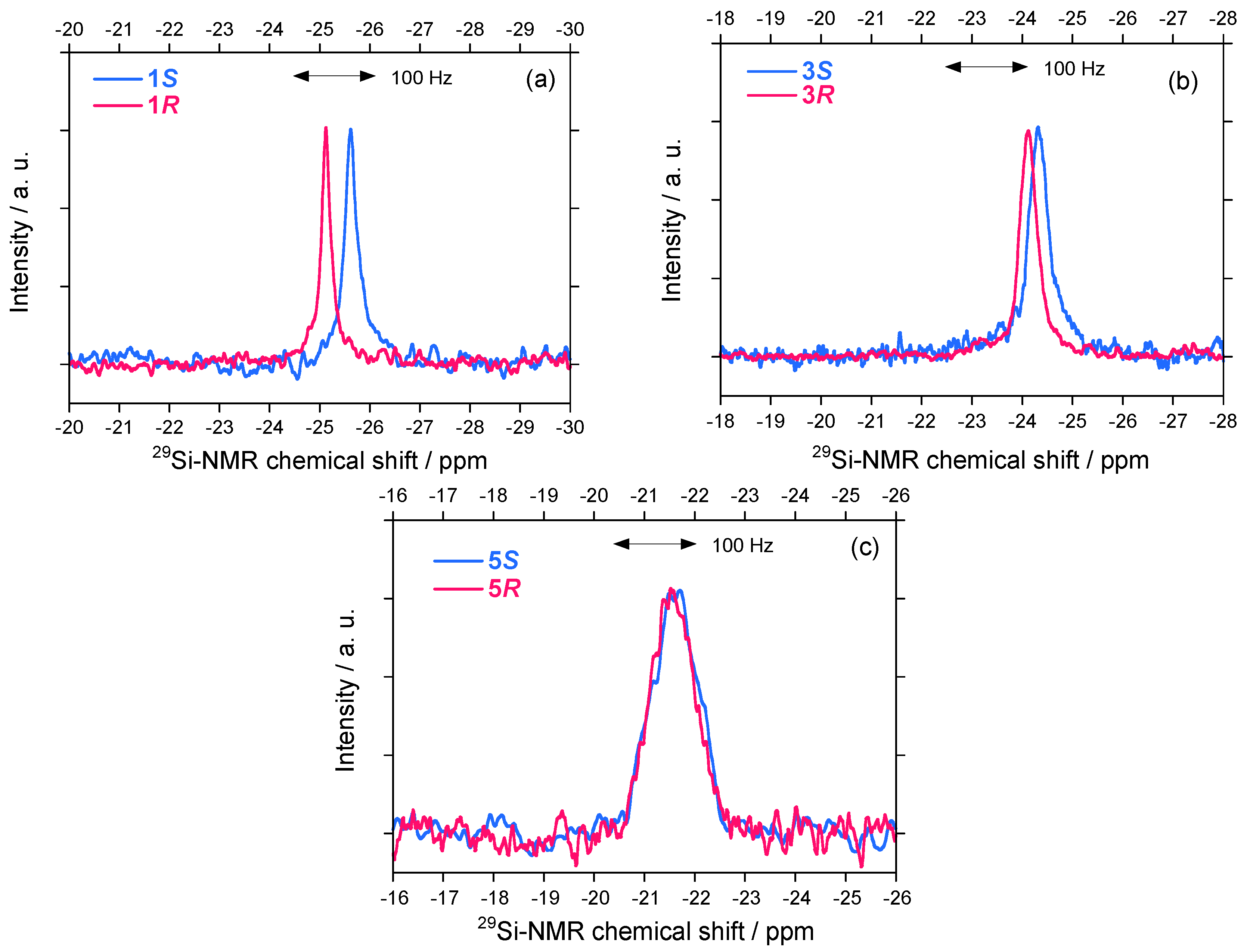
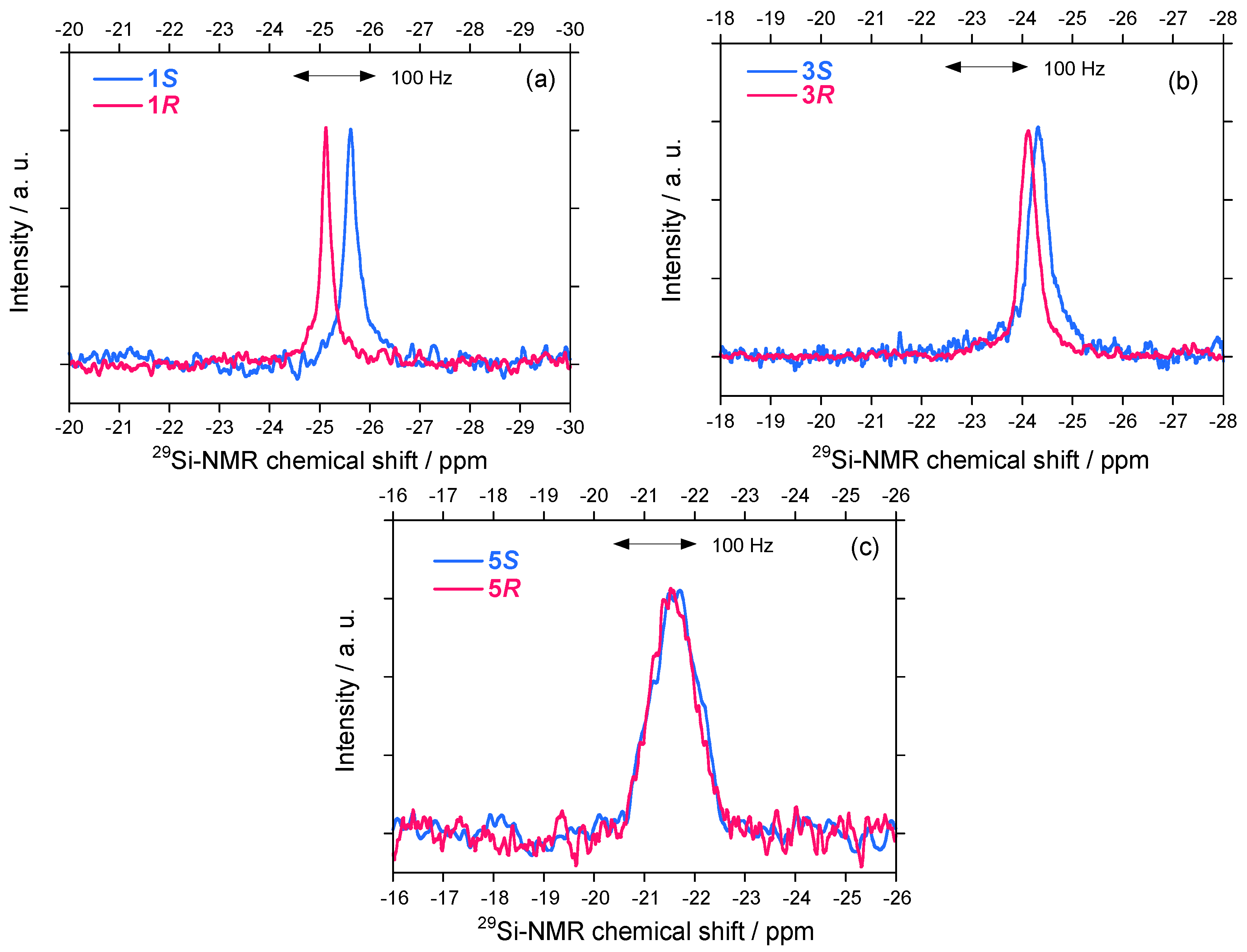
2.2.1. 29Si–NMR Spectroscopic Features
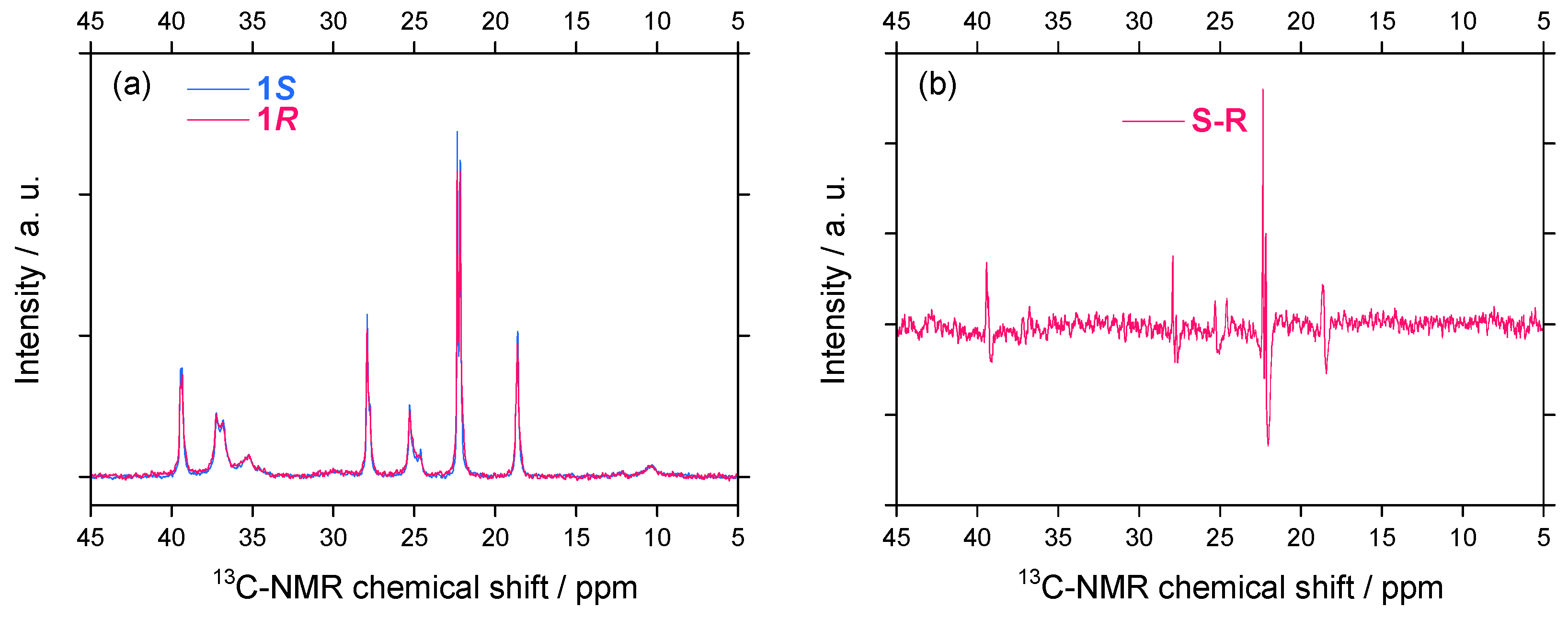
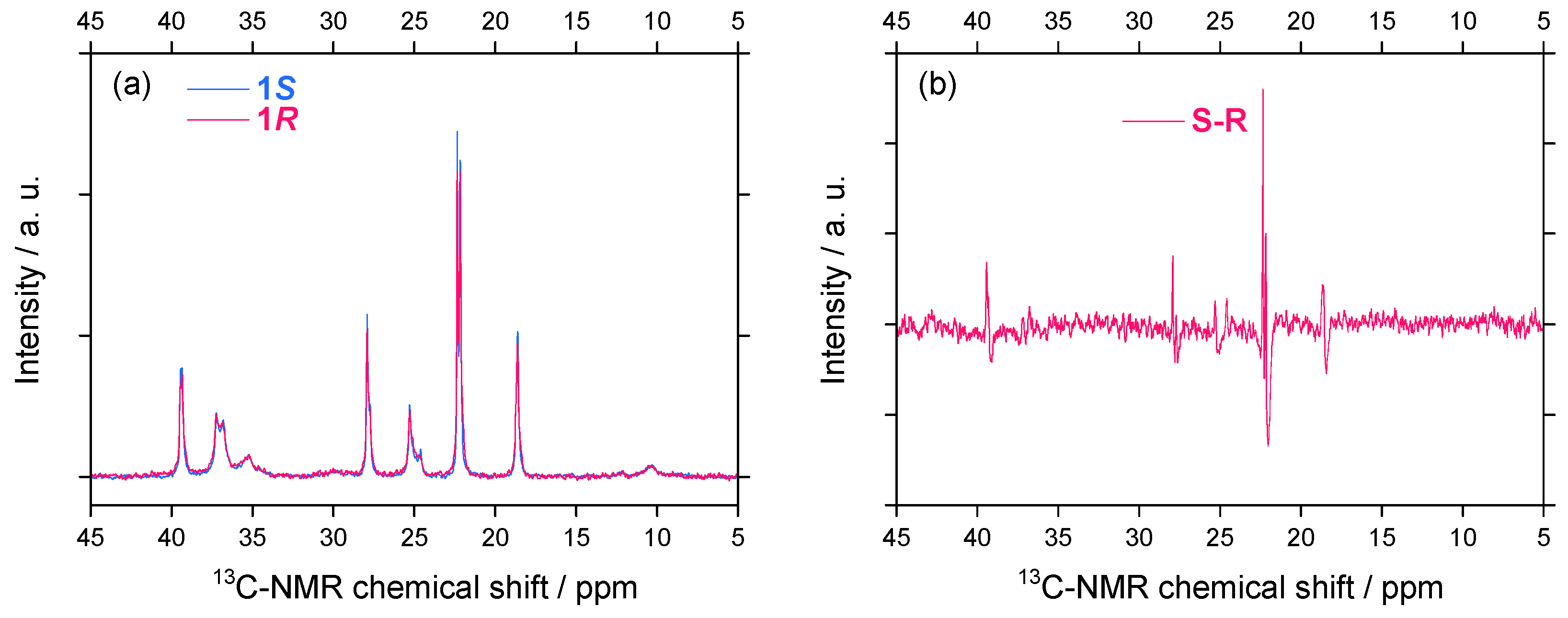
2.2.2. 13C–NMR Spectral Features
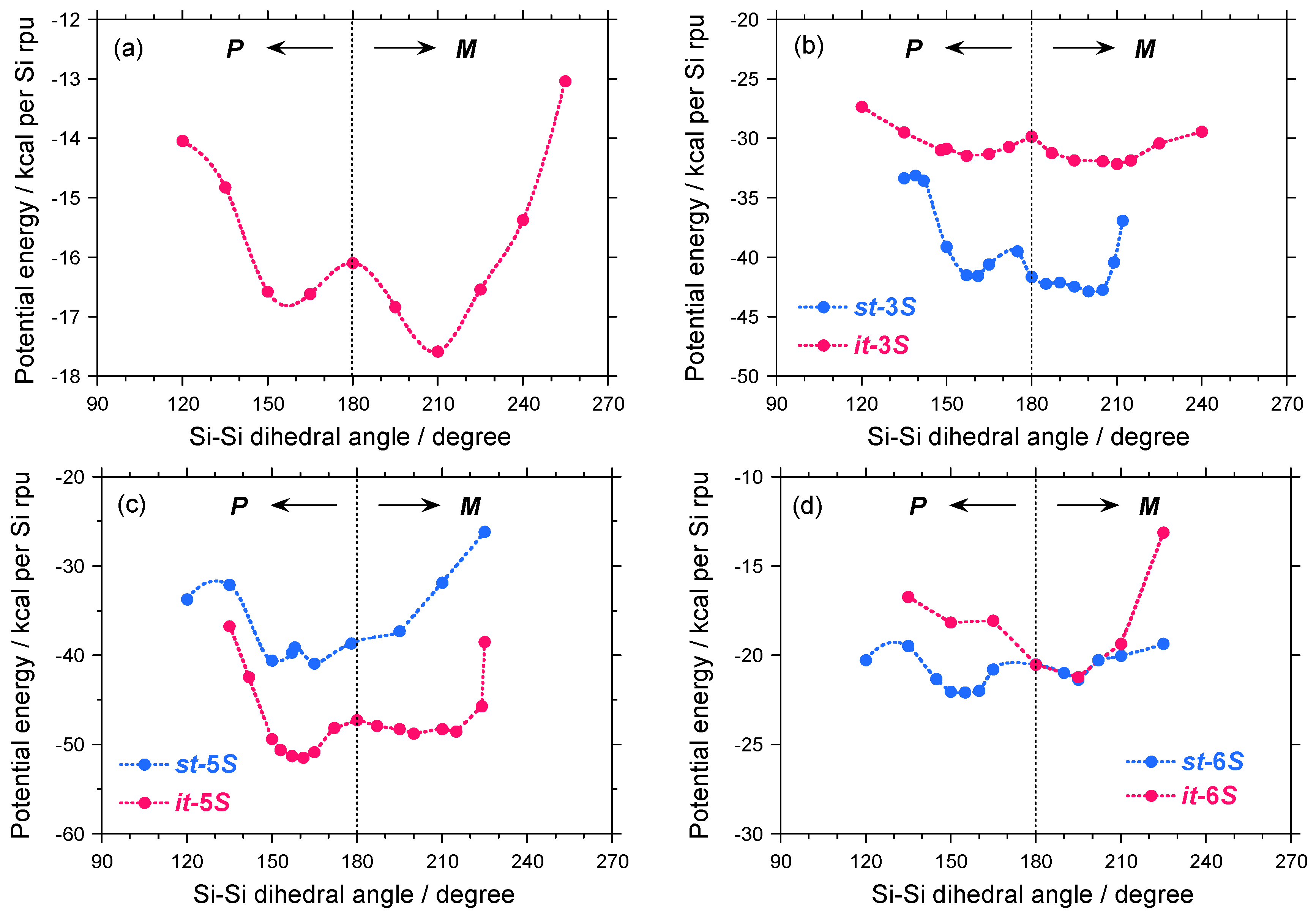
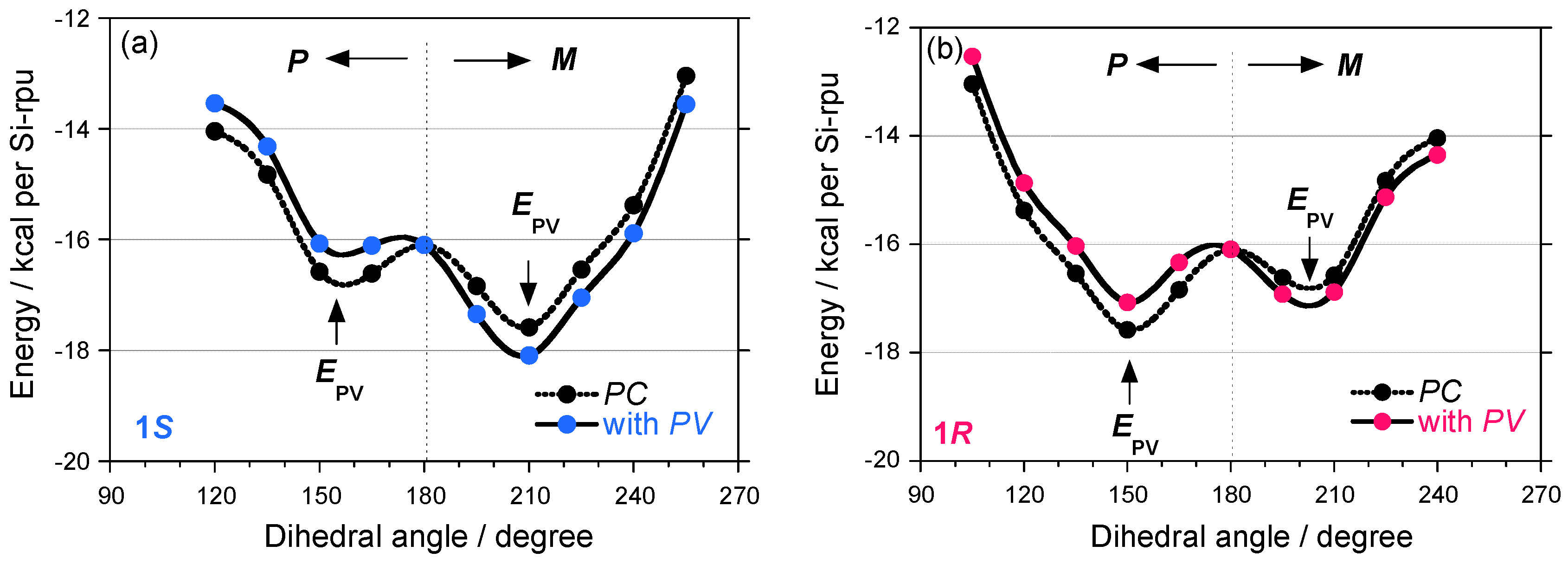
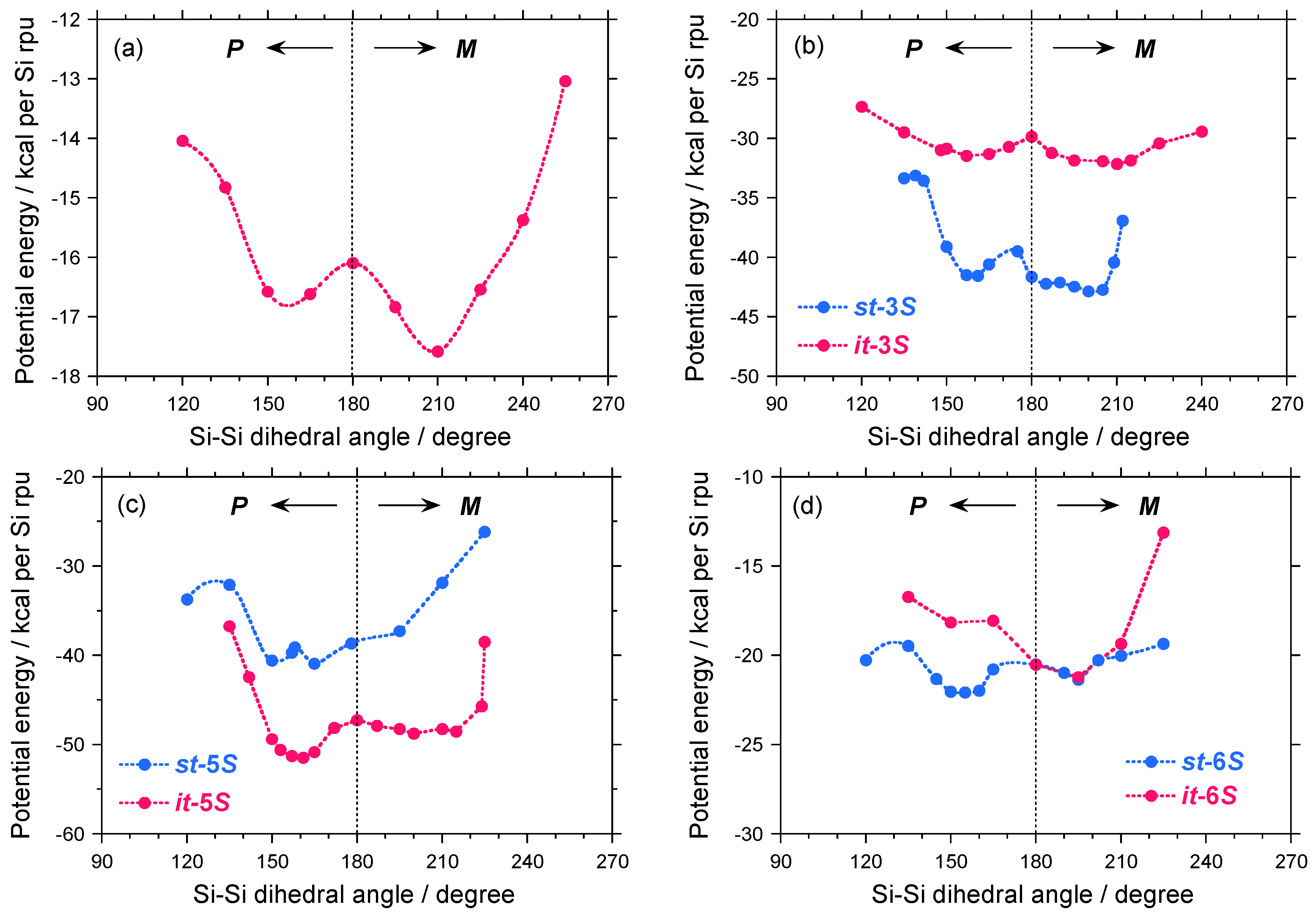
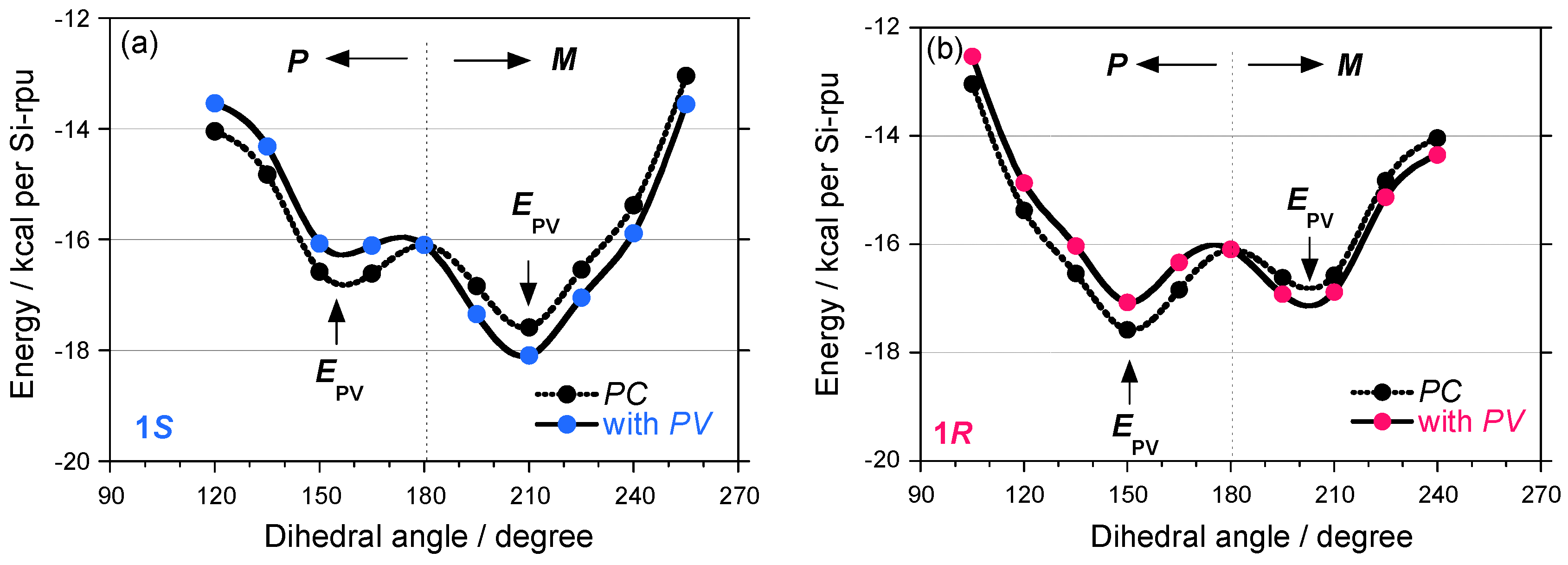
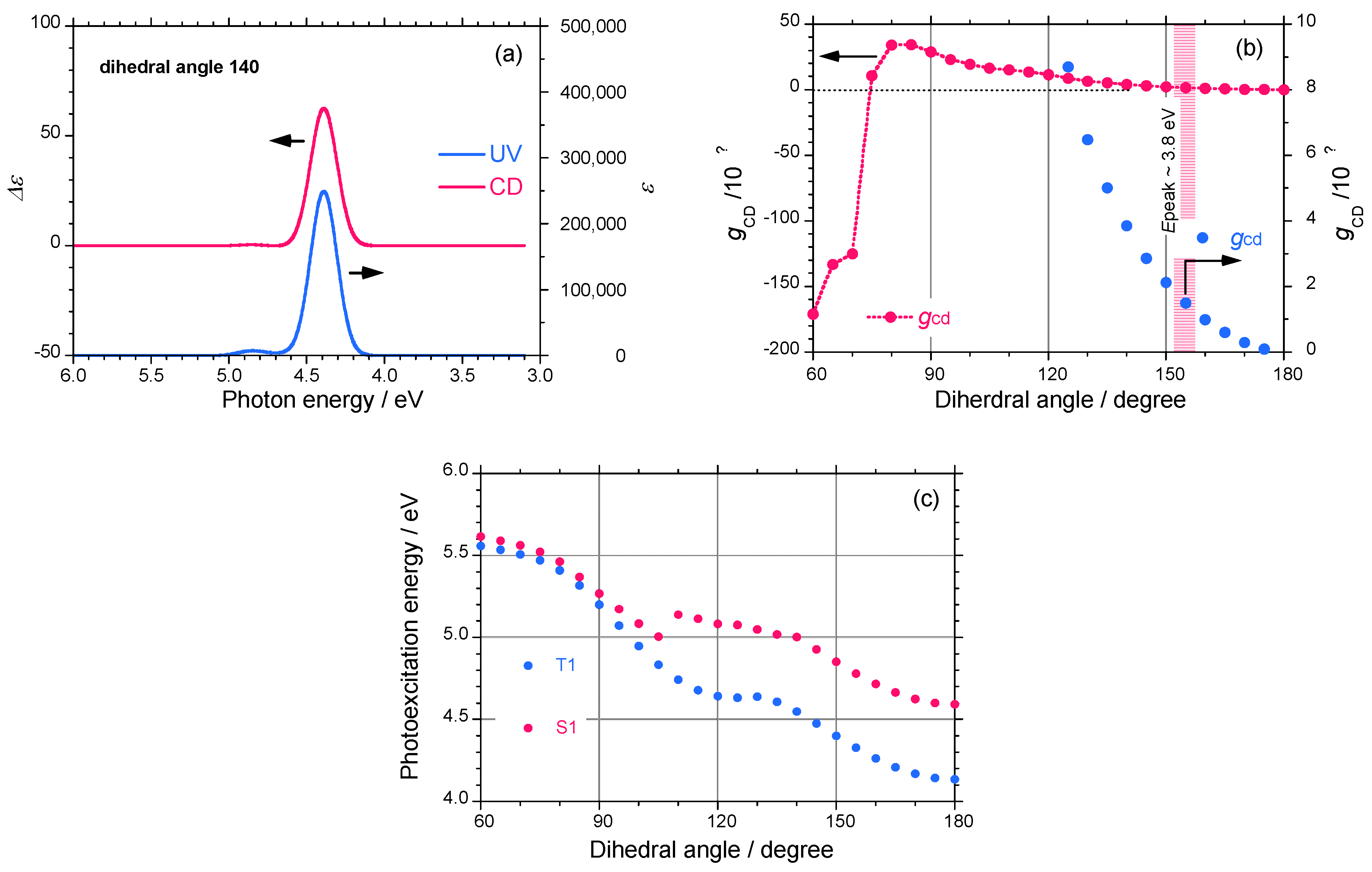
2.3. Electronic Calculation and Molecular Mechanics Calculation
2.4. Viscometric Measurements
3. Experimental Section
3.1. Measurements
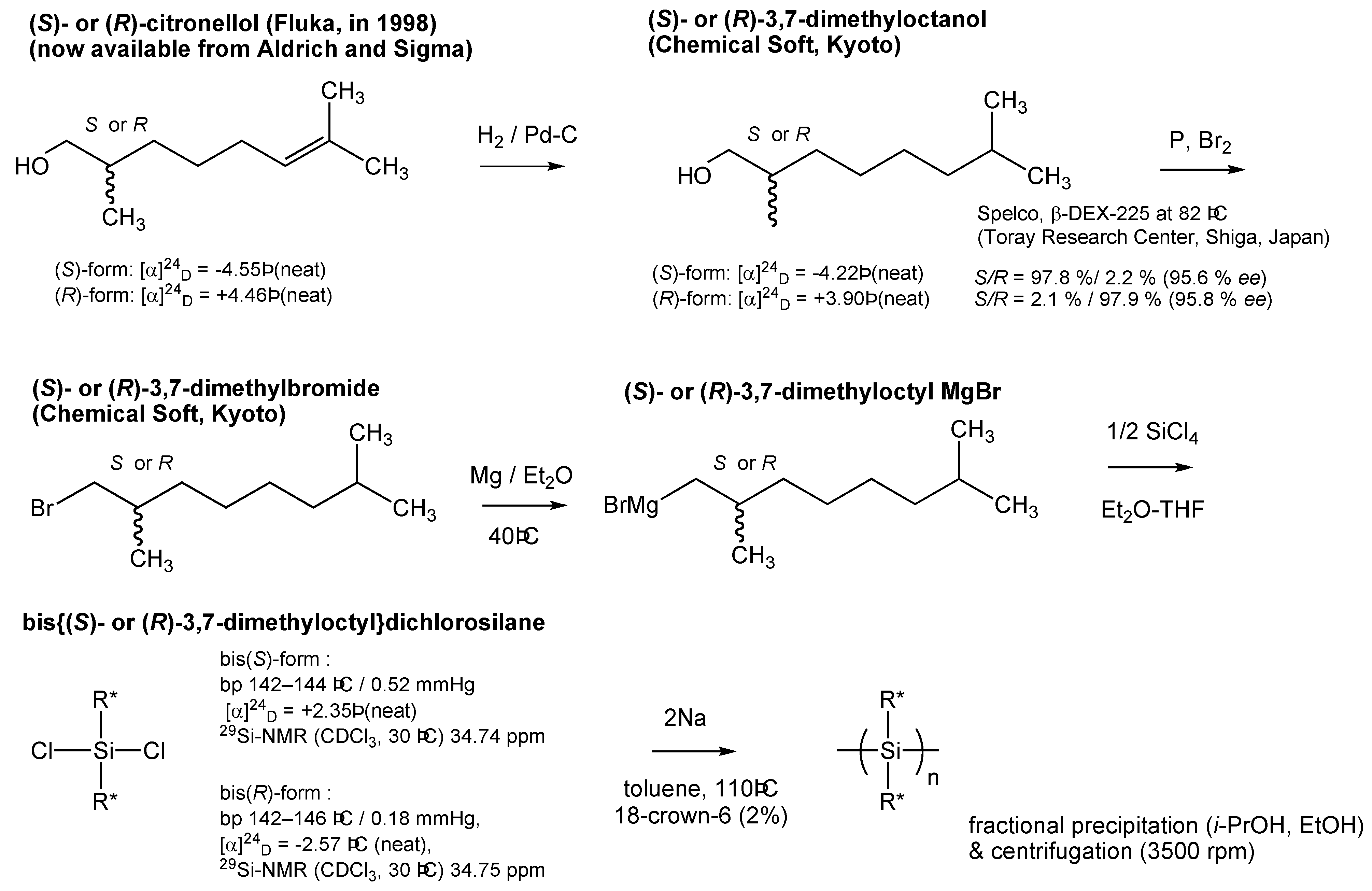
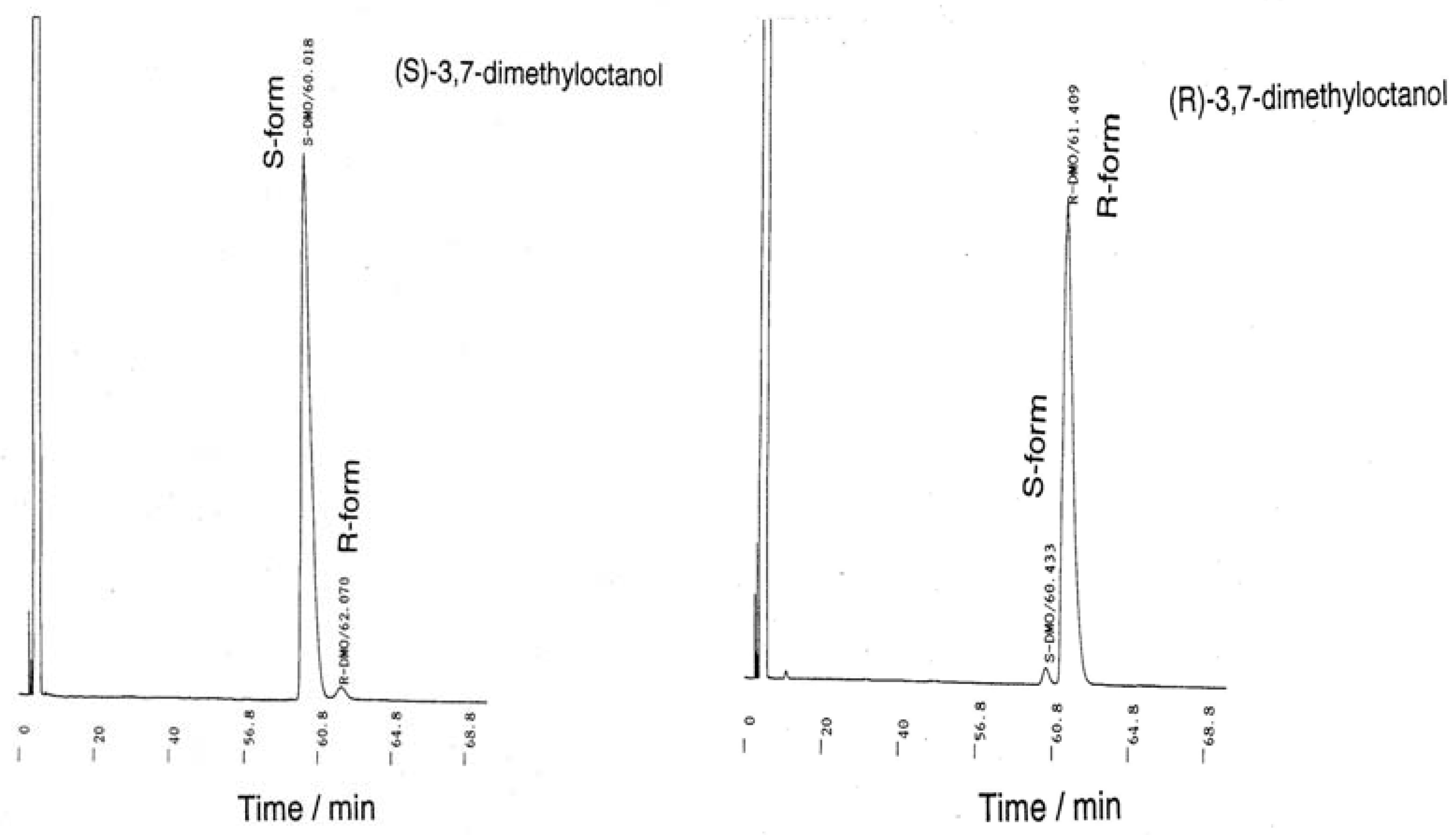
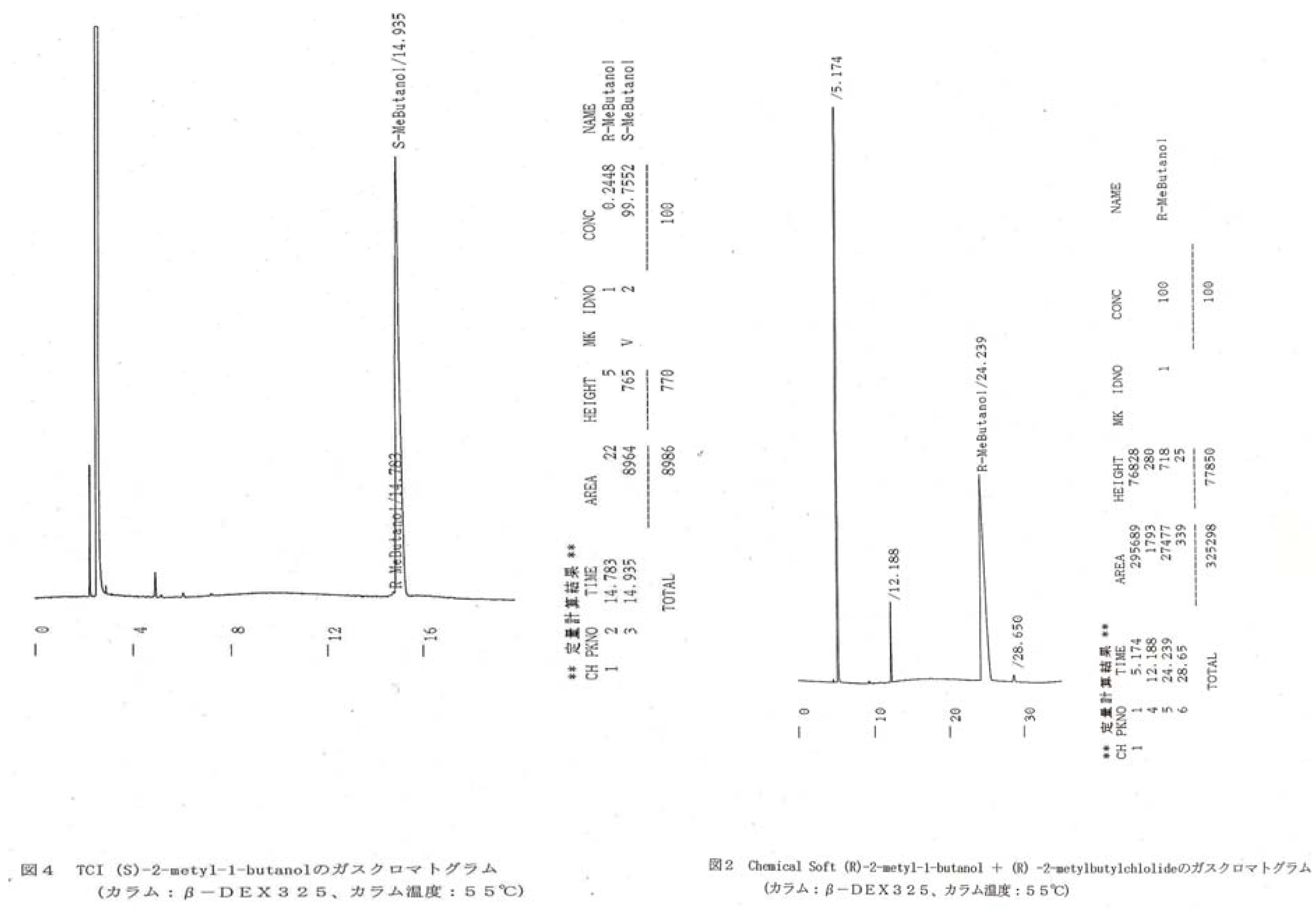
3.2. Monomer Synthesis
3.3. Polymer Preparation
3.4. Molecular Mechanics Calculations
3.5. Electronic State Calculation using Gaussian03 [78]
4. Conclusions
5. Acknowledgements
6. References
- Kipping, F.S.; Pope, W.J. Enantiomorphism. J. Chem. Soc. 1898, 73, 606–617. [Google Scholar] [CrossRef]
- Frank, F.C. On spontaneous asymmetric synthesis. Biochim. Biophys. Acta 1953, 11, 459–463. [Google Scholar] [CrossRef]
- Wald, G. The origin of optical activity. Ann. N.Y. Acad. Sci. 1957, 69, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, T.L.V.; Vester, F. Attempts to induce optical activity with polarized β-radiation. Tetrahedron 1962, 18, 629–637. [Google Scholar] [CrossRef]
- Yamagata, Y. A hypothesis for the asymmetric appearance of biomolecules on Earth. J. Theoret. Biol. 1966, 11, 495–498. [Google Scholar] [CrossRef]
- Thiemann, W.; Darge, W. Experimental attempts for the study of the origin of optical activity on earth. Orig. Life 1974, 5, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Rein, D. Some remarks on parity violating effects of intramolecular interactions. J. Mol. Evol. 1974, 4, 15–22. [Google Scholar] [CrossRef]
- Letokhov, V.S. On difference of energy levels of left and right molecules due to weak interactions. Phys. Lett. 1975, 53A, 275–276. [Google Scholar] [CrossRef]
