
A Stereoselective Entry to Enantiopure (S)-2-Amino-2-methyl-5-arylpent-4-ynoic Acids and Evaluation of Their Inhibitory Activity against Bacterial Collagenase G

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Instrumentation
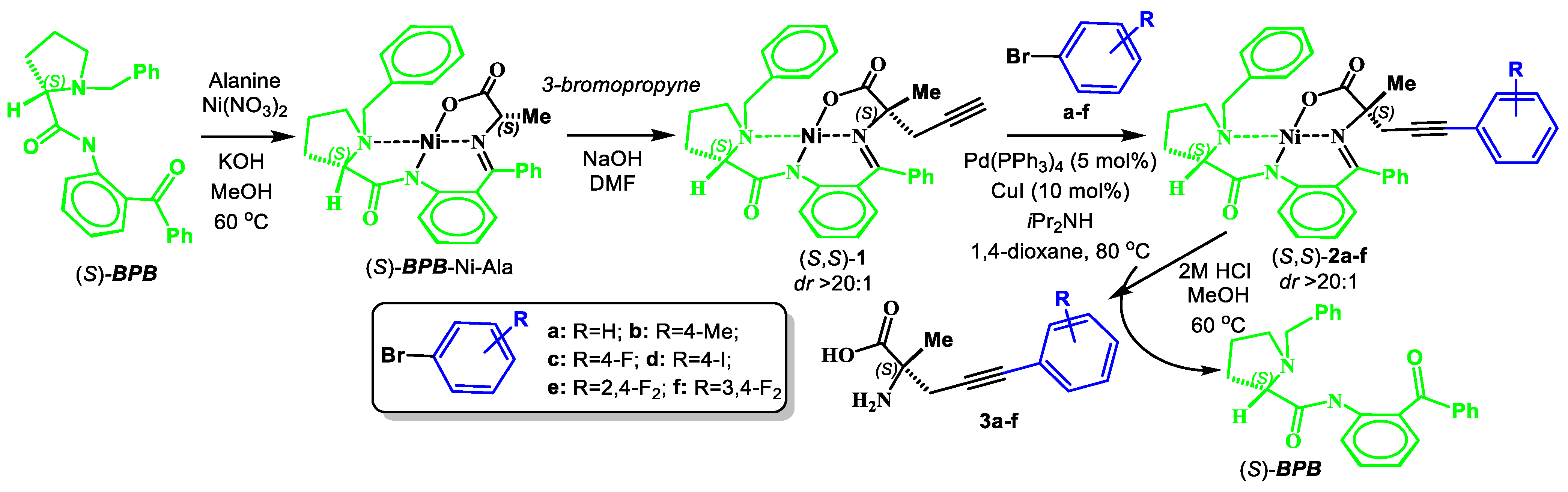
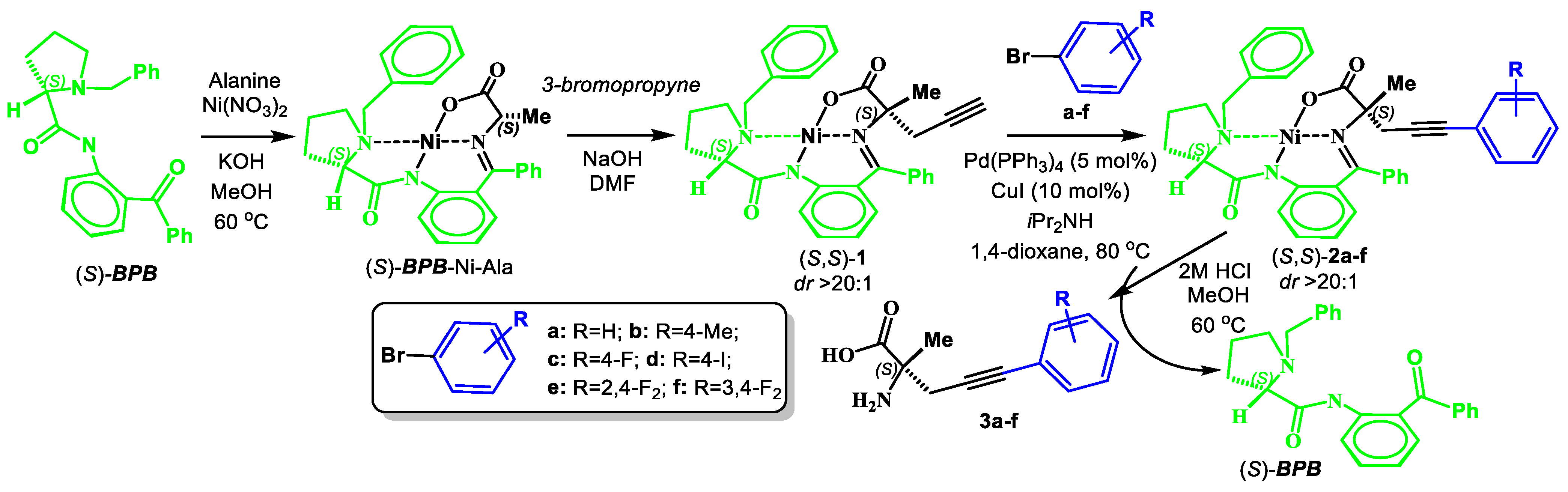
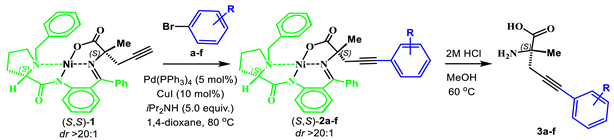
2.2. Procedure for Synthesis of Complex 1
2.3. General Procedure for Synthesis of Complexes 2a–f
- (S)-BPB-Ni-(S)-2-amino-2-methyl-5-phenylpent-4-ynoic acid complex (2a). The reaction of 1 (600 mg, 1.1 mmol), Pd(PPh3)4 (62.4 mg, 0.05 mmol), CuI (20.9 mg, 0.11 mmol) and bromobenzene (0.14 mL, 1.31 mmol) in iPr2NH (3 mL) and 1,4-dioxane (4.5 mL) for 6 h gave a red solid (513 mg, 75%); Mp. 120–122 °C. [α]D20 +1792.86 (c 0.07, MeOH). HRMS calculated for C37H33N3NiO3+: 626.1948; found: 626.1957. 