Diastereoselective Desymmetrization of Symmetric Dienes and its Synthetic Application

Abstract
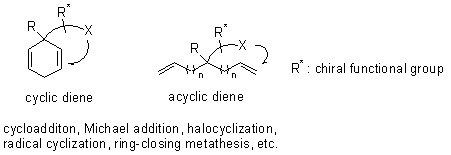
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Desymmetrization of diene substrates by diastereotopic group differentiating reactions
2.1. Cycloaddition
2.2. Michael addition
2.3. Iodocyclization
2.4. Radical cyclization / Epoxidation-cyclization
2.5. Ring-closing metathesis
2.6. Miscellaneous reactions
2.7. Asymmetric desymmetrization using C2-symmetric acetal or aminal
3. Conclusions
References
- Willis, M.C. Enantioselective desymmetrization. J. Chem. Soc. Perkin Trans. 1 1999, 1765–1784. [Google Scholar] [CrossRef]
- Hoffmann, R.W. Stereoselective synthesis using diastereotopic groups. Synthesis 2004, 2075–2090. [Google Scholar] [CrossRef]
- Abd Rahman, N.; Landais, Y. Desymmetrization of cyclic dienes. An efficient strategy for natural products synthesis. Curr. Org. Chem. 2002, 6, 1369–1395. [Google Scholar] [CrossRef]
- Studer, A.; Schleth, F. Desymmetrization and diastereotopic group selection in 1,4-cyclohexadienes. Synlett 2005, 3033–3041. [Google Scholar] [CrossRef]
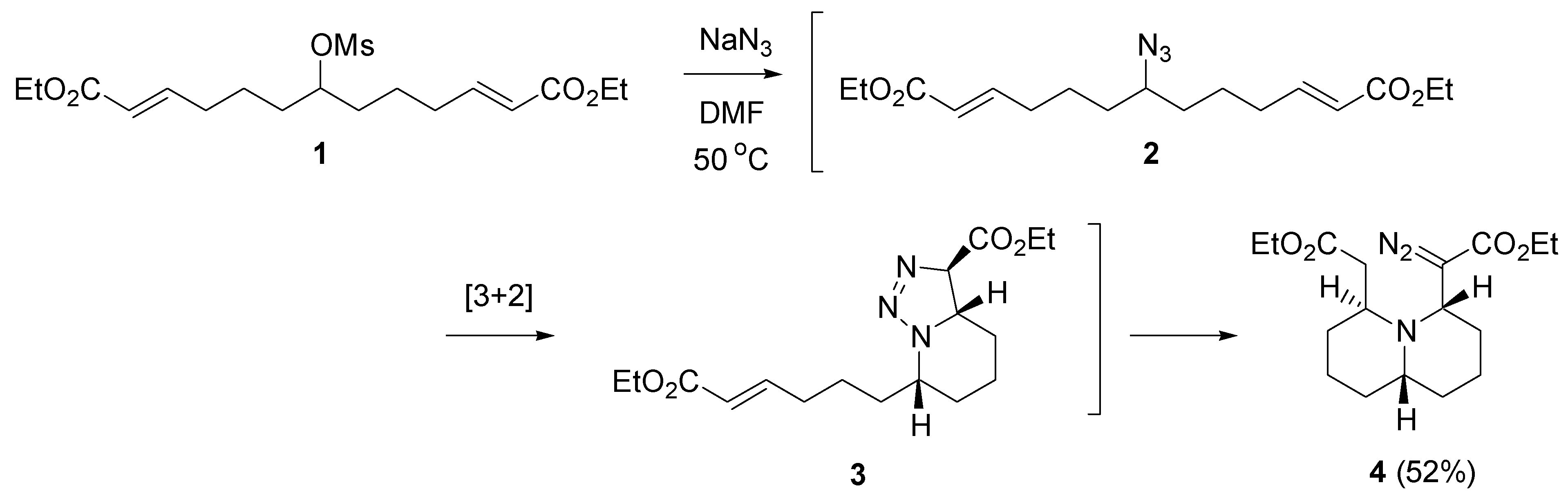
- Rejzek, M.; Stockman, R.A.; van Maarseveen, J.H.; Hughes, D.L. Combining two-directional synthesis and tandem reactions: Desymmetrisation by intramolecular cycloaddition/triazoline fragmentation. Chem. Commun. 2005, 4661–4662. [Google Scholar] [CrossRef] [PubMed]
- Azzi, N.; Griffen, E.; Light, M.; Linclau, B. An enantioselective desymmterisation approach to C9-substituted trans-hydrindene rings based on a diastereotopic group-selective intramolecular Diels-Alder reaction. Chem. Commun. 2006, 4909–4911. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.C.; El Sayed, N.N.E.; Ooi, L.-l. Sulfoxide-directed desymmetrisation of cyclohexa-1,4-dienes. Tetrahedron Lett. 2007, 48, 4561–4564. [Google Scholar] [CrossRef]