- Zel’dovich, B.Ya.; Saakyan, D.B.; Sobel’man, I.I. Energy difference between right- and left-hand molecules, due to parity nonconservation in weak interactions of electrons with nuclei. Sov. Phys. JETP. Lett. 1977, 25, 94–97. [Google Scholar]
- Keszthelyi, L. Origin of the asymmetry of biomolecules and weak interaction. Orig. Life 1977, 8, 299–340. [Google Scholar] [CrossRef]
- Harris, R.A.; Stodolsky, L. Quantum beats in optical activity and weak interactions. Phys. Lett. 1978, 78B, 313–317. [Google Scholar] [CrossRef]
- Hegstrom, R.A.; Rein, D.W.; Sandars, P.G.H. Calculation of the parity nonconserving energy difference between mirror-image molecules. J. Chem. Phys. 1980, 73, 2329–2341. [Google Scholar] [CrossRef]
- Mason, S.F. Origins of biomolecular handedness. Nature 1984, 311, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Hegstrom, R.A. Weak neutral current and β radiolysis effects on the origin of the biomolecular chirality. Nature 1985, 315, 749–750. [Google Scholar] [CrossRef]
- Kondepudi, D.K.; Nelson, G.W. Weak neutral currents and the origin of biomolecular chirality. Nature 1985, 314, 438–441. [Google Scholar] [CrossRef]
- Mason, S.F.; Tranter, G.E. The electroweak origin of biomolecular handedness. Proc. R. Soc. Lond. 1985, A397, 45–65. [Google Scholar] [CrossRef]
- Quack, M. On the measurement of the parity violating energy difference between enantiomers. Chem. Phys. Lett. 1986, 132, 147–153. [Google Scholar] [CrossRef]
- Barron, L.D. Symmetry and molecular chirality. Chem. Soc. Rev. 1986, 15, 189–223. [Google Scholar] [CrossRef]
- Applequist, J. Optical activity: Biot’s bequest. Amer. Sci. 1987, 75, 59–68. [Google Scholar]
- Wiesenfeld, L. Effect of atomic number on parity-violating energy differences between enantiomers, O, S, Se, Te. Mol. Phys. 1988, 64, 739–745. [Google Scholar] [CrossRef]
- Barra, A.L.; Robert, J.B.; Wiesenfeld, L. Possible observation of parity nonconservation by high-resolution NMR. Europhys. Lett. 1988, 5, 217–222. [Google Scholar] [CrossRef]
- Avetisov, V.A.; Goldanskii, V.I.; Kuz’min, V.V. Handedness, origin of life and evolution. Phys. Today 1991, 33–41. [Google Scholar] [CrossRef]