1H NMR (300 MHz, CDCl3): δ = 1.40 (s, 3H, CH3), 1.57–1.69 (m, 1H, γ-H Pro), 1.89–2.12 (m, 3H, β-Ha,b and δ-Ha Pro), 2.30 (d, 1H, J = 17.0, CH2), 3.01 (d, 1H, J = 17.0, CH2), 2.93–3.10 (m, 1H, γ-Hb Pro), 3.31 (dd, 1H, J = 10.4, 6.2, α-H Pro), 3.65 (d, 1H, J = 12.6, CH2Ph), 3.66–3.73 (m, 1H, δ-Hb Pro), 4.45 (d, 1H, J = 12.6, CH2Ph), 6.62–6.69 (m, 2H, 3,4-H Ph), 7.16 (ddd, 1H, J = 8.4, 5.6, 3.0, 5-H Ph), 7.26–7.53 (m, 13H, Ar), 8.04–8.12 (m, 3H, 6-H Ph and CH2Ph). 13C NMR (75.5 MHz, CDCl3): δ = 23.3 (γ-CH2), 29.1 (CH3), 30.2 (β-CH2), 31.1 (CH2), 57.6 (δ-CH2), 63.6 (CH2Ph), 69.9 (α-CH), 76.3 (CCH3), 85.5 (CH2C≡C), 86.0 (CH2C≡C), 120.7 (CHAr) 123.4 (CHAr), 124.3 (CHAr), 127.0 (CHAr), 128.0 (CHAr), 128.3 (CHAr), 128.4 (CHAr), 128.5 (CHAr), 128.53 (CHAr), 128.9 (CHAr), 129.0 (CHAr), 129.7 (CHAr), 130.1 (CHAr), 131.7 (CHAr), 131.8 (CHAr), 132.1 (CHAr), 133.5 (CHAr), 133.6 (CAr), 136.5 (CAr), 142.2 (CAr), 173.5 (C=N), 180.8 (COO), 182.2 (C=O).