- Merino, E.; Melo, R.P.A.; Ortega-Guerra, M.; Ribagorda, M.; Carreño, M.C. Stereocontrolled approach to phenyl cyclitols from (SR)-[(p-tolylsulfinyl)methyl]-p-quinol. J. Org. Chem. 2009, 74, 2824–2831. [Google Scholar] [CrossRef]
- Butters, M.; Elliott, M.C.; Hill-Cousins, J.; Paine, J.S.; Walker, J.K.E. Iodocyclization reactions for the desymmetrization of cyclohexa-1,4-dienes. Org. Lett. 2007, 9, 3635–3638. [Google Scholar] [CrossRef]
- Crich, D.; Krishnamurthy, V. Radical dearomatization of benzene leading to phenanthridine and phenanthridinone derivatives related to (±)-pancratistatin. Tetrahedron 2006, 62, 6830–6840. [Google Scholar] [CrossRef]
- Danishefsky, S.; Lee, J.Y. Total synthesis of (±)-pancratistatin. J. Am. Chem. Soc. 1989, 111, 4829–4837. [Google Scholar] [CrossRef]
- Elliott, M.C.; Paine, J.S. Studies towards the total synthesis of lycoposerramine A. Synthesis of a model for the tetracyclic core. Org. Biomol. Chem. 2009, 7, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.C.; El Sayed, N.N.E.; Paine, J.S. Concise synthesis of the tricyclic core of lycoposerramine S. Org. Biomol. Chem. 2008, 6, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
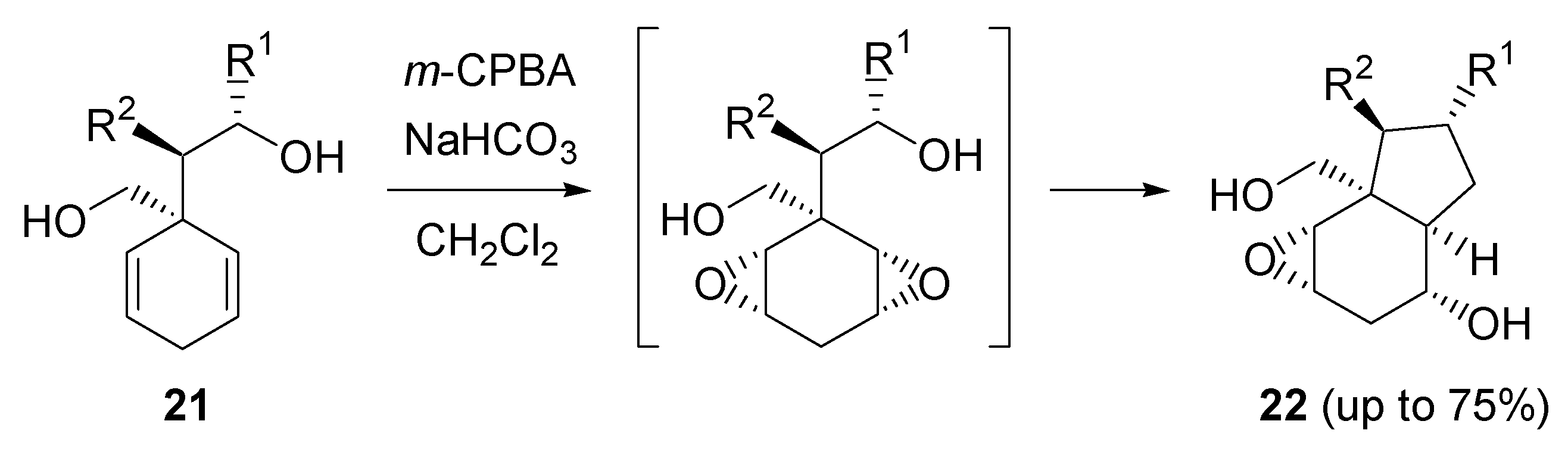
- Butters, M.; Beetstra, D.J.; Elliott, M.C.; Hill-Cousins, J.; Kariuki, B.M. Directed epoxidation of cyclohexa-1,4-dienes–stereoselective formation of up to six contiguous stereogenic centres. Org. Biomol. Chem. 2008, 6, 4426–4434. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Cowden, C.J.; Kennedy, D.J.; Ashwood, M.S.; Cottrell, I.F.; Dolling, U.-H. Stereoselective double ring closing metathesis reactions in the synthesis of spirocyclic compounds. Tetrahedron Lett. 2000, 41, 2027–2029. [Google Scholar] [CrossRef]