- Latel, H. Parity Violation in Atomic Physics. In Chirality - From Weak Bosons to the α-Helix; Janoschek, R., Ed.; Springer: Berlin, Germany, 1992; Chapter 1; pp. 1–17. [Google Scholar]
- Gardner, M. The new ambidextrous universe - Symmetry and asymmetry from mirror reflections to superstrings, 3rd ed.; Freeman: New York, NY, USA, 1990. [Google Scholar]
- Bonner, W.A. Terrestrial and extraterrestrial sources of molecular homochirality. Orig. Life Evol. Biosph. 1991, 21, 407–420. [Google Scholar] [CrossRef]
- Salam, A. The role of chirality in the origin of life. J. Mol. Evol. 1991, 33, 105–113. [Google Scholar] [CrossRef]
- Orgel, L. Molecular replication. Nature 1992, 358, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, O.; Kiyonaga, H. Parity-violating energy shift of helical n-alkane. J. Mol. Struct. 1994, 312, 271–274. [Google Scholar] [CrossRef]
- Bada, J.L. Origins of homochirality. Nature 1995, 374, 594–595. [Google Scholar] [CrossRef]
- Cline, D.B. Physical Origin of Homochirality in Life; AIP Conference Proceedings 379; AIP Press: Woodbury, NY, USA, 1996. [Google Scholar]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.; Palacios, J.; Barron, L.D. Absolute asymmetric synthesis under physical fields: Facts and fictions. Chem. Rev. 1998, 98, 2845–2874. [Google Scholar] [CrossRef]
- Bailey, J.; Chrysostomou, A.; Hough, J.H.; Gledhill, T.M.; McCall, A.; Clark, S.; Ménard, F.; Tamura, M. Circular polarization in star-formation regions: Implications for biomolecular homochirality. Science 1998, 281, 672–674. [Google Scholar] [CrossRef]
- Pályi, G.; Zucchi, C.; Caglioti, L. Advances in Biochemistry; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Szabó-Nagy, A.; Keszthelyi, L. Demonstration of the parity-violating energy difference between enantiomers. Proc. Natl. Acad. Sci. USA 1999, 96, 4252–4255. [Google Scholar] [CrossRef]
- Feringa, B.L.; van Delden, R.A. Absolute asymmetric synthesis: the origin, control, and amplification of chirality. Angew. Chem. Int. Ed. 1999, 38, 3418–3438. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.; Palacios, J. From parity to chirality: Chemical implications revisited. Tetrahedron Assym. 2000, 11, 2991–2404. [Google Scholar] [CrossRef]