- (S)-BPB-Ni-(S)-2-amino-2-methyl-5-(p-tolyl)pent-4-ynoic acid complex (2b). The reaction of 1 (600 mg, 1.1 mmol), Pd(PPh3)4 (62.4 mg, 0.05 mmol), CuI (20.9 mg, 0.11 mmol) and 1-bromo-4-methylbenzene (224.7 mg, 1.31 mmol) in iPr2NH (3 mL) and 1,4-dioxane (4.5 mL) for 6 h gave a red solid (491 mg, 70%); Mp. 130–132 °C. [α]D20 +936.67 (c 0.15, MeOH). HRMS calculated For C38H35N3NiO3+: 640.2105; found: 640.2119. 1H NMR (300 MHz, CDCl3): δ = 1.39 (s, 3H, CH3), 1.59–1.70 (m, 1H, γ-H Pro), 1.93–2.13 (m, 3H, β-CH2 and δ-Ha Pro), 2.29 (m, 1H, J = 17.2, CH2C≡C), 2.34 (s, 3H, CH3-Ar), 3.00 (d, 1H, J = 17.2, CH2C≡C), 2.99–3.14 (m, 1H, γ-Hb Pro), 3.31 (dd, 1H, J = 10.4, 6.4, α-H Pro), 3.66 (d, 1H, J = 12.6, CH2Ph), 3.70 (ddd, 1H, J = 11.0, 6.3, 2.5, δ-Hb Pro), 4.46 (d, 1H, J = 12.6, CH2Ph), 6.62–6.69 (m, 2H, 3,4-H, Ph), 7.09–7.13 (m, 1H PhCH3), 7.16 (ddd, 1H, J = 8.5, 5.5, 2.9, 5-H Ph), 7.26–7.32 (m, 1H, 4-Ph), 7.33–7.52 (m, 9H, Ar), 8.05–8.11 (m, 3H, 6-H Ph and 2-Ph). 13C NMR (75.5 MHz, CDCl3): δ = 21.6 (CH3-Ar), 23.4 (γ-CH2), 29.2 (CH3), 30.3 (β-CH2), 31.2 (CH2), 57.6 (δ-CH2), 63.5 (CH2Ph), 69.9 (α-CH), 76.3 (CCH3), 85.3 (CH2C≡C), 85.7 (CH2C≡C), 120.4 (CHAr), 120.7 (CHAr), 124.3 (CHAr), 126.9 (CHAr), 128.0 (CHAr), 128.3 (CHAr), 128.5 (CHAr), 128.9 (CHAr), 129.0 (CHAr), 129.3 (CHAr), 129.7 (CHAr), 130.1 (CHAr), 131.7 (CHAr), 131.8 (CHAr), 132.0 (CHAr), 133.5 (CHAr), 133.6 (CHAr), 136.6 (CAr), 138.5 (CAr), 142.2 (CAr), 173.5 (C=N), 180.7 (COO), 182.3 (C=O).
- (S)-BPB-Ni-(S)-2-amino-5-(4-fluorophenyl)-2-methylpent-4-ynoic acid complex (2c). The reaction of 1 (600 mg, 1.1 mmol), Pd(PPh3)4 (62.4 mg, 0.05 mmol), CuI (20.9 mg, 0.11 mmol) and 1-bromo-4-fluorobenzene (0.14 mL, 1.31 mmol) in iPr2NH (3 mL) and 1,4-dioxane (4.5 mL) for 6 h gave a red solid (531 mg, 75%); Mp. 120–122 °C. [α]D20 +1068.75 (c 0.32, MeOH). HRMS calculated for C37H32N3FNiO3+: 644.1854; found: 644.1857. 1H NMR (300 MHz, CDCl3): δ = 1.39 (s, 3H, CH3), 1.60–1.73 (m, 1H, γ-Ha Pro), 1.93–2.15 (m, 3H, β-Ha,b and δ-Ha Pro), 2.30 (d, 1H, J = 17.2, CH2), 3.00 (d, 1H, J = 17.2, CH2), 2.96–3.12 (m, 1H, γ-Hb Pro), 3.33 (dd, 1H, J = 10.6, 6.1, α-H Pro), 3.64–3.71 (m, 1H, δ-Hb Pro), 3.67 (d, 1H, J = 12.6, CH2Ph), 4.45 (d, 1H, J = 12.6, CH2Ph), 6.62–6.69 (m, 2H, 3,4-H Ph), 6.97–7.05 (m, 2H, C6H4F), 7.17 (ddd, 1H, J = 8.5, 4.7, 3.9, 5-H Ph), 7.26–7.56 (m, 10H, Ar), 8.04–8.11 (m, 3H, 6-H, C6H4 and 2-Ph). 13C NMR (75.5 MHz, CDCl3): δ = 23.4 (γ-CH2), 29.1 (CH3), 30.3 (β-CH2), 31.1 (CH2), 57.5 (δ-CH2), 63.6 (CH2Ph), 69.9 (α-CH), 76.2 (CCH3), 84.4 (CH2C≡C), 85.8 (d, JCF = 1.8, CH2C≡C), 115.9 (d, JCF = 22.0, CFAr), 120.8 (CHAr), 124.3 (CHAr), 127.0 (CHAr), 127.9 (CHAr), 128.3 (CHAr), 128.4 (CHAr), 129.0 (CHAr), 129.1 (CHAr), 129.8 (CHAr), 130.1 (CHAr), 131.8 (CHAr), 131.8 (CHAr), 133.5 (CHAr), 133.6 (CHAr), 134.0 (d, JCF = 8.4, CFAr), 136.5 (CAr), 138.5 (CAr), 142.1 (CAr), 162.6 (d, JCF = 246.0, CFAr), 173.5 (C=N), 180.7 (COO), 182.2 (C=O).