- Wallace, D.J.; Goodman, J.M.; Kennedy, D.J.; Davies, A.J.; Cowden, C.J.; Ashwood, M.S.; Cottrell, I.F.; Dolling, U.-H.; Reider, P.J. A double ring closing metathesis reaction in the rapid, enantioselective synthesis of NK-1 receptor antagonists. Org. Lett. 2001, 3, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J. On the mechanism of a double ring-closing metathesis reaction. Tetrahedron Lett. 2005, 46, 591–594. [Google Scholar] [CrossRef]
- Wallace, D.J. Exploiting catalyst characteristics: A protocol for increasing diastereoselectivity in a double ring-closing metathesis reaction. J. Mol. Catal. A: Chem. 2006, 254, 78–84. [Google Scholar] [CrossRef]
- Grimm, J.B.; Lee, D. Efficient synthesis of alkynylsilyl ethers and silaketals via base-induced alkynylsilane alcoholysis. J. Org. Chem. 2004, 69, 8967–8970. [Google Scholar] [CrossRef]
- Grimm, J.B.; Otte, R.D.; Lee, D. Tandem dienyne ring-closing metathesis of alkynyl silaketals for the formation of bicyclic siloxanes. J. Organomet. Chem. 2005, 690, 5508–5516. [Google Scholar] [CrossRef]
- Dunne, K.S.; Bisaro, F.; Odell, B.; Paris, J.-M.; Gouverneur, V. Diastereoselective ring-closing metathesis: Synthesis of P-stereogenic phosphinates from prochiral phosphinic acid derivatives. J. Org. Chem. 2005, 70, 10803–10809. [Google Scholar] [CrossRef] [PubMed]
- Böhrsch, V.; Neidhöfer, J.; Blechert, S. Diastereoselective ring-rearrangement metathesis. Angew. Chem., Int. Ed. 2006, 45, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Hooper, A.M.; Dufour, S.; Willaert, S.; Pouvreau, S.; Pickett, J.A. Synthesis of (2S,7S)-dibutyroxynonane, the sex pheromone of the orange wheat blossom midge, Sitodiplosis mosellana (Géhin) (Diptera: Cecidomyiidae), by diastereoselective silicon-tethered ring-closing metathesis. Tetrahedron Lett. 2007, 48, 5991–5994. [Google Scholar] [CrossRef]