- MacDermott, A.J. The ascent of parity-violation: Exochirality in the solar system and beyond. Enantiomer 2000, 5, 153–168. [Google Scholar] [PubMed]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C. Chiral autocatalysis: Where stereochemistry meets the origin of life. Chem. Commun. 2000, 887–892. [Google Scholar] [CrossRef]
- Frank, P.; Bonner, W.A.; Zare, R. On One Hand But Not the Other: The Challenge of the Origin and Survival of Homochirality in Prebiotic Chemistry. In Chemistry for the 21st Century; Keinan, E., Schechter, E., Eds.; Wiley-VCH: Weinheim, German, 2001; Chapter 11; pp. 175–208. [Google Scholar]
- Lough, L.W.; Wainer, I.W. Chirality in Natural and Applied Science; Blackwell: Oxford, UK, 2002. [Google Scholar]
- Quack, M. How important is parity violation for molecular and biomolecular chirality? Angew. Chem. Int. Ed. 2002, 41, 4618–4630. [Google Scholar] [CrossRef] [PubMed]
- Shinitzky, M.; Nudelman, F.; Barda, Y.; Haimovitz, R.; Chen, E.; Deamer, D.W. Unexpected differences between D- and L-tyrosine lead to chiral enhancement in racemic mixtures. Orig. Life Evol. Biosph. 2002, 32, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Crassous, J.; Chardonnet, C.; Saue, T.; Schwerdtfeger, P. Recent experimental and theoretical developments towards the observation of parity violation (PV) effects in molecules by spectroscopy. Org. Biomol. Chem. 2005, 3, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Wagniére, G.H. On Chirality and the Universal Asymmetry—Reflections in Image and Mirror Image; Wiley-VCH: Weinheim, German, 2007. [Google Scholar]
- Soai, K.; Kawasaki, T. Asymmetric autocatalysis with amplification of chirality. Top. Curr. Chem. 2008, 284, 1–33. [Google Scholar]
- Guijarro, A.; Yus, M. The Origin of Chirality in The Molecules of Life; RSC: London, UK, 2009. [Google Scholar]
- Fujiki, M. Mirror symmetry breaking of silicon polymers—From weak bosons to artificial helix. Chem. Rec. 2009, 9, 271–298. [Google Scholar] [CrossRef]