- (S)-BPB-Ni-(S)-2-amino-5-(4-bromophenyl)-2-methylpent-4-ynoic acid complex (2d). The reaction of 1 (600 mg, 1.1 mmol), Pd(PPh3)4 (62.4 mg, 0.05 mmol), CuI (20.9 mg, 0.11 mmol) and 1-bromo-4-iodobenzene (371.7 mg, 1.31 mmol) in iPr2NH (3 mL) and 1,4-dioxane (4.5 mL) for 6 h gave a red solid (525 mg, 68%); Mp. 123–125 °C. [α]D20 +417.24 (c 0.29, MeOH). HRMS calculated For C37H32N3BrNiO3+: 704.1053; found: 704.1065. 1H NMR (300 MHz, CDCl3): δ = 1.38 (s, 3H, CH3), 1.63–1.75 (m, 1H, γ-Ha Pro), 1.93–2.14 (m, 3H, β-CH2, δ-Ha Pro), 2.30 (d, 1H, J = 17.2, CH2C≡C), 3.00 (d, 1H, J = 17.2, CH2C≡C), 2.96–3.12 (m, 1H, γ-Hb Pro), 3.35 (dd, 1H, J = 10.5, 6.1, α-H Pro), 3.63–3.69 (m, 1H, δ-Hb Pro), 3.67 (d, 1H, J = 12.6, CH2Ph), 4.44 (d, 1H, J = 12.6, CH2Ph), 6.60–6.70 (m 2H, 3,4-H Ph), 7.12–7.21 (m, 2H, C6H4F), 7.26–7.53 (m, 12H, Ar), 8.05 (br. d, 1H, J = 8.6, 6-H Ph), 8.07–8.12 (m, 2H, 2-Ph). 13C NMR (75.5 MHz, CDCl3): δ = 23.4 (γ-CH2 Pro), 29.1 (CH3), 30.3 (β-CH2), 31.1 (CH2), 57.5 (δ-CH2), 63.6 (CH2Ph), 76.2 (CCH3), 84.4 (CH2C≡C), 87.4 (CH2C≡C), 120.8 (CHAr), 122.3, 122.7 (Ar), 124.3 (CHAr), 127.1 (CHAr), 127.9 (CHAr), 128.3 (CHAr), 128.4 (Ar), 129.0 (2∙CHAr), 129.1 (CHAr), 129.8 (CHAr), 130.1 (CHAr), 131.8 (2∙CHAr), 131.8 (2∙CHAr), 131.9 (CHAr), 133.5 (CHAr), 133.6 (CAr), 136.5 (CAr), 142.1 (CAr), 173.6 (C=N), 180.7 (COO), 182.1 (C=O).
- (S)-BPB-Ni-(S)-2-amino-5-(2,4-difluorophenyl)-2-methylpent-4-ynoic acid complex (2e). The reaction of 1 (600 mg, 1.1 mmol), Pd(PPh3)4 (62.4 mg, 0.05 mmol), CuI (20.9 mg, 0.11 mmol) and 1-bromo-2,4-difluorobenzene (0.15 mL, 1.31 mmol) in iPr2NH (3 mL) and 1,4-dioxane (4.5 mL) for 6 h gave a red solid (507 mg, 70%); Mp. 118–120 °C. [α]D20 +1061.67 (c 0.30, MeOH). HRMS calculated for C37H31N3F2NiO3+: 662.1760; found: 662.1771. 1H NMR (300 MHz, CDCl3): δ = 1.43 (s, 3H, CH3), 1.69–1.80 (m, 1H, γ-Ha Pro), 1.94–2.10 (m, 2H, β-Ha and δ-Ha Pro), 2.12–2.20 (m, 2H, β-Hβ Pro), 2.23 (d, 1H, J = 17.2, CH2C≡C), 3.02 (d, 1H, J = 17.2, CH2C≡C), 3.04–3.20 (m, 1H, γ-Hb Pro), 3.34 (dd, 1H, J = 10.6, 6.1, α-H Pro), 3.66–3.73 (m, 1H, δ-Hb Pro), 3.69 (d, 1H, J = 12.6, CH2Ph), 4.46 (d, 1H, J = 12.6, CH2Ph), 6.62–6.68 (m, 2H, 3,4-H Ph), 6.82–6.89 (m, 2H, Ar), 7.17 (ddd, 1H, J = 8.4, 5.0, 3.6, 5-H Ph), 7.27–7.36 (m, 2H, Ar), 7.39–7.54 (m, 7H, Ar), 8.03–8.11 (m, 3H, 6-H Ph and 2-Ph). 