

Symmetries of Confined H2+ Molecule



Abstract
1. Introduction
2. H-Confinement in Polyhedral and Cylindrical Wells
3. Numerical Method
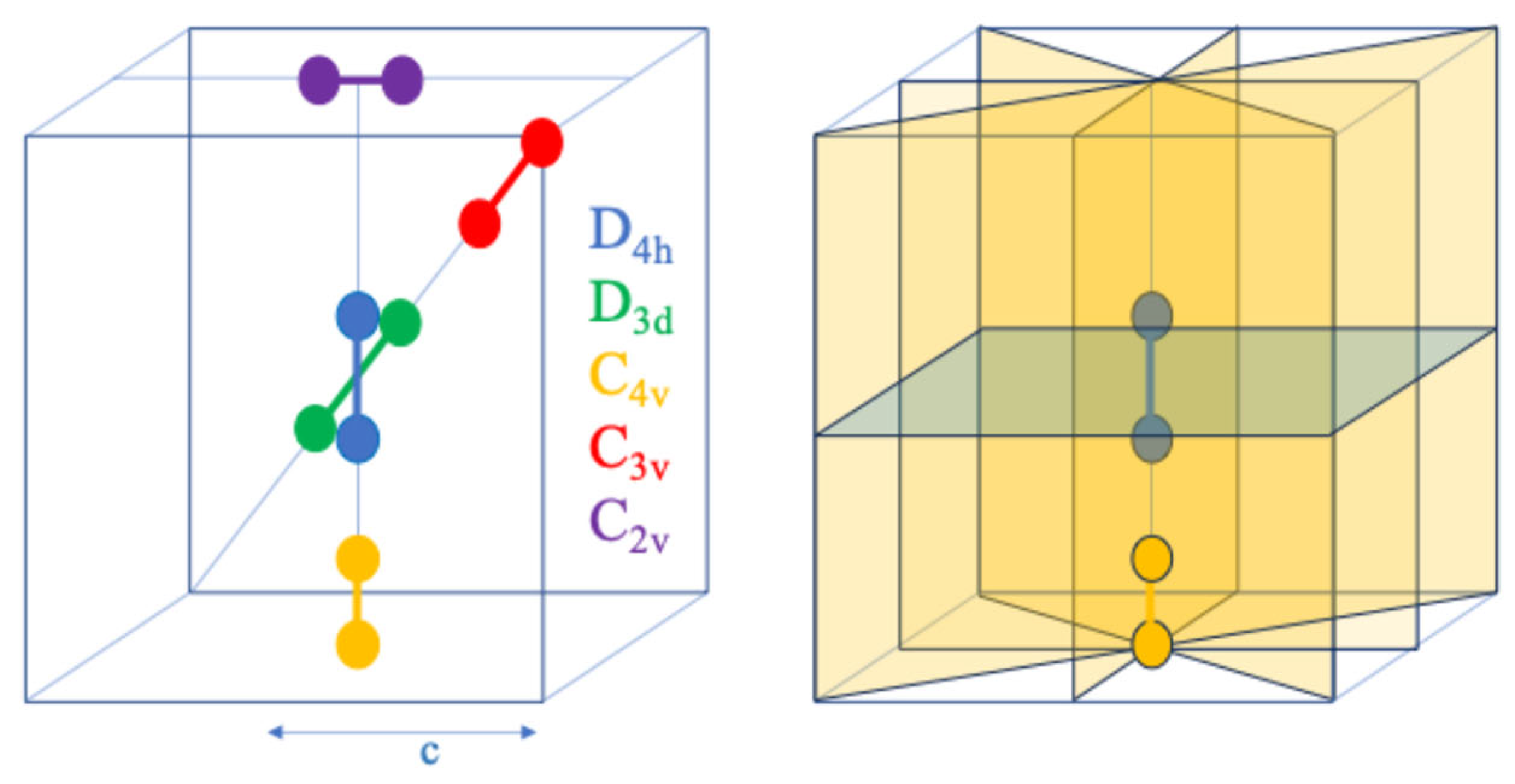
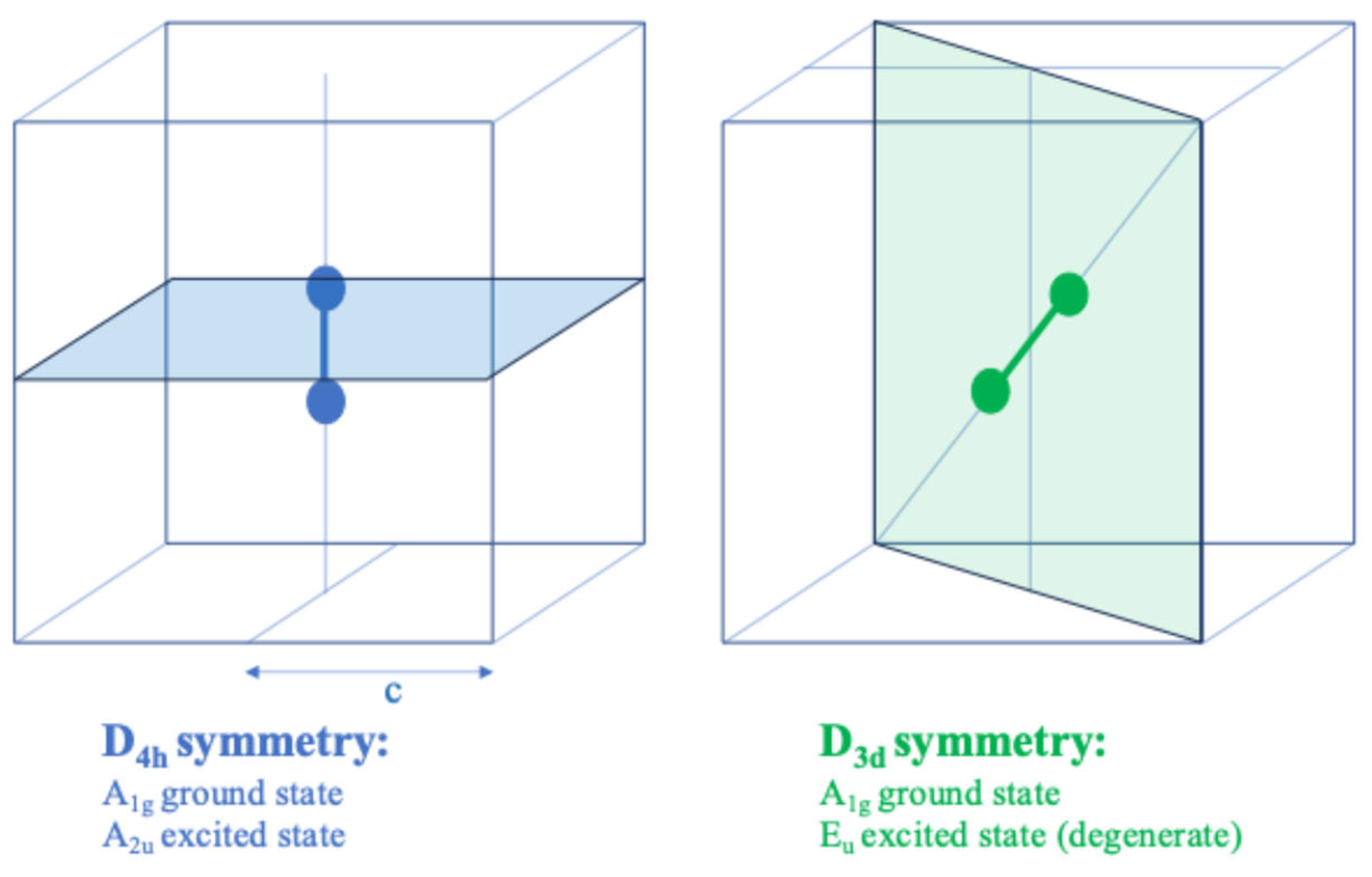
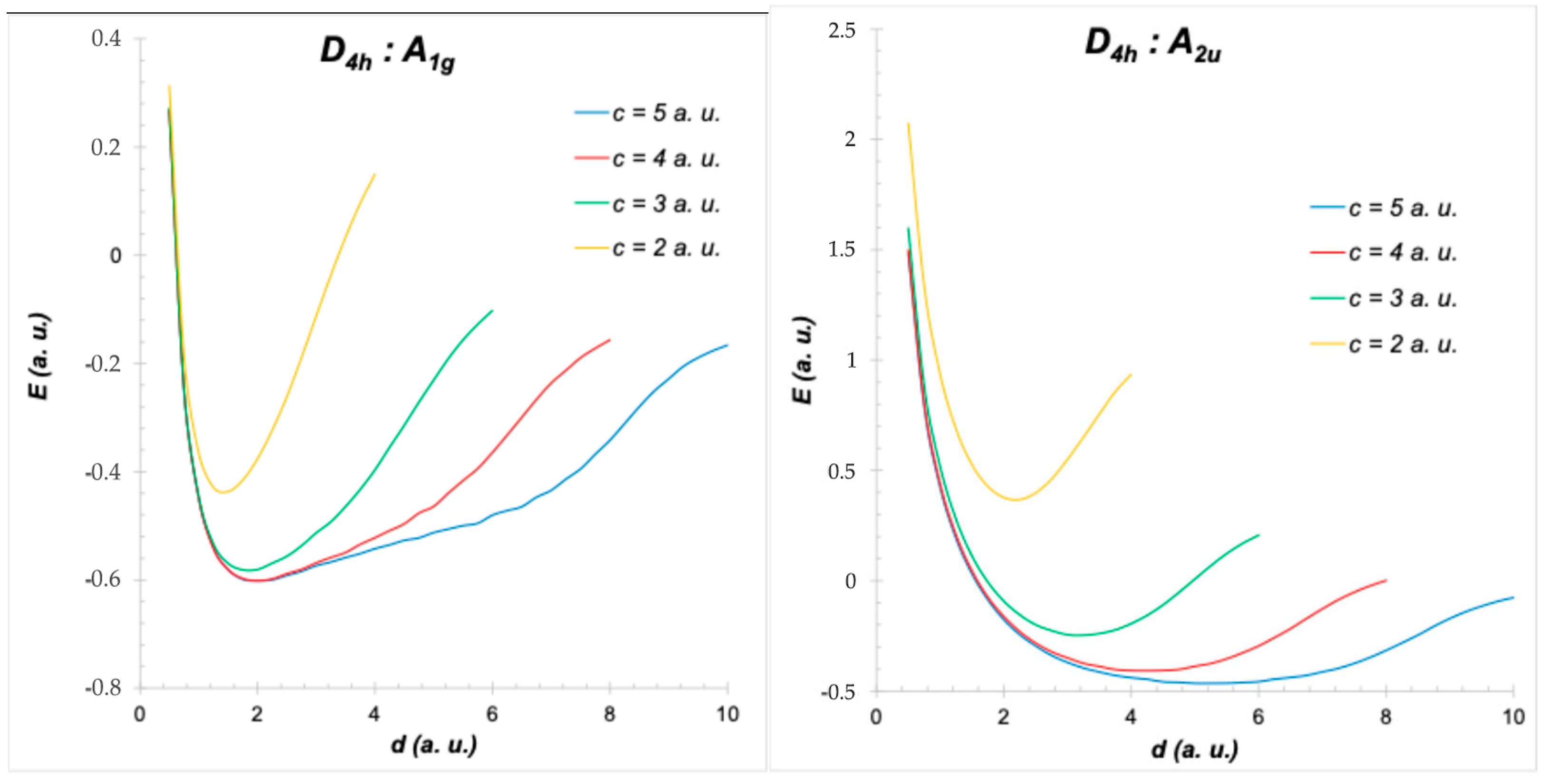
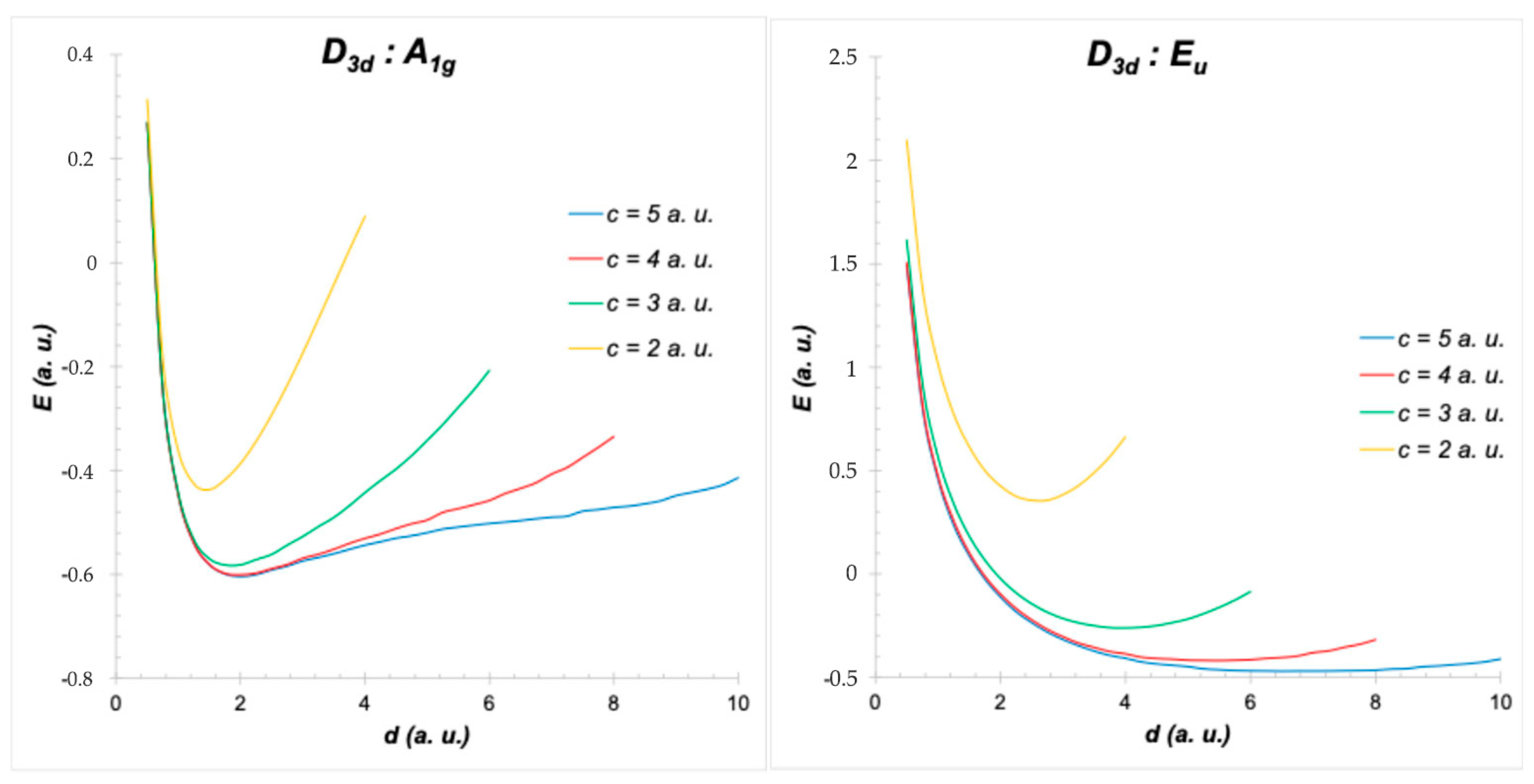
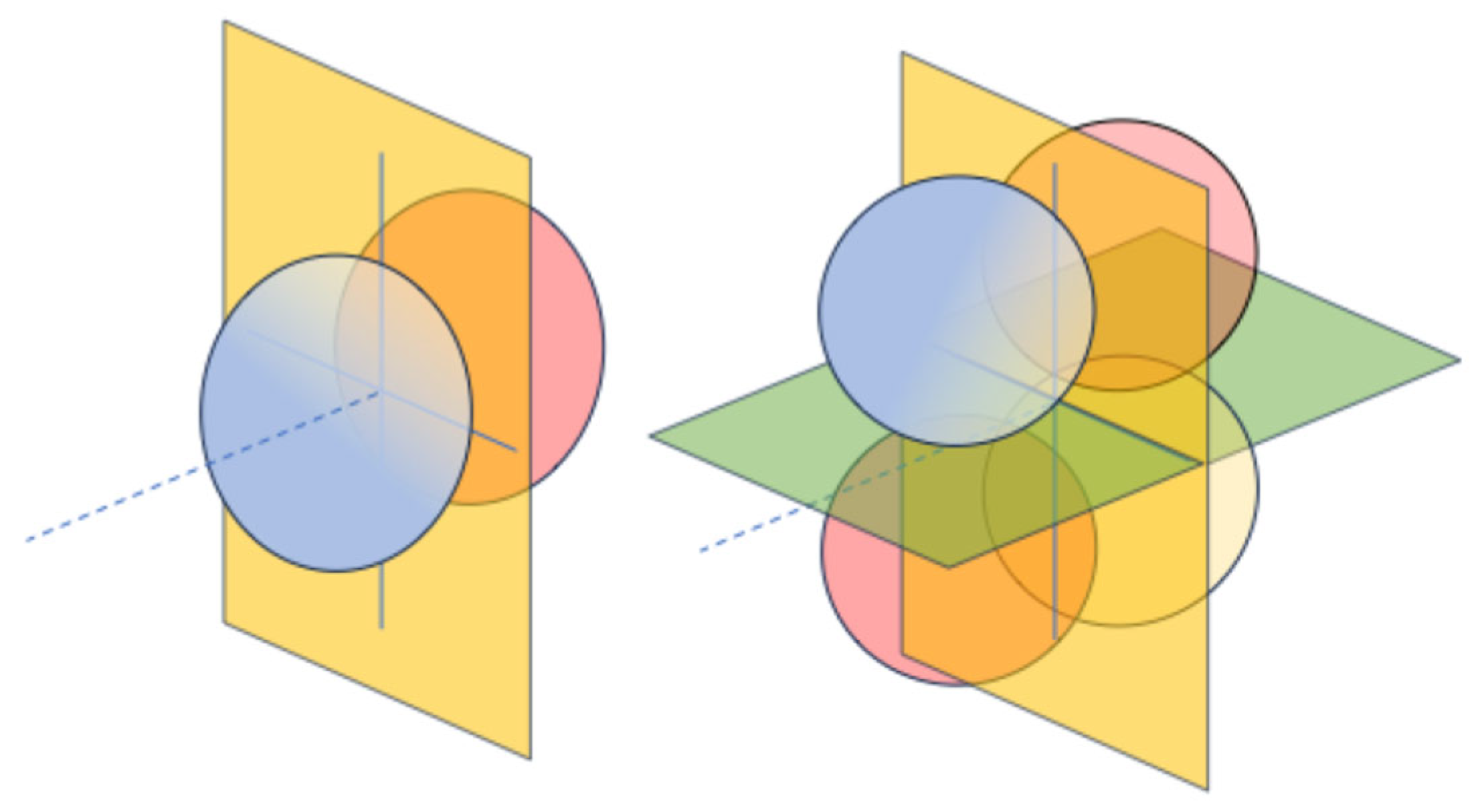
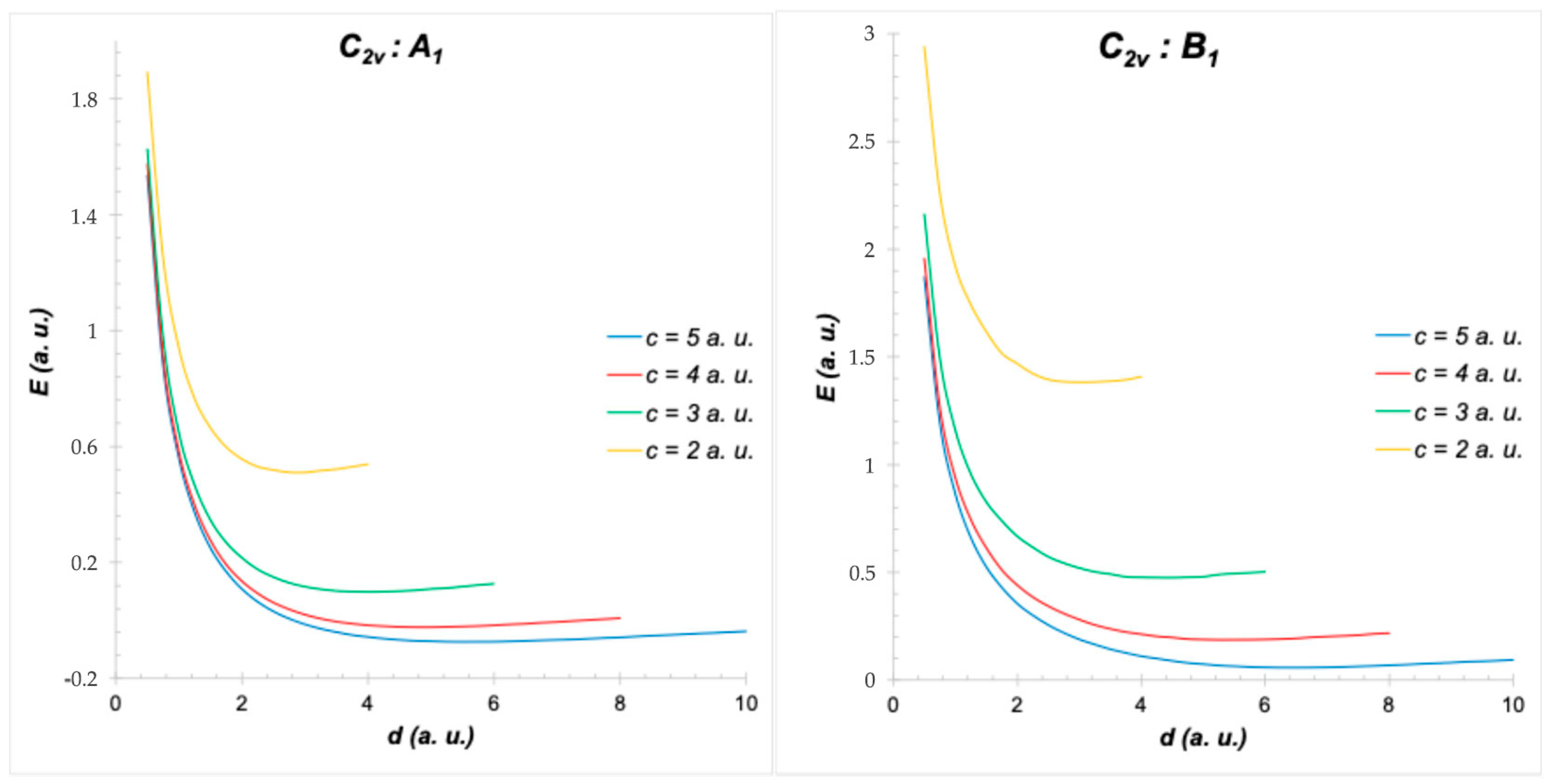
4. Symmetry of H2+ in Different Positions of a Cubic Cavity
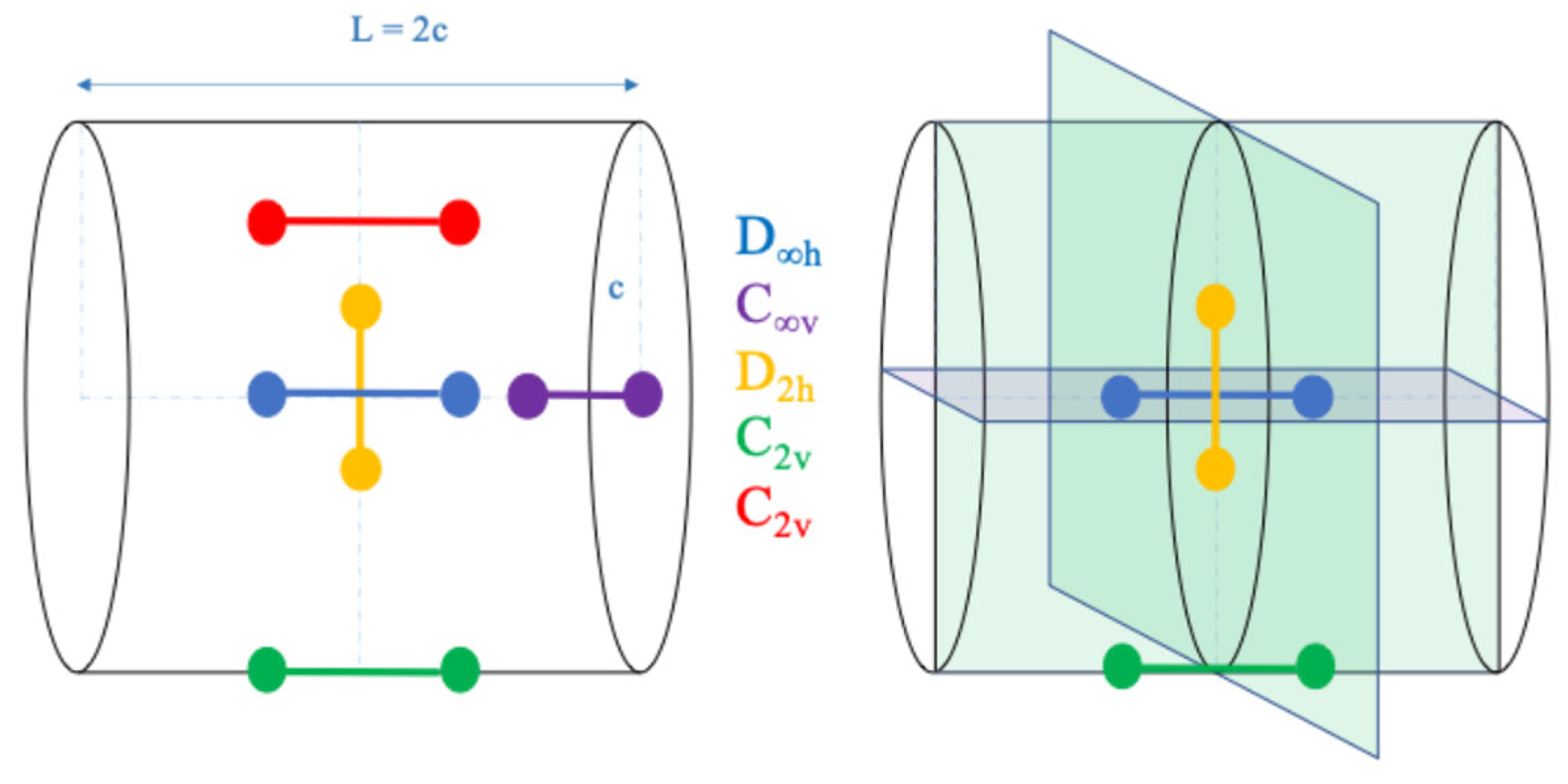
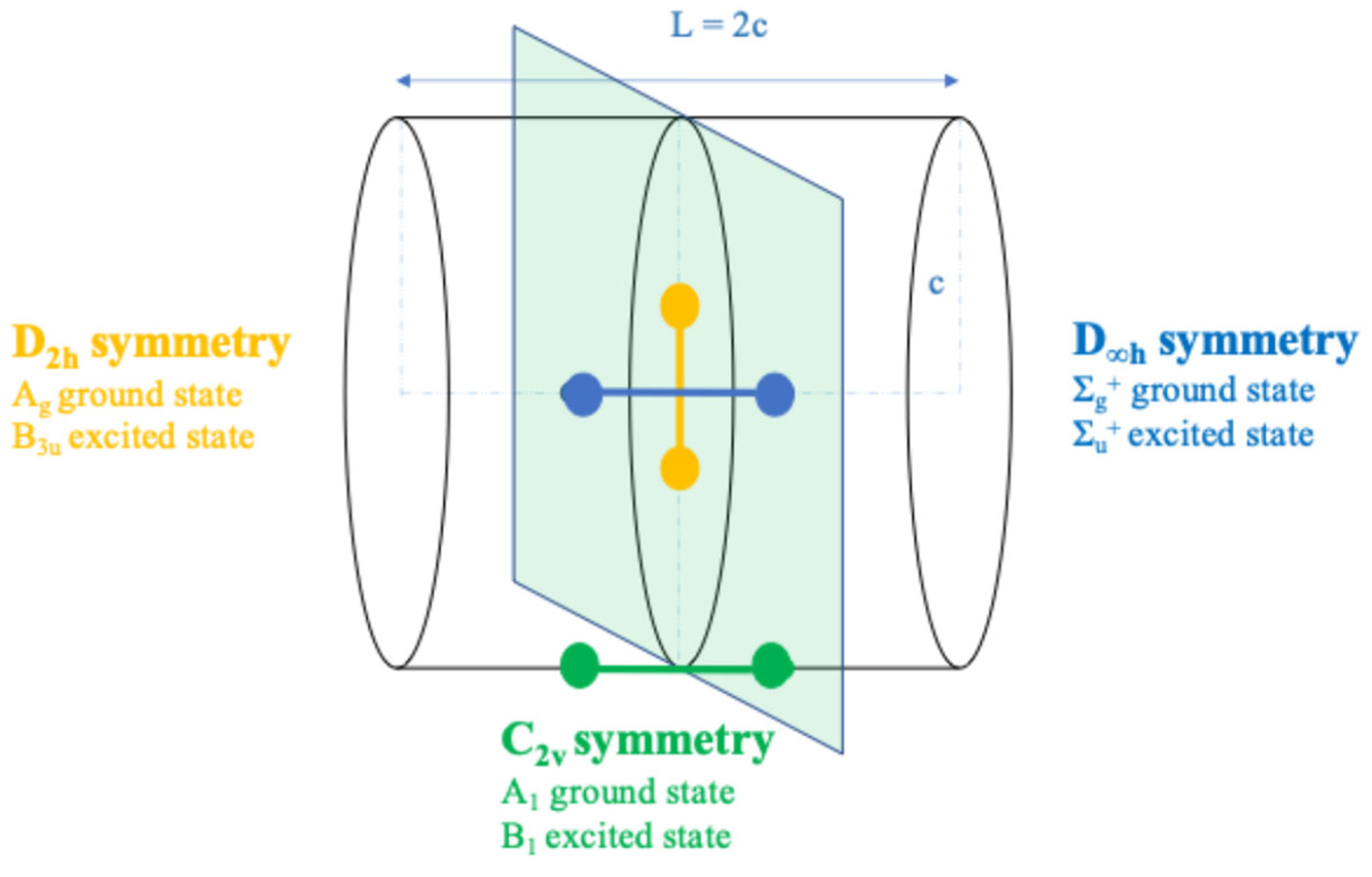
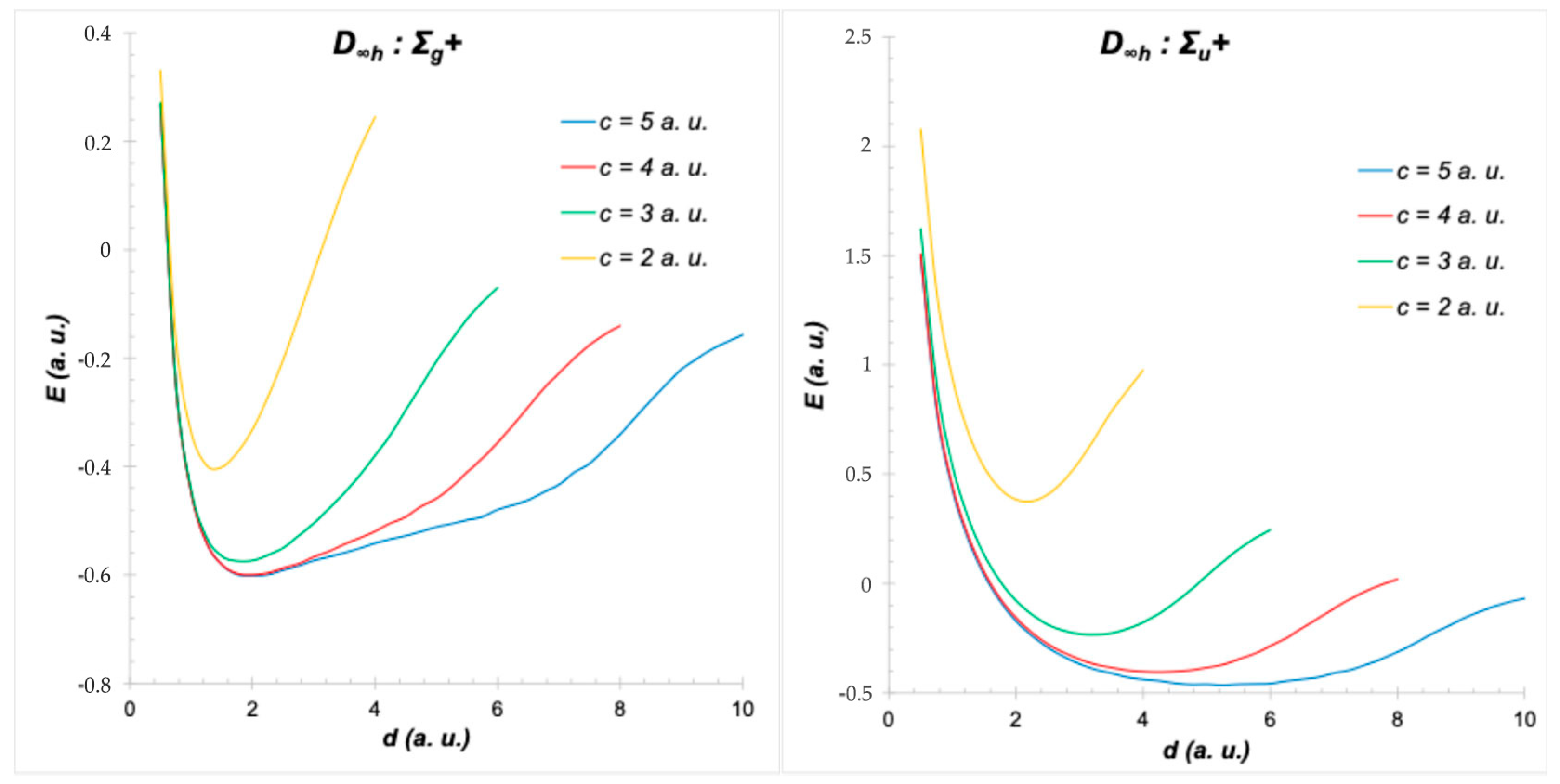
5. Cylindrical Confined H2+
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dmitriev, I.S. Symmetry in the World of Molecules; Mir Publishers: Moscow, Russia, 1979. [Google Scholar]
- King, R.B. Atomic orbitals, symmetry, and coordination polyhedra. Coord. Chem. Rev. 2000, 197, 141–168. [Google Scholar] [CrossRef]
- Sugano, S.; Tanabe, Y.; Kamimura, H. Multiplets of Transition Metal Ions in Crystals; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Harris, D.C.; Bertolucci, M.D. Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy; Courier Corporation: North Chelmsford, MA, USA, 1989. [Google Scholar]