- Sozzi, M.S. Discrete symmetries and CP violation: From experiment to theory; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Wigner, E. Einige folgerungen aus der schroedingerschen theorie fur die termstrukturen. Z. Phys. 1927, 43, 624–652. [Google Scholar] [CrossRef]
- Wigner, E.P. Symmetries and Reflections: Scientific Essays; MIT Press: Cambridge, MA, USA, 1970. [Google Scholar]
- Wigner, E.P. Violations of symmetry in physics. Sci. Am. 1965, 213, 28–36. [Google Scholar] [CrossRef]
- Adair, R.K. A flaw in a universal mirror. Sci. Am. 1988, 258, 30–36. [Google Scholar] [CrossRef]
- Wu, C.S.; Ambler, E.; Hayward, R.W.; Hoppes, D.D.; Hudson, R.P. Experimental test of parity conservation in beta decay. Phys. Rev. 1957, 105, 1413–1415. [Google Scholar] [CrossRef]
- Lee, T.D.; Yang, C.N. Question of parity conservation in weak interactions. Phys. Rev. 1956, 105, 254–258. [Google Scholar] [CrossRef]
- Nambu, Y.; Jona-Lasinio, G. Dynamical model of elementary particles based on an analogy with superconductivity. I. Phys. Rev. 1961, 122, 345–358. [Google Scholar] [CrossRef]
- Nambu, Y.; Jona-Lasinio, G. Dynamical model of elementary particles based on an analogy with superconductivity. II. Phys. Rev. 1961, 124, 246–254. [Google Scholar] [CrossRef]
- Weinberg, S. Conceptual foundations of the unified theory of weak and electromagnetic interactions. Rev. Mod. Phys. 1980, 52, 515–523. [Google Scholar] [CrossRef]
- Salam, A. Gauge unification of fundamental forces. Rev. Mod. Phys. 1980, 52, 525–538. [Google Scholar] [CrossRef]
- Glashow, S.L. Towards a unified theory: Threads in a tapestry. Rev. Mod. Phys. 1980, 52, 539–543. [Google Scholar] [CrossRef]
- Rubbia, C. Experimental observation of the intermediate vector bosons W+, W-, and Z0. Rev. Mod. Phys. 1985, 57, 699–722. [Google Scholar] [CrossRef]
- Bouchiat, M.A.; Pottier, L. Optical experiments and weak interactions. Science 1986, 234, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Frois, B.; Bouchiat, M.A. Parity violation in atoms and polarized electron scattering; World Scientific: London, UK, 1999. [Google Scholar]