- Schleth, F.; Studer, A. Desymmetrization of metalated cyclohexadienes and application to the synthesis of nephrosteranic acid. Angew. Chem., Int. Ed. 2004, 43, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Schleth, F.; Vogler, T.; Harms, K.; Studer, A. Stereoselective synthesis of (+)-nephrosteranic acid, (+)-trans-cognac lactone, and (+)-trans-whisky lactone using a chiral cyclohexadienyl Ti compound. Chem. Eur. J. 2004, 10, 4171–4185. [Google Scholar] [CrossRef] [PubMed]
- Maji, M.S.; Fröhlich, R.; Studer, A. Desymmetrization of metallated cyclohexadienes with chiral N-tert-butanesulfinyl imines. Org. Lett. 2008, 10, 1847–1850. [Google Scholar] [CrossRef] [PubMed]
- Gromov, A.; Enev, V.; Mulzer, J. A desymmetrization approach toward highly oxygenated cis-decalins. Org. Lett. 2009, 11, 2884–2886. [Google Scholar] [CrossRef]
- Lebeuf, R.; Robert, F.; Schenk, K.; Landais, Y. Desymmetrization of cyclohexa-2,5-dienes through a diastereoselective protonation-hydroamination cascade. Org. Lett. 2006, 8, 4755–4758. [Google Scholar] [CrossRef]
- Beniazza, R.; Dunet, J.; Robert, F.; Schenk, K.; Landais, Y. Efficient synthetic approaches to the common scaffold of indole alkaloids. Org. Lett. 2007, 9, 3913–3916. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Yang, G.; Hu, Y.; Khire, U.R. Prins desymmetrization of a C2-symmetric diol: Application to the synthesis of 17-deoxyroflamycoin. J. Org. Chem. 1997, 62, 3022–3023. [Google Scholar] [CrossRef]