- Sen, K.D.; Sen, K.D. (Eds.) Electronic Structure of Quantum Confined Atoms and Molecules; Springer: Cham, Switzerland, 2014; Volume 10, pp. 978–983. [Google Scholar]
- Ley-Koo, E.; Cruz, S.A. The hydrogen atom and the H2+ and HeH++ molecular ions inside prolate spheroidal boxes. J. Chem. Phys. 1981, 74, 4603–4610. [Google Scholar] [CrossRef]
- Olivares-Pilón, H.; Cruz, S.A. The H, H2+, and HeH2+ systems confined by an impenetrable spheroidal cavity: Revisited study via the Lagrange-mesh approach. Int. J. Quantum Chem. 2017, 117, e25399. [Google Scholar] [CrossRef]
- Michels, A.; De Boer, J.; Bijl, A. Remarks concerning molecural interaction and their influence on the polarisability. Physica 1937, 4, 981–994. [Google Scholar] [CrossRef]
- De Groot, S.R.; Ten Seldam, C.A. On the energy levels of a model of the compressed hydrogen atom. Physica 1946, 12, 669–682. [Google Scholar] [CrossRef]
- Laughlin, C.; Burrows, B.L.; Cohen, M. A hydrogen-like atomconfined within an impenetrable spherical box. J. Phys. B At. Mol. Opt. Phys. 2002, 35, 701. [Google Scholar] [CrossRef]
- Mateos-Cortés, S.; Ley-Koo, E.; Cruz, S.A. Hydrogen molecular ion inside penetrable prolate spheroidal boxes: Electronic and vibrational properties. Int. J. Quantum Chem. 2002, 86, 376–389. [Google Scholar] [CrossRef]