13C NMR (75.5 MHz, CDCl3): δ = 23.4 (γ-CH2), 29.2 (CH3), 30.4 (β-CH2 Pro), 31.0 (CH2), 57.5 (δ-CH2 Pro), 63.6 (CH2Ph), 70.0 (α-CH Pro), 75.4, 76.1 (CCH3), 76.6 (CH2C≡C), 91.4 (dd, JC,F = 4.3, 1.8, CH2C≡C), 104.5 (t, JC,F = 25.3, 3-CH, C6H3F2), 111.7 (dd, JCF = 21.9, 4.0, 5-CH, CFAr), 120.8 (4-CH, Ar), 124.4 (6-CH, Ar), 127.0 (CHAr), 128.07 (CHAr), 128.3 (CHAr), 128.5, 129.0 (2∙CHAr), 129.1 (CHAr), 129.7 (CHAr), 129.8 (CHAr), 130.1 (CHAr), 131.8 (CHAr), 131.8 (2∙CHAr), 133.5 (CHAr), 133.6 (CAr), 134.6 (dd, JCF = 9.7, 2.7, 1C, CFAr), 136.5 (CAr), 142.1 (CAr), 162.7 (dd, JCF = 252.2, 11.5 CFAr), 163.7 (dd, JCF = 254.2, 12.0, CFAr), 173.5 (C=N), 180.7 (COO), 182.1 (C=O).
- (S)-BPB-Ni-(S)-2-amino-5-(3,4-difluorophenyl)-2-methylpent-4-ynoic acid complex (2f). The reaction of 1 (600 mg, 1.1 mmol), Pd(PPh3)4 (62.4 mg, 0.05 mmol), CuI (20.9 mg, 0.11 mmol) and 4-bromo-1,2-difluorobenzene (0.15 mL, 1.31 mmol) in iPr2NH (3 mL) and 1,4-dioxane (4.5 mL) for 6 h gave a red solid (457 mg, 63%); Mp. 118–120 °C. [α]D20 +1297.65 (c 0.09, MeOH). HRMS calculated For C37H31N3F2NiO3+: 662.1760; found: 662.1769. 1H NMR (300 MHz, CDCl3): δ = 1.38 (s, 3H, CH3), 1.69–1.81 (m, 1H, γ-Ha Pro), 1.95–2.22 (m, 3H, β-CH2, δ-Ha Pro), 2.30 (d, 1H, J = 17.2, CH2C≡C), 2.99 (d, 1H, J = 17.2, CH2C≡C), 2.99–3.15 (m, 1H, γ-Hb Pro), 3.37 (dd, 1H, J = 10.5, 6.1, α-H Pro), 3.65–3.72 (m, 1H, δ-Hb Pro), 3.69 (d, 1H, J = 12.6, CH2Ph), 4.44 (d, 1H, J = 12.6, CH2Ph), 6.63–6.70 (m, 2H, 3,4-H Ph), 7.05–7.24 (m, 3H, Ar), 7.28–7.37 (m, 4H, Ar), 7.39–7.48 (m, 3H, Ar), 7.49–7.54 (m, 2H, Ar), 8.05–8.11 (m, 3H, Ar). 13C NMR (75.5 MHz, CDCl3): δ = 23.3 (γ-CH2), 29.1(CH3), 30.4 (β-CH2 Pro), 31.0 (CH2), 57.5 (δ-CH2 Pro), 63.7 (CH2Ph), 70.0 (α-CH Pro), 76.1 (CCH3), 83.2 (t, JCF = 2.3, CH2C≡C), 86.8 (d, JCF = 2.0, CH2C≡C), 117.8 (d, JCF = 17.6, CFAr), 120.2 (dd, JCF = 7.6, 4.2, 1-C, CFAr), 120.8 (d, JCF = 18.4, 4-CH, Ar), 120.9 (CHAr), 124.3 (CHAr), 127.1 (CHAr), 127.8 (CHAr), 128.3 (CHAr), 128.3 (CHAr), 128.8 (dd, JCF = 6.4, 3.5, Ar), 129.0 (CHAr), 129.1 (CHAr), 129.8 (CHAr), 130.0 (CHAr), 131.8 (CHAr), 131.9 (CHAr), 133.5 (CHAr), 133.6 (CAr), 136.5 (CAr), 142.1 (CAr), 150.1 (dd, JCF = 249.6, 12.8, CFAr), 150.7 (dd, JCF = 251.6, 12.3 CFAr), 173.6 (C=N), 180.7 (COO), 182.1 (C=O).