- Elliott, M.C.; El Sayed, N.N.E.; Paine, J.S. Diastereospecific tandem Prins cyclisation/rearrangement reactions for the desymmetrisation of cyclohexa-1,4-dienes. Eur. J. Org. Chem. 2007, 792–803. [Google Scholar] [CrossRef]
- Butters, M.; Elliott, M.C.; Hill-Cousins, J.; Paine, J.S.; Westwood, A.W.J. An improved protocol for the Prins desymmetrisation of cyclohexa-1,4-dienes. Tetrahedron Lett. 2008, 49, 4446–4448. [Google Scholar] [CrossRef]
- Rousseau, G.; Robert, F.; Schenk, K.; Landais, Y. Rearrangement of spirocyclic oxindoles with lithium amide bases. Org. Lett. 2008, 10, 4441–4444. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, G.; Robert, F.; Landais, Y. Functionalization and rearrangement of spirocyclohexadienyl oxindoles: Experimental and theoretical investigations. Chem. Eur. J. 2009, 15, 11160–11173. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Grant, D.; Wink, D.J. Expedient two-step synthesis of phenolic cyclitols from benzene. J. Org. Chem. 2006, 71, 4521–4524. [Google Scholar] [CrossRef] [PubMed]
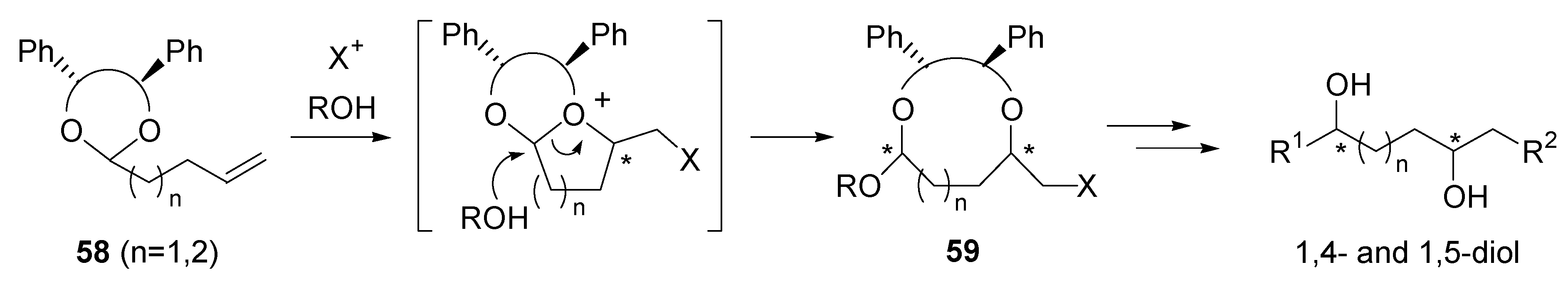
- Fujioka, H.; Kitagawa, H.; Matsunaga, N.; Nagatomi, Y.; Kita, Y. Remote asymmetric induction for chiral 1,4-diols using a chiral acetal derived from chiral hydrobenzoin. Tetrahedron Lett. 1996, 37, 2245–2248. [Google Scholar] [CrossRef]
- Fujioka, H.; Kitagawa, H.; Nagatomi, Y.; Kita, Y. Asymmetric induction via an intramolecular haloetherification reaction of chiral ene acetals: A novel approach to optically active 1,4- and 1,5-diols. J. Org. Chem. 1996, 61, 7309–7315. [Google Scholar] [CrossRef]
- Fujioka, H.; Ohba, Y.; Hirose, H.; Murai, K.; Kita, Y. Mild and efficient removal of hydroxyethyl unit from 2-hydroxyethyl ether derivatives leading to alcohols. Org. Lett. 2005, 7, 3303–3306. [Google Scholar] [CrossRef]