- Aquino, N.; Campoy, G.; Montgomery, H.E., Jr. Highly accurate solutions for the confined hydrogen atom. Int. J. Quantum Chem. 2007, 107, 1548–1558. [Google Scholar] [CrossRef]
- Colín-Rodríguez, R.; Díaz-García, C.; Cruz, S.A. The hydrogen molecule and the H2+ molecular ion inside padded prolate spheroidal cavities with arbitrary nuclear positions. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 241001. [Google Scholar] [CrossRef]
- Micca Longo, G.; Longo, S.; Giordano, D. Quantum states of confined hydrogen plasma species: Monte Carlo calculations. Plasma Sources Sci. Technol. 2015, 24, 065019. [Google Scholar] [CrossRef]
- Sveshnikov, K.A.; Tolokonnikov, A.V. H2+ in a lattice of cavities: Ammonia-like splitting of the lowest level. Mosc. Univ. Phys. Bull. 2015, 70, 181–189. [Google Scholar] [CrossRef]
- Strobel, T.A.; Kim, Y.; Andrews, G.S.; Ferrell Iii, J.R.; Koh, C.A.; Herring, A.M.; Sloan, E.D. Chemical–clathrate hybrid hydrogen storage: Storage in both guest and host. J. Am. Chem. Soc. 2008, 130, 14975–14977. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Bandaru, S.; Mondal, S. Hydrogen storage in clathrate hydrates. J. Phys. Chem. A 2011, 115, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Bačić, Z. Perspective: Accurate treatment of the quantum dynamics of light molecules inside fullerene cages: Translation-rotation states, spectroscopy, and symmetry breaking. J. Chem. Phys. 2018, 149, 100901. [Google Scholar] [CrossRef]
- Powers, A.; Scribano, Y.; Lauvergnat, D.; Mebe, E.; Benoit, D.M.; Bačić, Z. The effect of the condensed-phase environment on the vibrational frequency shift of a hydrogen molecule inside clathrate hydrates. J. Chem. Phys. 2018, 148, 144304. [Google Scholar] [CrossRef]
- Lauvergnat, D.; Felker, P.; Scribano, Y.; Benoit, D.M.; Bačić, Z. H2, HD, and D2 in the small cage of structure II clathrate hydrate: Vibrational frequency shifts from fully coupled quantum six-dimensional calculations of the vibration-translation-rotation eigenstates. J. Chem. Phys. 2019, 150, 154303. [Google Scholar] [CrossRef]
- Wang, J.; Lu, H.; Ripmeester, J.A. Raman spectroscopy and cage occupancy of hydrogen clathrate hydrate from first-principle calculations. J. Am. Chem. Soc. 2009, 131, 14132–14133. [Google Scholar] [CrossRef]
- Wang, J.; Lu, H.; Ripmeester, J.A.; Becker, U. Molecular-dynamics and first-principles calculations of raman spectra and molecular and electronic structure of hydrogen clusters in hydrogen clathrate hydrate. J. Phys. Chem. C 2010, 114, 21042–21050. [Google Scholar] [CrossRef]