- Wang, W.; Yi, F.; Ni, Y.; Zhao, Z.; Jin, X.; Tang, Y. Parity violation of electroweak force in phase transitions of single crystals of D- and L-alanine and valine. J Biol. Phys. 2000, 26, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Pyda, M.; Pak, J.; Wunderlich, B.; Thompson, J.R.; Pagni, R.; Pan, H.; Barnes, C.; Schwerdtfeger, P.; Compton, R. Search for electroweak interactions in amino acid crystals. II. The Salam hypothesis. J. Phys. Chem. A 2003, 107, 6674–6680. [Google Scholar] [CrossRef]
- Wilson, C.C.; Myles, D.; Ghosh, M.; Johnson, L.N.; Wang, W. Neutron diffraction investigations of L- and D-alanine at different temperatures: The search for structural evidence for parity violation. New J. Chem. 2005, 29, 1318–1322. [Google Scholar] [CrossRef]
- Figgen, D.; Koers, A.; Schwerdtfeger, P. NWHClI: A small and compact chiral molecule with large parity-violation effects in the vibrational spectrum. Angew. Chem. Int. Ed. 2010, 49, 2941–2943. [Google Scholar] [CrossRef] [PubMed]
- Scolnik, Y.; Portnaya, I.; Cogan, U.; Tal, S.; Haimovitz, R.; Fridkin, M.; Elitzur, A.C.; Deamer, D.W.; Shinitzky, M. Subtle differences in structural transitions between poly-L- and poly-D-amino acids of equal length in water. Phys. Chem. Chem. Phys. 2006, 8, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Lahav, M.; Weissbuch, I.; Shavit, E.; Reiner, C.; Nicholson, G.J.; Schurig, V. Parity violating energetic difference and enantiomorphous crystals-caveats; Reinvestigation of tyrosine crystallization. Orig. Life Evol. Biosph. 2006, 36, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Figgen, D.; Schwerdtfeger, P. Structures, inversion barriers, and parity violation effects in chiral SeOXY molecules (X,Y=H, F, Cl, Br, or I). J. Chem. Phys. 2009, 130, 054306. [Google Scholar] [CrossRef]
- Kodona, E.K.; Alexopoulos, C.; Panou-Pomonis, E.; Pomonis, P.J. Chirality and helix stability of polyglutamic acid enantiomers. J. Colloid Interf. Sci. 2008, 319, 71–80. [Google Scholar] [CrossRef]