- Fujioka, H.; Hirose, H.; Ohba, Y.; Murai, K.; Nakahara, K.; Kita, Y. Cerium ammonium nitrate (CAN) for mild and efficient reagent to remove hydroxyethyl units from 2-hydroxyethyl ethers and 2-hydroxyethyl amines. Tetrahedron 2007, 63, 625–637. [Google Scholar] [CrossRef]
- Fujioka, H.; Kotoku, N.; Sawama, Y.; Nagatomi, Y.; Kita, Y. Concise asymmetric synthesis of a model compound, (4S,5S,6S)-6-(2,2-dimethoxy)ethyl-4,5-epoxy-6-hydroxy-2-cyclohexenone, for the cyclohexenone core of scyphostatin. Tetrahedron Lett. 2002, 43, 4825–4828. [Google Scholar] [CrossRef]
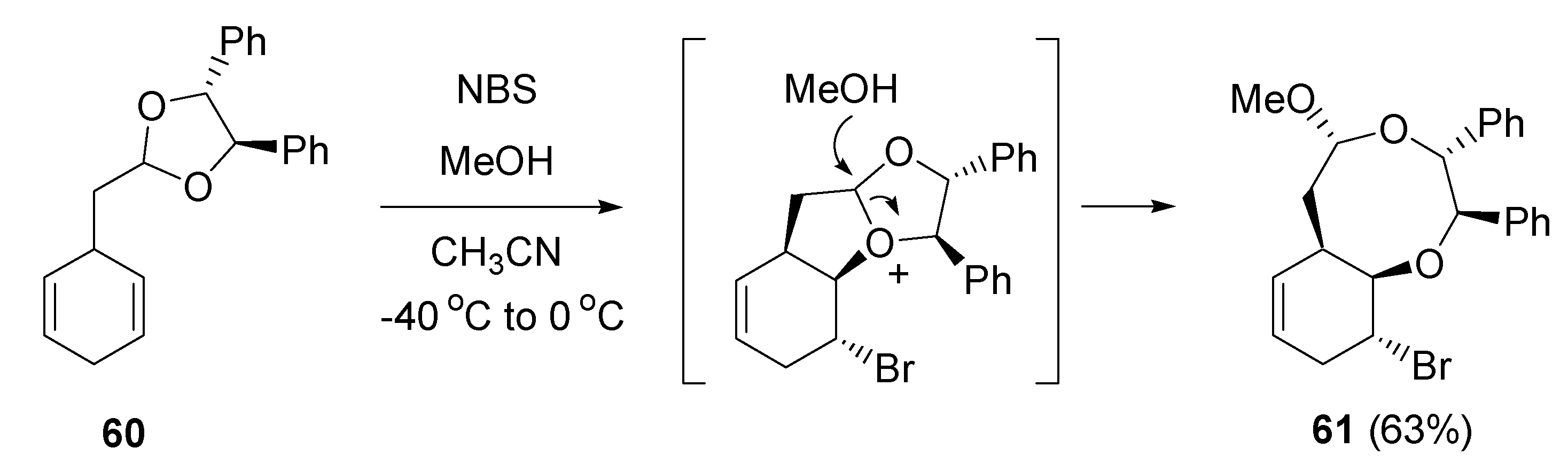
- Fujioka, H.; Kotoku, N.; Sawama, Y.; Kitagawa, H.; Ohba, Y.; Wang, T.-L.; Nagatomi, Y.; Kita, Y. Asymmetric synthesis by the intramolecular haloetherification reaction of ene acetal: Discrimination of prochiral dienes in cyclohexane systems. Chem. Pharm. Bull. 2005, 53, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Ohba, Y.; Nakahara, K.; Takatsuji, M.; Murai, K.; Ito, M.; Kita, Y. New perspective for natural products synthesis: Concise synthesis of (+)-Sch 642305 by chiral auxiliary multiuse methodology. Org. Lett. 2007, 9, 5605–5608. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Sawama, Y.; Kotoku, N.; Ohnaka, T.; Okitsu, T.; Murata, N.; Kubo, O.; Li, R.; Kita, Y. Concise asymmetric total synthesis of scyphostatin, a potent inhibitor of neutral sphingomyelinase. Chem. Eur. J. 2007, 13, 10225–10238. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Sawama, Y.; Murata, N.; Okitsu, T.; Kubo, O.; Matsuda, S.; Kita, Y. Unexpected highly chemoselective deprotection of the acetals from aldehydes and not ketones: TESOTf–2,6-lutidine combination. J. Am. Chem. Soc. 2004, 126, 11800–11801. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Okitsu, T.; Sawama, Y.; Murata, N.; Li, R.; Kita, Y. Reaction of the acetals with TESOTf–base combination; Speculation of the intermediates and efficient mixed acetal formation. J. Am. Chem. Soc. 2006, 128, 5930–5938. [Google Scholar] [CrossRef] [PubMed]