- Ranieri, U.; Del Rosso, L.; Bove, L.E.; Celli, M.; Colognesi, D.; Gaal, R.; Hansen, T.C.; Koza, M.M.; Ulivi, L. Large-cage occupation and quantum dynamics of hydrogen molecules in sII clathrate hydrates. J. Chem. Phys. 2024, 160, 164706. [Google Scholar] [CrossRef]
- Kang, S.; Liu, Y.M.; Shi, T.Y. The characteristics for H2+-like impurities confined by spherical quantum dots. Eur. Phys. J. B 2008, 63, 37–42. [Google Scholar] [CrossRef]
- Henderson, B.; Imbusch, G.F. Optical Spectroscopy of Inorganic Solids; Oxford University Press: Oxford, UK, 2006; Volume 44. [Google Scholar]
- Al, E.B.; Sari, H.; Kasapoglu, E.; Sakiroglu, S.; Sökmen, I. Photoionization cross section of a complex in quantum dots: The role of donor atoms configuration. Eur. Phys. J. Plus 2022, 137, 919. [Google Scholar] [CrossRef]
- Wang, D.H.; Tang, T.T.; An, Z.K.; Chu, B.H.; Zhao, G. Quantum Rényi entropy of hydrogenic impurity states in the GaAsxP1-x semiconductor quantum dot. Micro Nanostruct. 2025, 204, 208167. [Google Scholar] [CrossRef]
- Juodkazis, K.; Juodkazytė, J.; Grigucevičienė, A.; Juodkazis, S. Hydrogen species within the metals: Role of molecular hydrogen ion H2+. Appl. Surf. Sci. 2011, 258, 743–747. [Google Scholar] [CrossRef]