2.4. General Procedure for the Isolation of AAs 3a–f
- (S)-2-amino-2-methyl-5-phenylpent-4-ynoic acid (3a). The decomposition of 2a (486 mg, 0.78 mmol) gave 3a as a white solid (147 mg, 94%). Mp. 282–284 °C. [α]D20 –5.12 (c 0.078, MeOH). HRMS calculated for C12H13NO2+: 204.1019; found: 204.1026. 1H NMR (300 MHz, DMSO-d6+CCl4+CF3COOD): δ = 1.60 (s, 3H, CH3), 2.99 (d, 1H, J = 17.3, CH2), 3.02 (d, 1H, J = 17.3, CH2), 7.28–7.33 (m, 3H), 7.41–7.47 (m, 2H, Ph), 8.70 (br. s, 2H, NH2), 10.20 (COOH). 13C NMR (75.5 MHz, DMSO+CCl4+CF3COOD): δ = 21.2 (CH3), 27.7 (CH2), 58.0 (CCH3), 82.4 (CH2C≡C), 84.3 (CH2C≡C), 122.5 (Cipso), 127.8 (CHAr), 127.9 (CHAr), 131.4 (CHAr), 171.2 (COOH).
- (S)-2-amino-2-methyl-5-(p-tolyl)pent-4-ynoic acid (3b). The decomposition of 2b (477 mg, 0.74 mmol) gave 3b as a white solid (147 mg, 91%). Mp. 282–284 °C. [α]D20 –3.80 (c 0.105, MeOH). HRMS calculated For C13H15NO2+: 218.1176; found: 218.1182. 1H NMR (300 MHz, D2O+CF3COOD): δ = 1.66 (s, 3H, CH3), 2.28 (s, 3H, PhCH3), 2.99 (d, 1H, J = 17.7, CH2), 3.14 (d, 1H, J = 17.3, CH2), 7.14–7.20 (m, 2H, Ph), 7.31–7.37 (m, 2H, C6H4). 13C NMR (75.5 MHz, D2O+CF3COOD): δ = 19.8 (CH3-Ar), 20.3 (CH3), 27.0 (CH2), 58.6 (CCH3), 80.0 (CH2C≡C), 84.7 (CH2C≡C), 117.7 (CHAr), 120.5 (CHAr), 131.0 (CHAr), 139.1 (CAr), 172.0 (COOH).
- (S)-2-amino-5-(4-fluorophenyl)-2-methylpent-4-ynoic acid (3c). The decomposition of 2c (531 mg, 0.82 mmol) gave 3c as a white solid (174 mg, 96%). Mp. 280–282 °C. [α]D20 –6.05 (c 0.09, MeOH). HRMS calculated for C12H12FNO2+: 222.0925; found: 222.0934. 1H NMR (300 MHz DMSO-d6+CF3COOD): δ = 1.63 (s, 3H, CH3), 3.01 (d, 1H, J = 17.2, CH2), 3.08 (d, 1H, J = 17.2, CH2), 6.69–7.07 (m, 2H, C6H4F), 7.47–7.53 (m, 2H, C6H4F). 13C NMR (75.5 MHz, DMSO+CF3COOD): δ = 21.1 (CH3), 27.6 (CH2), 58.1 (CCH3), 82.4 (CH2C≡C), 83.2 (CH2C≡C), 115.1 (d, JCF = 21.9, CHAr), 133.62 (d, JCF = 8.2, CHAr), 161.8 (d, JCF = 248.6, CFAr), 171.0 (COOH).
- (S)-2-amino-5-(4-bromophenyl)-2-methylpent-4-ynoic acid (3d). The decomposition of 2d (525 mg, 0.74 mmol) gave 3d as a white solid (150 mg, 71%). Mp. 283–285 °C. [α]D20 –4.80 (c 0.095, MeOH). HRMS calculated for C12H12BrNO2+: 282.0124; found: 282.0133. 1H NMR (300 MHz, D2O+CF3COOD): δ = 1.69 (s, 3H, CH3), 3.04 (d, 1H, J = 17.7, CH2), 3.18 (d, 1H, J = 17.7, CH2), 7.35 (d, J = 8.5, 2H, C6H4Br), 7.53 (d, J = 8.5, 2H, C6H4Br). 13C NMR (75.5 MHz, D2O+CF3COOD): δ = 20.4 (CH3), 27.0 (CH2), 58.5 (CCH3), 81.8 (CH2C≡C), 83.7 (CH2C≡C), 120.0 (CHAr), 121,9 (CHAr), 130.9 (CHAr), 132.6 (CHAr), 172.0 (COOH).