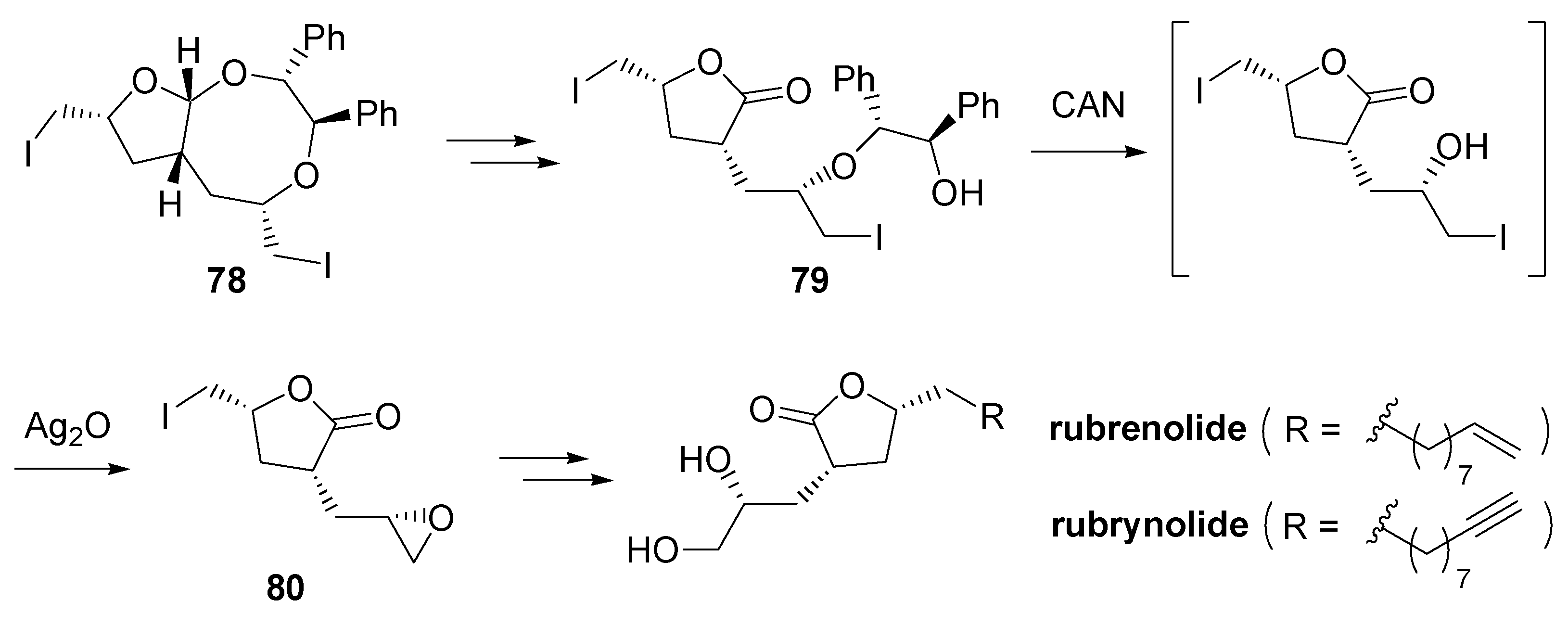
- Fujioka, H.; Ohba, Y.; Hirose, H.; Murai, K.; Kita, Y. A double iodoetherification of σ-symmetric diene acetals for installing four stereogenic centers in a single operation: Short asymmetric total synthesis of rubrenolide. Angew. Chem., Int. Ed. 2005, 44, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Ohba, Y.; Hirose, H.; Nakahara, K.; Murai, K.; Kita, Y. Facile formation of tetrahydrofurans with multiple chiral centers using double iodoetherification of σ-symmetric diene acetals: Short asymmetric total synthesis of rubrenolide and rubrynolide. Tetrahedron 2008, 64, 4233–4245. [Google Scholar] [CrossRef]
- Takano, S.; Murakata, C.; Imamura, Y.; Tamura, N.; Ogasawara, K. Chiral route to some alkaloids through asymmetric iodolactonization. Heterocycles 1981, 16, 1291–1294. [Google Scholar] [CrossRef]
- Hart, D.J.; Huang, H.-C.; Krishnamurthy, R.; Schwartz, T. Free-radical cyclizations: Application to the total synthesis of dl-pleurotin and dl-dihydropleurotin acid. J. Am. Chem. Soc. 1989, 111, 7507–7519. [Google Scholar] [CrossRef]
- Fuji, K.; Node, M.; Naniwa, Y.; Kawabata, T. Enantioselective iodolactonization through diastereotopic group differentiation. Tetrahedron Lett. 1990, 31, 3175–3178. [Google Scholar]