- Juodkazytė, J.; Šebeka, B.; Juodkazis, S. Reversible hydrogen evolution and oxidation on Pt electrode mediated by molecular ion. Appl. Surf. Sci. 2014, 290, 13–17. [Google Scholar] [CrossRef]
- Micca Longo, G.; Longo, S.; Giordano, D. Confined H(1s) and H(2p) under different geometries. Phys. Scr. 2015, 90, 085402. [Google Scholar] [CrossRef]
- Longo, S.; Micca Longo, G.; Giordano, D. Monte Carlo calculation of the potential energy surface for octahedral confined H2+. Rend. Lincei. Sci. Fis. E Nat. 2018, 29, 173–177. [Google Scholar] [CrossRef]
- Longo, S.; Lonigro, D.; Lerario, G.; Stripoli, C.; Micca Longo, G. Quantum states of H2+ and H2 in an icosahedral potential well. Eur. Phys. J. D 2023, 77, 29. [Google Scholar] [CrossRef]
- Pupyshev, V.I. Electronic states of hydrogen atom in tetrahedral and similar polyhedral cavities. Int. J. Quantum Chem. 2011, 111, 2510–2518. [Google Scholar] [CrossRef]
- Pupyshev, V.I.; Scherbinin, A.V. Symmetry reduction and energy levels splitting of the one-electron atom in an impenetrable cavity. In Electronic Structure of Quantum Confined Atoms and Molecules; Springer: Cham, Switzerland, 2014; pp. 31–58. [Google Scholar]