- Fujiki, M.; Koe, J.R.; Terao, K.; Sato, T.; Teramoto, A.; Watanabe, J. Optically active polysilanes. Ten years of progress and new polymer twist for nanoscience and nanotechnology. Polym. J. 2003, 35, 297–344. [Google Scholar] [CrossRef]
- Fujiki, M. Switching handedness in optically active polysilanes. J. Organomet. Chem. 2003, 685, 15–34. [Google Scholar] [CrossRef]
- Fujiki, M. Experimental tests of parity violation at helical polysilylene level. Macromol. Rapid Commun. 2001, 22, 669–674. [Google Scholar] [CrossRef]
- Dekkers, H.P.J.M. Circularly Polarized Luminescence: A probe for Chirality in the Excited State. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N., Nakanishi, K., Woody, R.W., Eds.; Wiley-VCH: New York, NY, USA, 2000; Chapter 7. [Google Scholar]
- Sato, T.; Terao, K.; Teramoto, A.; Fujiki, M. Molecular properties of helical polysilylenes in solution. Polymer 2003, 44, 5477–5495. [Google Scholar] [CrossRef]
- Laubender, G.; Berger, R. Ab initio calculation of parity-violating chemical shifts in NMR spectra of chiral molecules. ChemPhysChem 2003, 4, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Weijo, V.; Manninen, P.; Vaara, J. Perturbational calculations of parity-violating effects in nuclear-magnetic-resonance parameters. J. Chem. Phys. 2005, 123, 054501. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Kamide, K.; Kataoka, A. Theoretical relationships between parameters in the Kuhn-Mark- Houwink-Sakurada equation. Macromol. Chem. Phys. 2003, 128, 217–228. [Google Scholar] [CrossRef]
- Carter, A.B.; Sanda, A.I. CP violation in B-meson decays. Phys. Rev. D 1981, 23, 1567–1579. [Google Scholar] [CrossRef]
- Aubert, B.; Boutigny, D.; Gaillard, J.M.; Hicheur, A.; Karyotakis, Y.; Lees, J.P.; Robbe, P.; Tisserand, V.; Palano, A.; Chen, G.P.; Chen, J.C.; Qi, N.D.; Rong, G.; et al. Observation of CP violation in the B0 meson system. Phys. Rev. Lett. 2001, 87, 091801. [Google Scholar] [CrossRef]
- Abe, K.; Abe, R.; Adachi, I.; Aihara, H.; Akatsu, M.; Alimonti, G.; Asai, K.; Asai, M.; Asano, Y.; Aso, T.; Aulchenko, V.; et al. Observation of large CP violation in the neutral B meson system. Phys. Rev. Lett. 2001, 87, 091802. [Google Scholar] [CrossRef]