- Yokomatsu, T.; Iwasawa, H.; Shibuya, S. Highly enantioselective creation of quaternary carbons in a halolactonization of bis-γ,δ-unsaturated carboxylic imides derived from a camphorsultam: Enantioselective synthesis of (+)-mesembrine. Tetrahedron Lett. 1992, 33, 6999–7002. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Iwasawa, H.; Shibuya, S. Enantioselective halolactonisation of bis-γ,δ-unsaturated carboxylic acid derivatives: Use of a sultam and oxazolidine-2-ones as chiral auxiliary. J. Chem. Soc. Chem. Commun. 1992, 728–729. [Google Scholar] [CrossRef]
- Kitagawa, O.; Momose, S.; Fushimi, Y.; Taguchi, T. Efficient synthesis of various atropisomeric amides in optically pure forms and their application to asymmetric reactions. Tetrahedron Lett. 1999, 40, 8827–8831. [Google Scholar] [CrossRef]
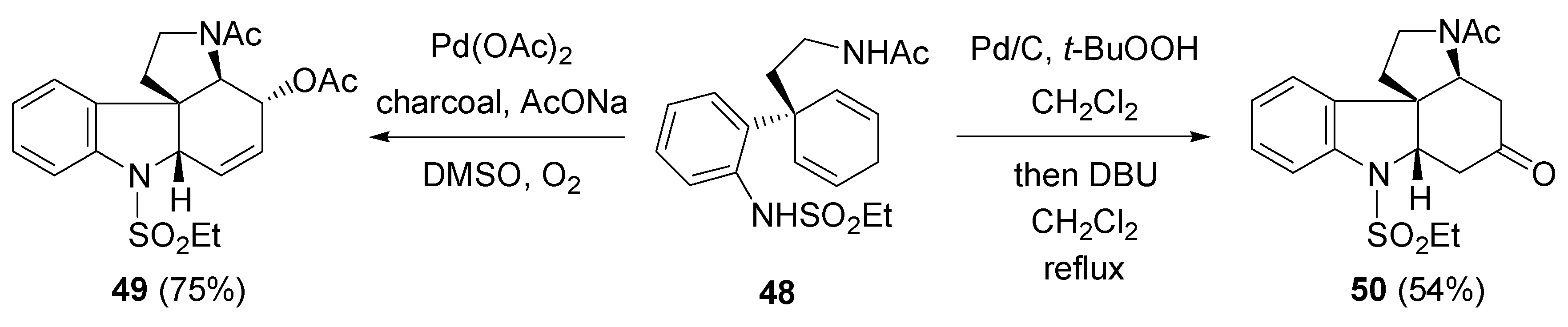
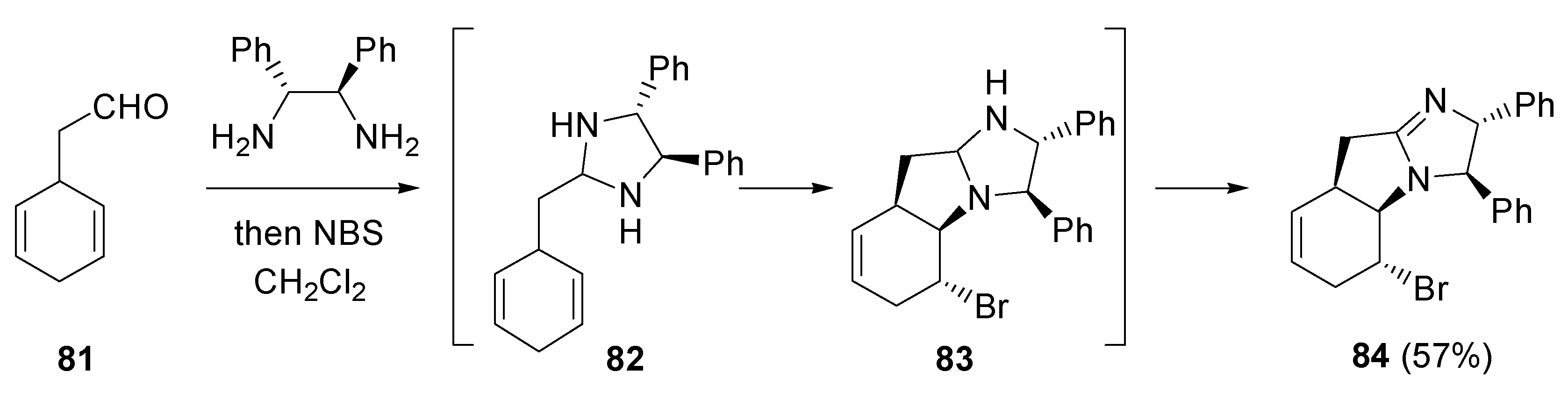
- Fujioka, H.; Murai, K.; Ohba, Y.; Hirose, H.; Kita, Y. Intramolecular bromo-aminaion of 1,4-cyclohexadiene aminal: one-pot discrimination of two olefins and concise asymmetric synthesis of (–)-γ-lycorane. Chem. Commun. 2006, 832–834. [Google Scholar] [CrossRef] [PubMed]

































© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakahara, K.; Fujioka, H. Diastereoselective Desymmetrization of Symmetric Dienes and its Synthetic Application. Symmetry 2010, 2, 437-454. https://doi.org/10.3390/sym2020437
Nakahara K, Fujioka H. Diastereoselective Desymmetrization of Symmetric Dienes and its Synthetic Application. Symmetry. 2010; 2(2):437-454. https://doi.org/10.3390/sym2020437
Chicago/Turabian StyleNakahara, Kenji, and Hiromichi Fujioka. 2010. "Diastereoselective Desymmetrization of Symmetric Dienes and its Synthetic Application" Symmetry 2, no. 2: 437-454. https://doi.org/10.3390/sym2020437
APA StyleNakahara, K., & Fujioka, H. (2010). Diastereoselective Desymmetrization of Symmetric Dienes and its Synthetic Application. Symmetry, 2(2), 437-454. https://doi.org/10.3390/sym2020437