- Van Gorder, R.A. Compressed hydrogen atoms confined within generic boxes. Proc. R. Soc. A 2022, 478, 20220467. [Google Scholar] [CrossRef]
- Yurenev, P.V.; Scherbinin, A.V.; Pupyshev, V.I. Energy levels of the hydrogen atom in a cylindrical cavity. Int. J. Quantum Chem. 2006, 106, 2201–2207. [Google Scholar] [CrossRef]
- Micca Longo, G.; Longo, S. Quantum states and static field ionization of a cylindrical confined hydrogen atom: A diffusion Monte Carlo study. Int. J. Quantum Chem. 2024, 124, e27392. [Google Scholar] [CrossRef]
- Foulkes, W.M.; Mitas, L.; Needs, R.J.; Rajagopal, G. Quantum Monte Carlo simulations of solids. Rev. Mod. Phys. 2001, 73, 33. [Google Scholar] [CrossRef]
- Annarelli, A.; Alfè, D.; Zen, A. A brief introduction to the diffusion Monte Carlo method and the fixed-node approximation. J. Chem. Phys. 2024, 161, 241501. [Google Scholar] [CrossRef] [PubMed]
- Micca Longo, G.; Coppola, C.M.; Giordano, D.; Longo, S. The unbiased diffusion Monte Carlo: A versatile tool for two-electron systems confined in different geometries. Eur. Phys. J. D 2021, 75, 83. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Well Shape | Symmetry | Low-Lying Molecular and Asymptotic States |
|---|---|---|
| Spherical (K) | D∞h | 2Σg+ (2P), 2Σu+ (2P), 2Πu (2D), 2Πg (2D) |