- (S)-2-amino-5-(2,4-difluorophenyl)-2-methylpent-4-ynoic acid (3e). The decomposition of 2e (507 mg, 0.77 mmol) gave 3e as a white solid (180 mg, 95%). Mp. 284–286 °C. [α]D20 –6.67 (c 0.12, MeOH). HRMS calculated for C12H11F2NO2+: 240.0831; found: 240.0842. 1H NMR (300 MHz, DMSO-d6+CF3COOD): δ = 1.65 (s, 3H, CH3), 3.02 (d, 1H, J = 17.7, CH2), 3.16 (d, 1H, J = 17.7, CH2), 7,01–7.63 (m, 1H, C6H3). 13C NMR (75 MHz, DMSO): δ = 21.0 (CH3), 27.6 (CH2), 57.8 (CCH3), 76.6 (C≡C), 87.5 (CH2C≡), 103.6 (t, JCF = 25.3 Hz, CHAr), 107.3 (dd, JCF = 15.9, 4.1 Hz, CHAr), 111.2 (dd, JCF = 21.8, 3.2 Hz, CHAr), 134.9 (dd, JCF = 9.6, 2.3 Hz, CAr), 161.9 (dd, JCF = 251.4, 11.4 Hz, CFAr), 162.4 (dd, JCF = 253.3, 12.2 Hz, CFAr), 171.0 (COOH).
- (S)-2-amino-5-(3,4-difluorophenyl)-2-methylpent-4-ynoic acid (3f). The decomposition of 2f (501 mg, 0.76 mmol) gave 3f as a white solid (129 mg, 72%). Mp. 278–280 օC. [α]D20 –7.2 (c 0.10, MeOH). HRMS calculated For C12H11F2NO2+: 240.0831; found: 240.0844. 1H NMR (300 MHz, D2O+CF3COOD): δ = 1.67 (s, 3H, CH3), 3.02 (d, 1H, J = 17.8, CH2), 3.18 (d, 1H, J = 17.8, CH2), 7,15–7.26 (m, 2H), 7.28–7.37 (m, 1H, C6H3). 13C NMR (75.5 MHz, D2O+CF3COOD): δ = 20.4 (CH3), 26.8 (CH2), 58.5 (CCH3), 81.1 (CH2C≡C), 82.6 (CH2C≡C), 116.8 (d, JCF = 17.9, CHAr), 117.9 (dd, JCF = 8.2, 4.0, CHAr), 119.8 (d, JCF = 18.8, CHAr), 128.1 (dd, JCF = 6.8, 3.5, CFAr), 148.8 (dd, JCF = 246.2, 13.1, CFAr), 149.8 (dd, JCF = 249.3, 12.5, CFAr), 171.8 (COOH).
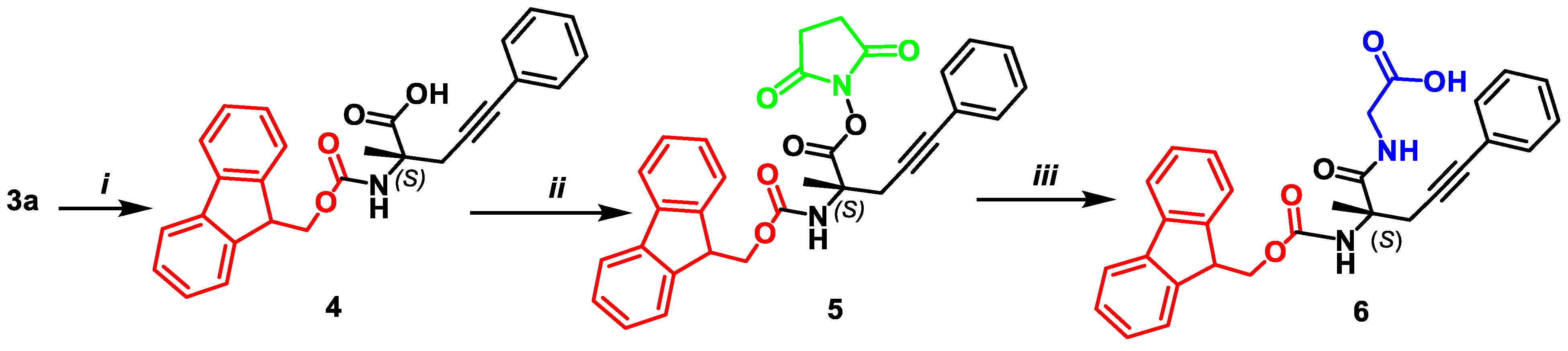
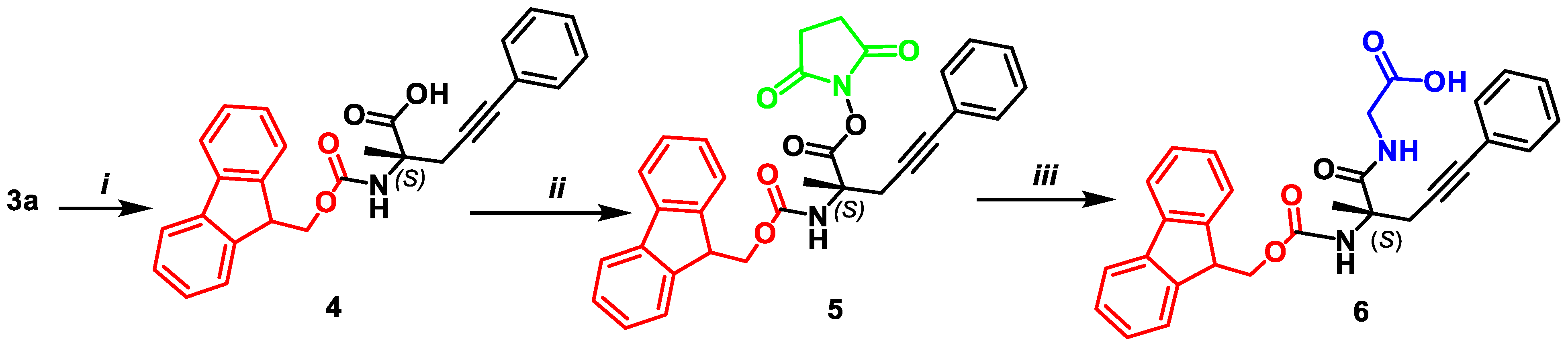