7. Appendix

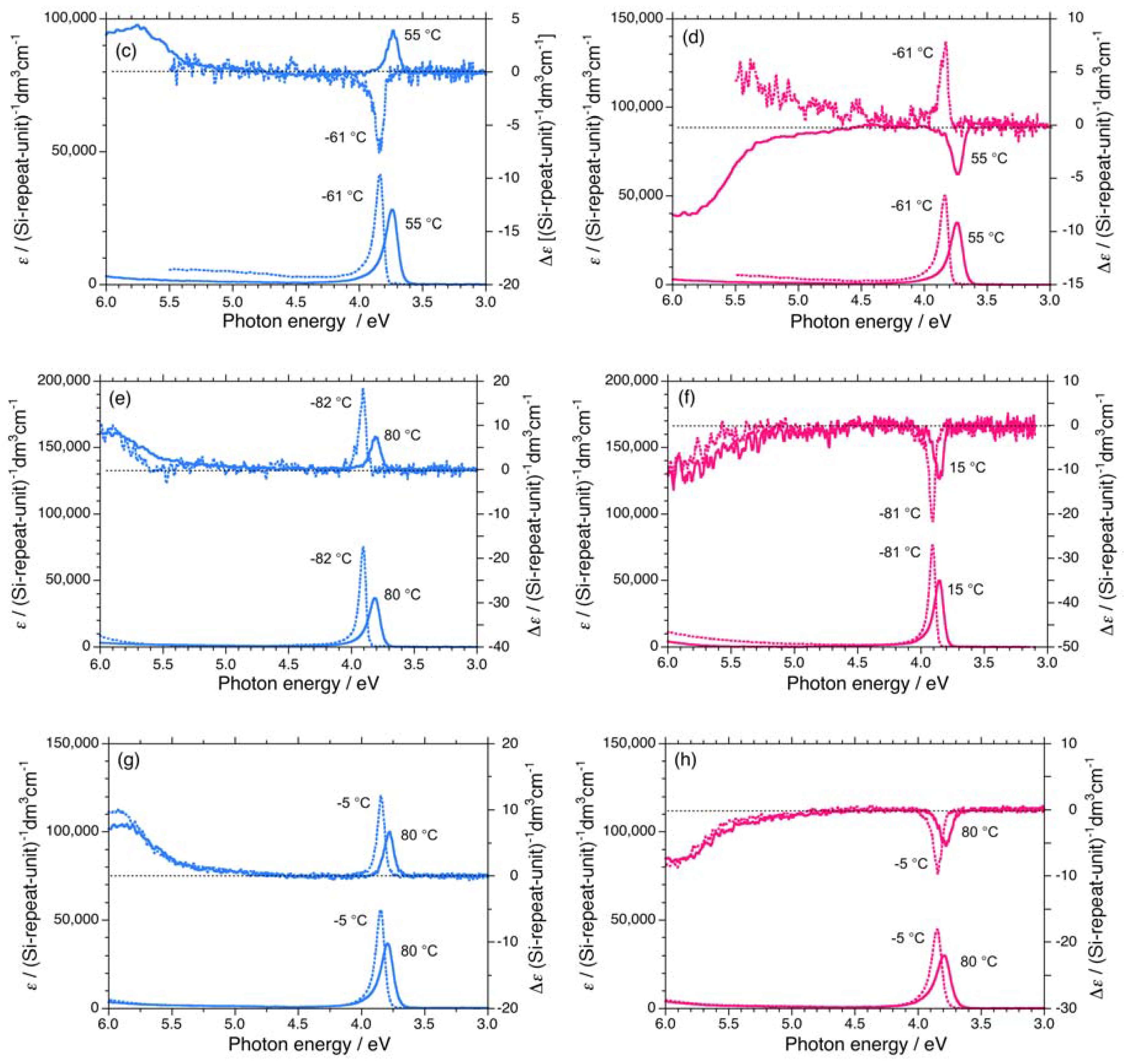
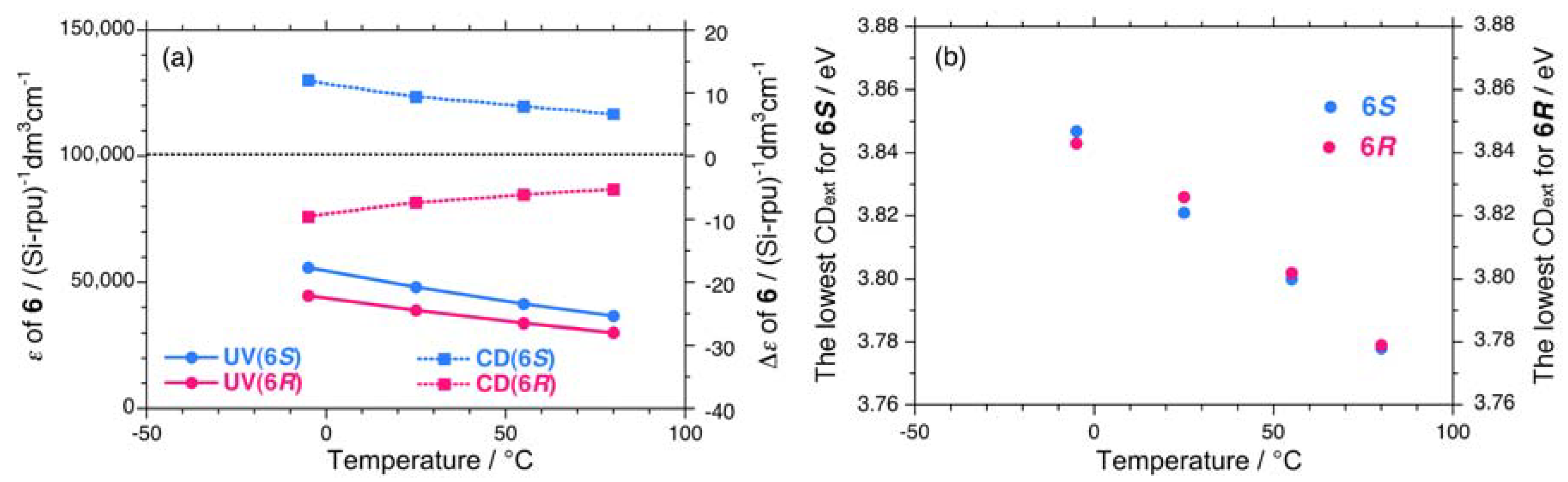
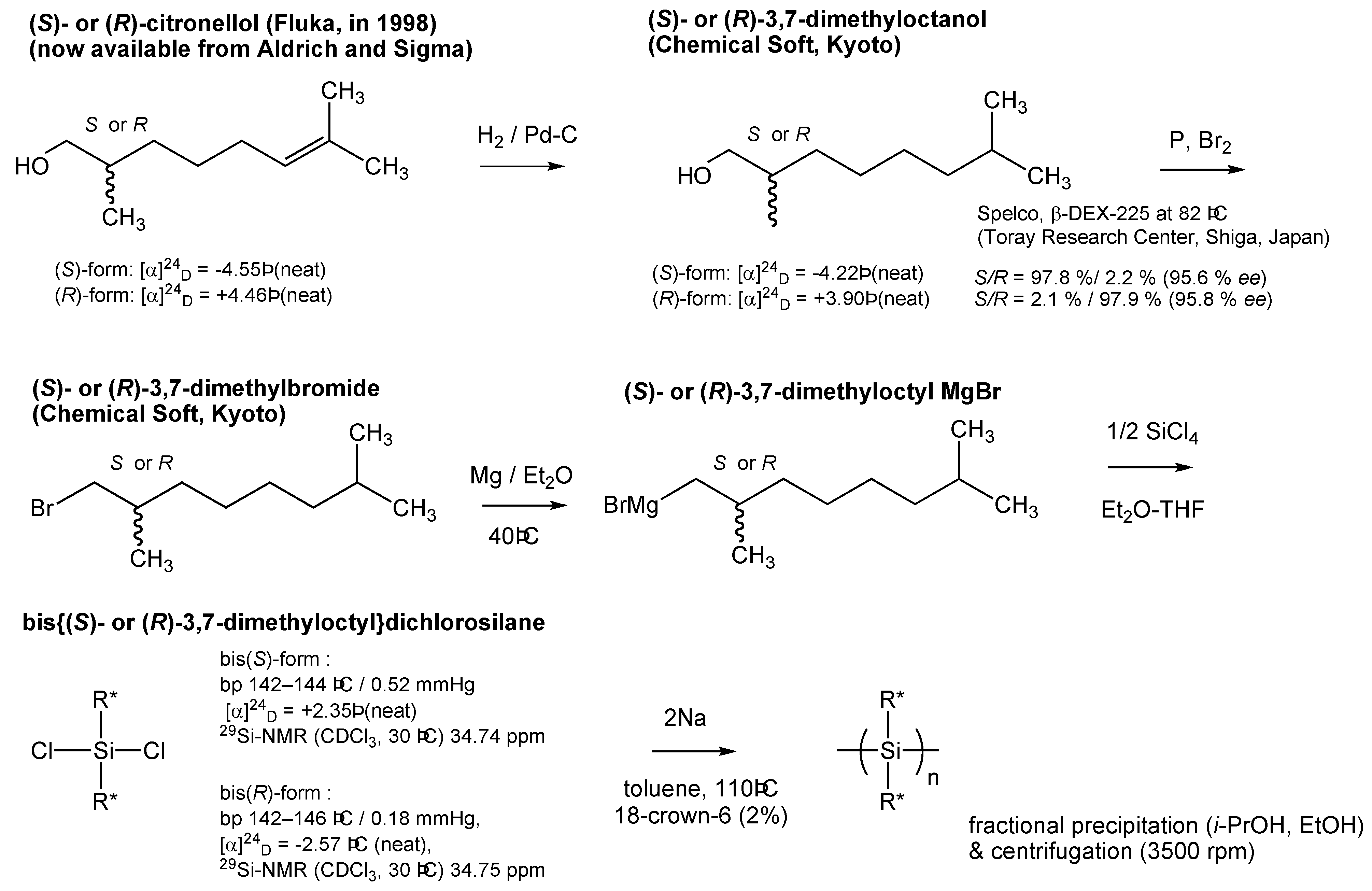
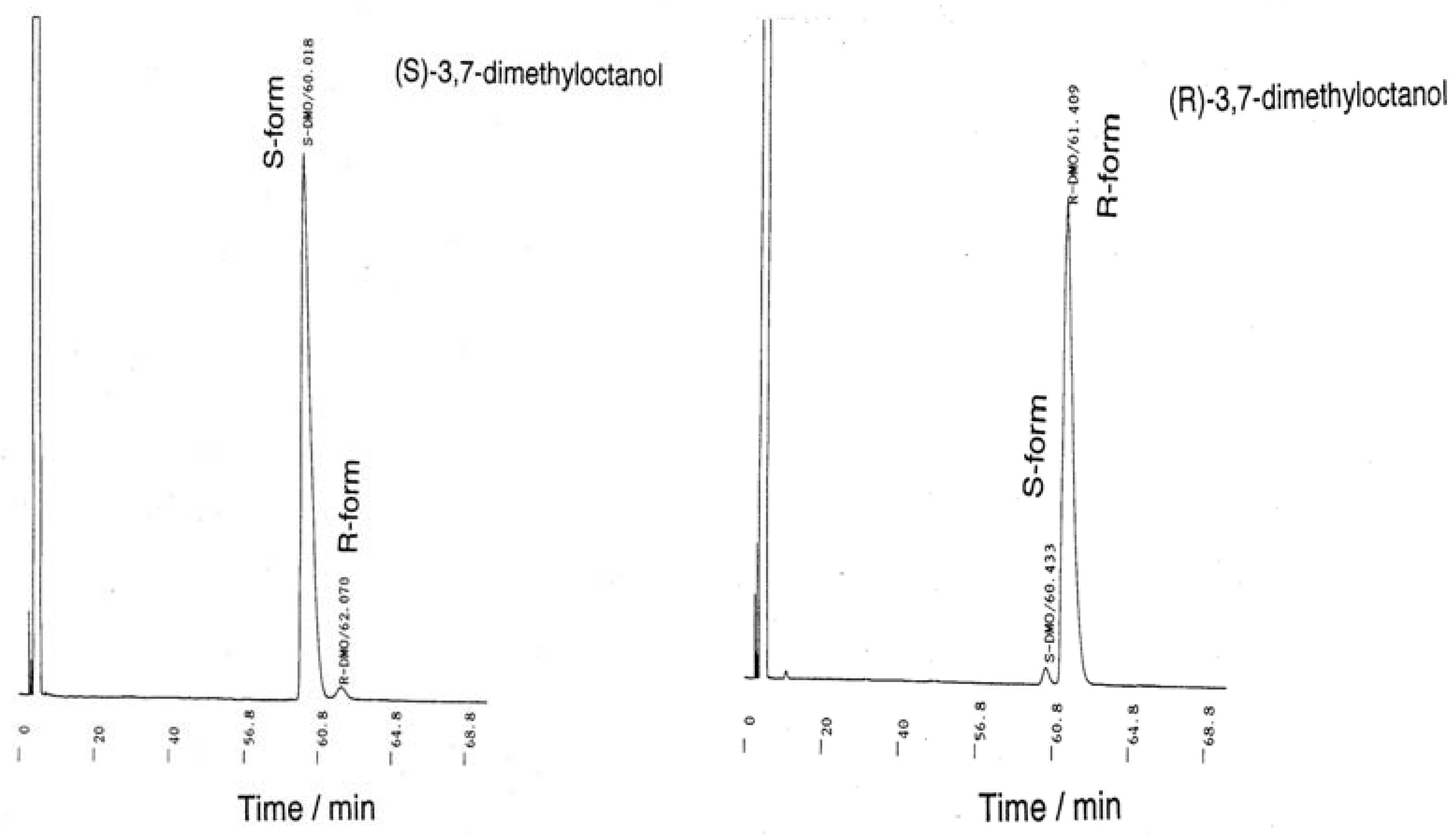

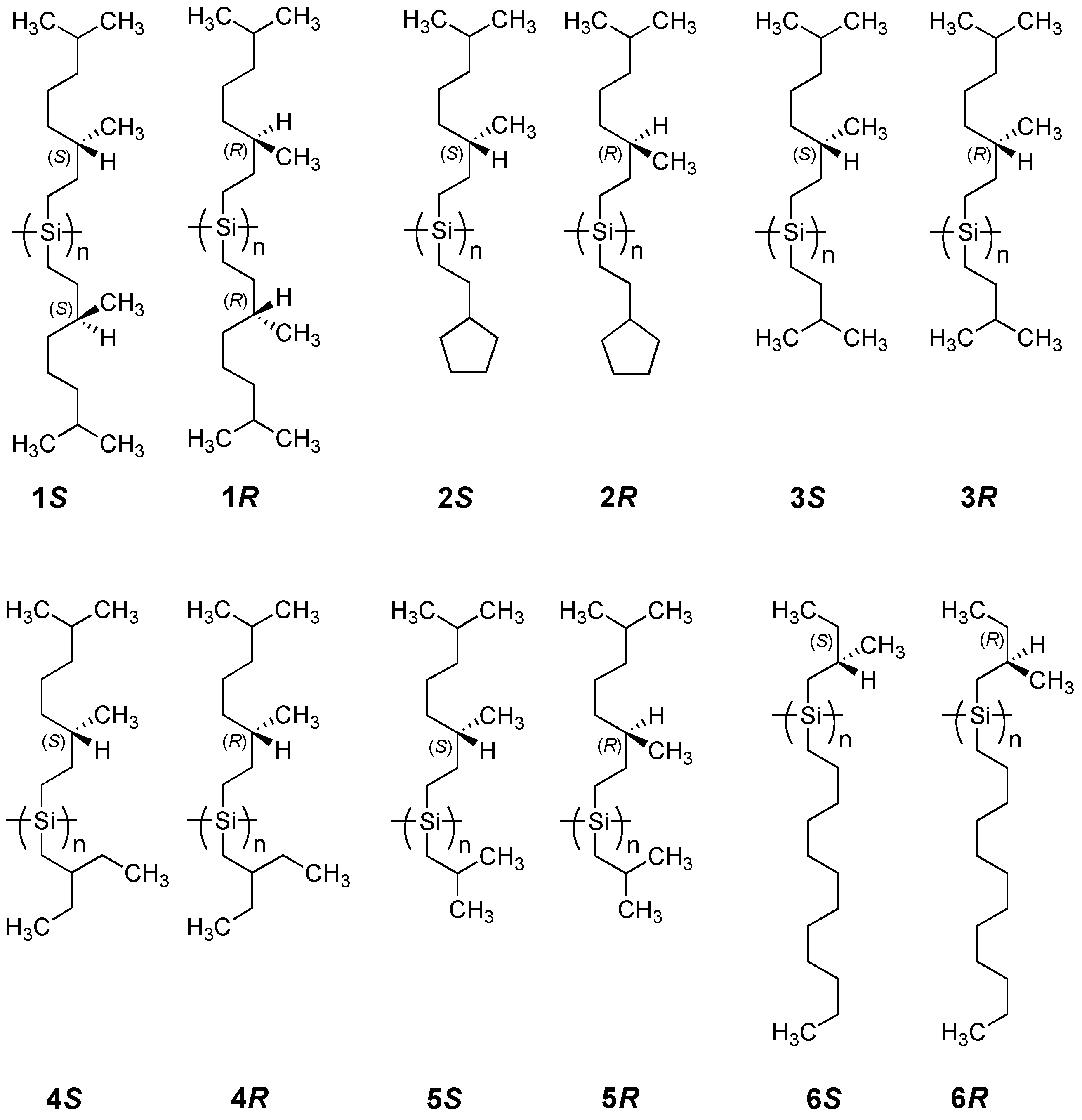
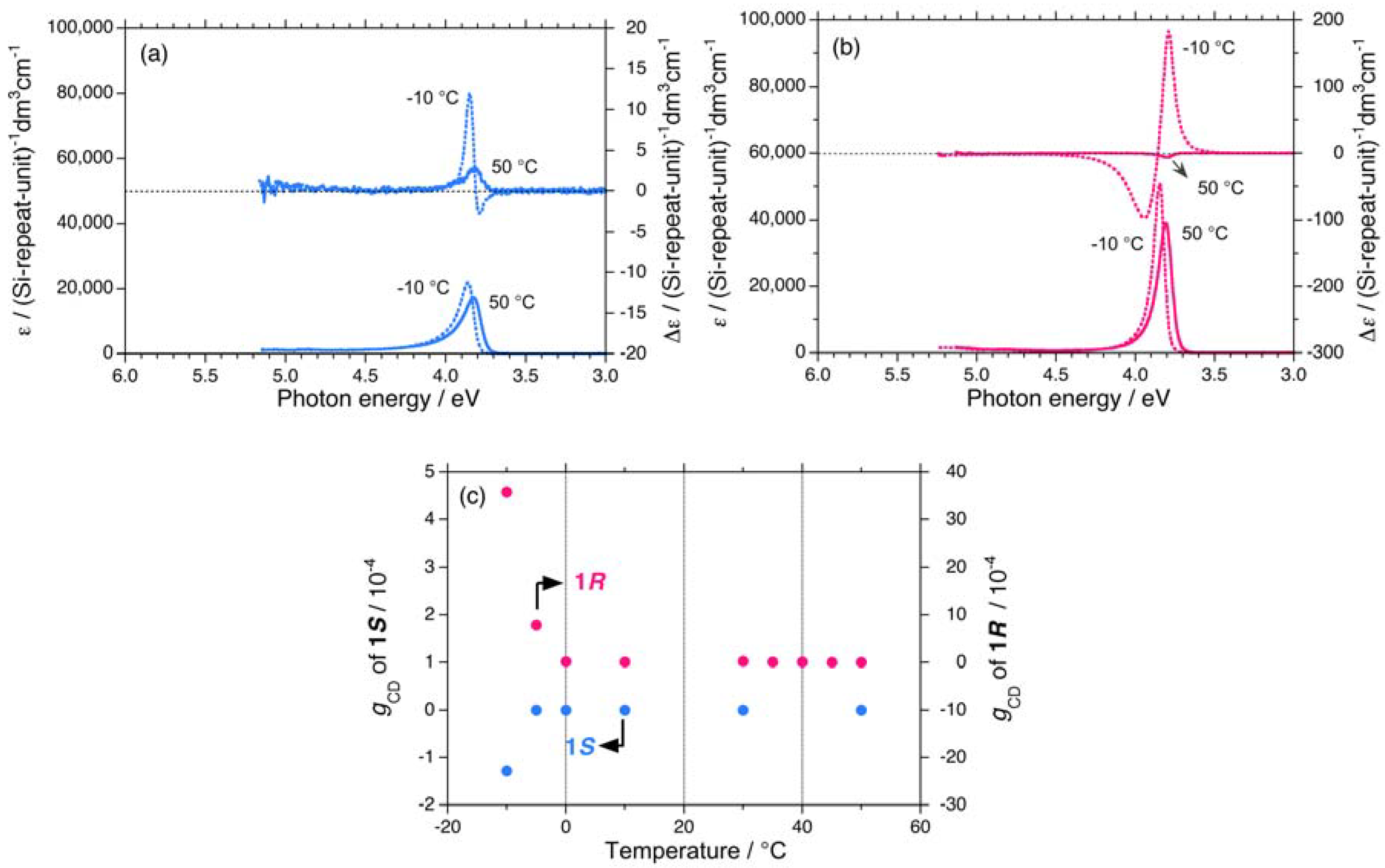
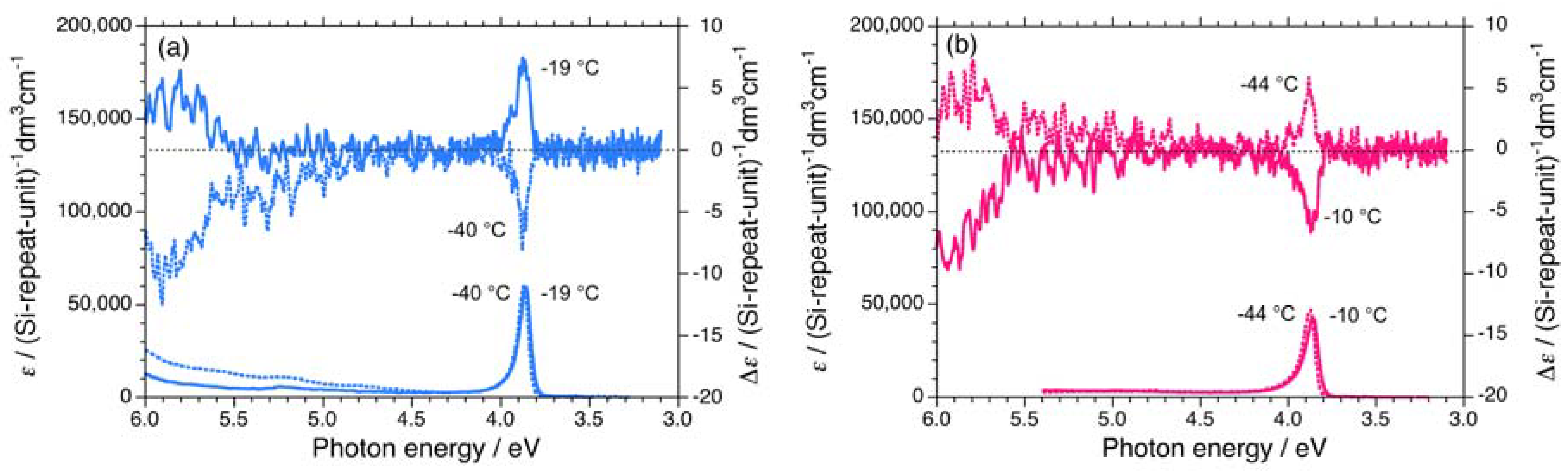
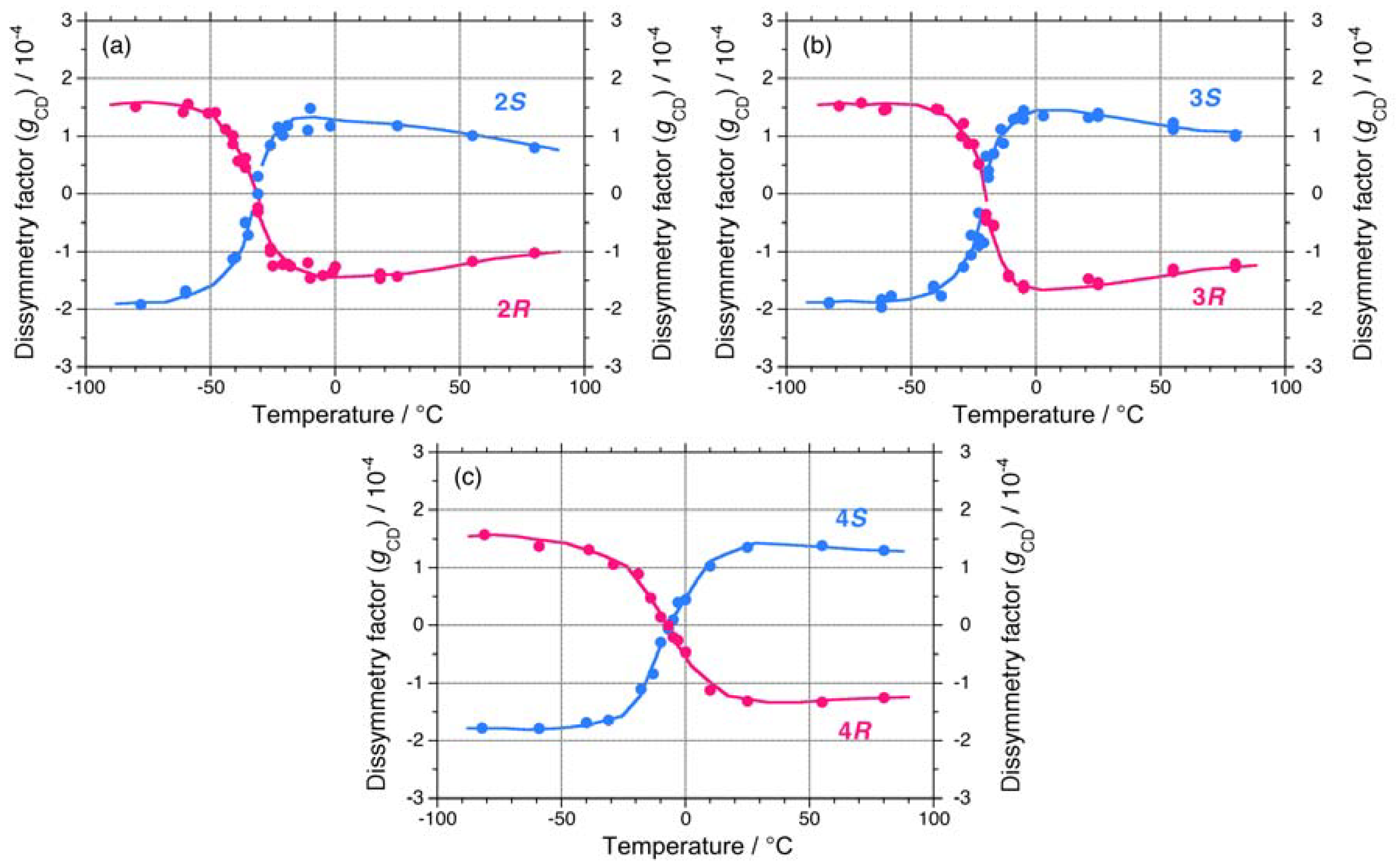
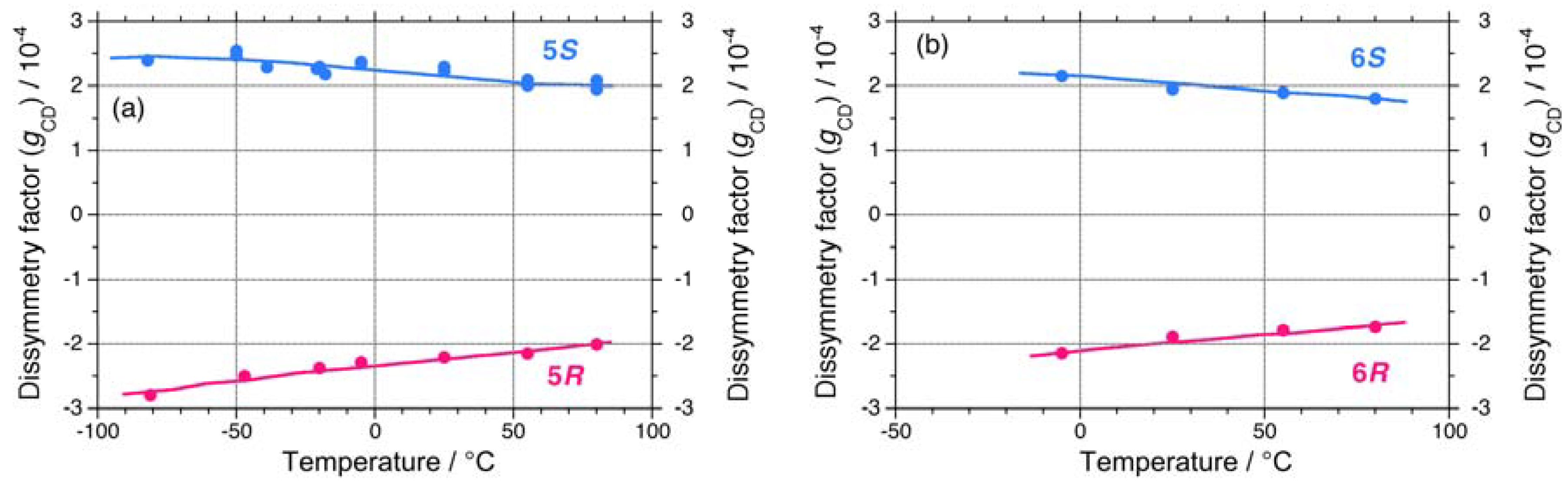


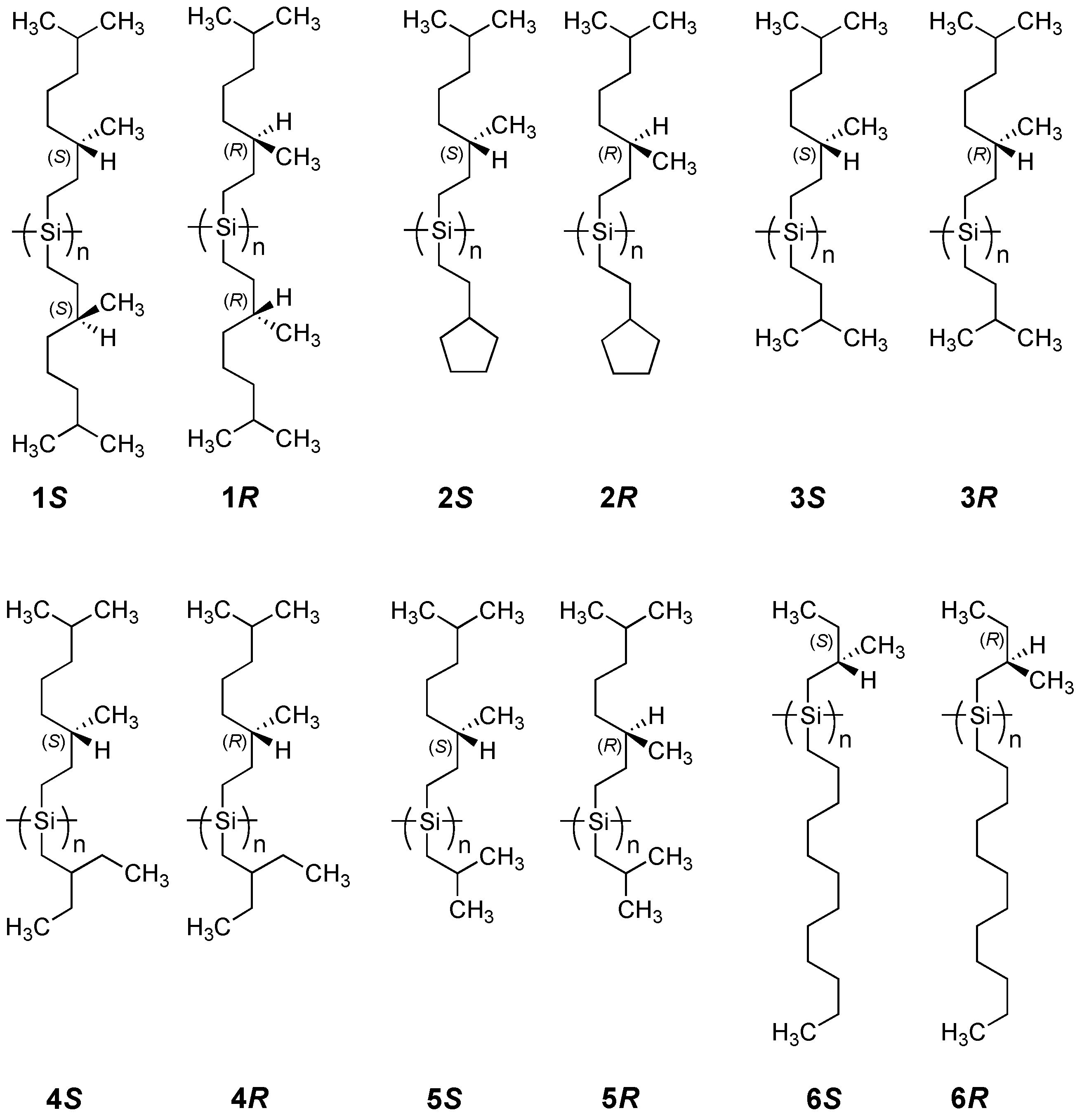{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tc / °C | gCD at –80 °C | gCD at +80 °C | |gPC| / gPV at –80 °C | |gPC| / gPV at +80 °C |
|---|---|---|---|---|---|
| 1S | –65 | –2.11 | +1.52 | 1.82 / –0.30 | 1.63 / –0.11 |
| 1R | –65 | +1.52 | –1.74 | ||
| 2S | –33 | –1.95 | +0.81 | 1.74 / –0.21 | 0.94 / –0.13 |
| 2R | –33 | +1.53 | –1.07 | ||
| 3S | –22 | –1.90 | +1.04 | 1.72 / –0.18 | 1.13 / –0.08 |
| 3R | –22 | +1.53 | –1.21 | ||
| 4S | –7 | –1.78 | +1.33 | 1.68 / –0.10 | 1.30 / +0.03 |
| 4R | –7 | +1.58 | –1.26 | ||
| 5S | – | +2.36 | +1.95 | 2.57 / –0.21 | 1.98 / –0.02 |
| 5R | – | –2.78 | –2.00 | ||
| 6S | – | – | +1.79 | – | – |
| 6R | – | – | –1.71 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fujiki, M. Mirror Symmetry Breaking in Helical Polysilanes: Preference between Left and Right of Chemical and Physical Origin. Symmetry 2010, 2, 1625-1652. https://doi.org/10.3390/sym2031625
Fujiki M. Mirror Symmetry Breaking in Helical Polysilanes: Preference between Left and Right of Chemical and Physical Origin. Symmetry. 2010; 2(3):1625-1652. https://doi.org/10.3390/sym2031625
Chicago/Turabian StyleFujiki, Michiya. 2010. "Mirror Symmetry Breaking in Helical Polysilanes: Preference between Left and Right of Chemical and Physical Origin" Symmetry 2, no. 3: 1625-1652. https://doi.org/10.3390/sym2031625
APA StyleFujiki, M. (2010). Mirror Symmetry Breaking in Helical Polysilanes: Preference between Left and Right of Chemical and Physical Origin. Symmetry, 2(3), 1625-1652. https://doi.org/10.3390/sym2031625