| Cylinder (D∞h) | D∞h | 2Σg+ (2P), 2Σu+ (2P), 2Πu (2D), 2Πg (2D) |
| D2h | 2Ag (2P), 2B1u (2D), 2B2u (2F), 2B3u (2F) | |
| Cubic (Oh) | D4h | 2A1g (2P), 2A2u (2P), 2E2u (2D) |
| D3d | 2A1g (2F), 2A2u (2F), 2E2u | |
| D2h | 2Ag (2D), 2B1u (2D), 2B2u (2F) | |
| Octahedral (Oh) | D4h | 2A1g (~2F), 2A2u (~2F) |
| D3d | 2A1g (2P), 2A2u (2P), 2E2u (2D) | |
| D2h | 2Ag (2D), 2B1u (2D), 2B2u (2F) | |
| Icosahedral (Ih) | D5d | 2A1g (~2D), 2A2u (~2D), 2E1u |
| D3d | 2A1g (2P), 2A2u (2P), 2E2u (2D) | |
| D2h | 2Ag (~2P), 2B1u (~2P), 2B2u (~2D), 2B3u (~2D) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micca Longo, G.; Bonasia, G.; Longo, S. Symmetries of Confined H2+ Molecule. Symmetry 2025, 17, 1169. https://doi.org/10.3390/sym17081169
Micca Longo G, Bonasia G, Longo S. Symmetries of Confined H2+ Molecule. Symmetry. 2025; 17(8):1169. https://doi.org/10.3390/sym17081169
Chicago/Turabian StyleMicca Longo, Gaia, Grazia Bonasia, and Savino Longo. 2025. "Symmetries of Confined H2+ Molecule" Symmetry 17, no. 8: 1169. https://doi.org/10.3390/sym17081169
APA StyleMicca Longo, G., Bonasia, G., & Longo, S. (2025). Symmetries of Confined H2+ Molecule. Symmetry, 17(8), 1169. https://doi.org/10.3390/sym17081169