2.5. Synthesis of N-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-(S)-2-methyl-5-phenylpent-4-ynoic acid 4
- Mp. 173–175 °C. HRMS calculated for C27H23NO4+: 426.1700; found: 426.1715. 1H NMR (300 MHz, DMSO-d6): δ = 1.58 (s, 3H), 2.95 (d, 1H, J = 17.0, CH2C≡), 3.21 (d, 1H, J = 17.0, CH2C≡), 4.36–4.19 (m, 3H, OCH2 and CH2CH), 7.45–7.18 (m, 10H, Ar), 7.68 (d, 1H, J = 7.5, Ar), 7.74 (d, 1H, J = 7.5, Ar). 13C NMR (75 MHz, DMSO): δ = 22.7 (CH3), 26.8 (CH2C≡), 46.6 (CH2CH), 57.6 (CH2CH), 65.5 (CCH3), 82.3 (≡CPh), 86.1 (C≡CPh), 119.3 (CHAr), 123.3 (CAr), 124.9, 126.56, 127.0, 127.1, 127.7, 131.1 (CHAr), 140.5, 143.6 (CAr), 154.3 (C=O), 174.0 (COOH).
2.6. Synthesis of N-9-fluorenylmethyloxycarbonyl-(S)-2-amino-2-methyl-5-phenylpent-4-ynoic Acid Succinimide Ester 5
2.7. Synthesis of Dipeptide N-9-fluorenylmethyloxycarbonyl-α-phenyl-(S)-propargylalanylglycine 6
- Mp. 98–100 °C. [α]D20 –81.4 (c 1.0, MeOH). HRMS calculated for C29H26N2O5+: 515.1635; found: 515.1645. 1H NMR (300 MHz, DMSO-d6): δ = 1.56 (s, 1H, CH3), 2.97 (d, 1H, J = 17.9, CH2C≡), 3.25 (d, 1H, J = 17.9, CH2C≡), 3.74 (dd, 1H, J = 17.7, 5.4, NHCH2), 3.85 (dd, 1H, J = 17.7, 5.4, NHCH2), 4.33–4.21 (m, 3H, CHCH2O), 7.40–7.16 (m, 10H, CHAr and 2NH), 7.76–7.61 (m, 5H, CHAr). 13C NMR (75 MHz, DMSO): δ = 24.5 (CH3), 46.7 (CH2), 58.1 (CH2), 72.2 (CCH3), 82.3 (C≡C), 86.4 (CH2C≡), 119.3 (CHAr), 123.5, 125.0, 126.5, 127.0, 127.1, 127.7, 131.2, 140.6 (CAr), 143.7, 154.3, 170.9 (C=O), 172.7, 174.4 (COOH).
2.8. Determination of Collagenase Activity
3. Results and Discussion
3.1. Syntheses of Complexes 2, AAs 3 and Dipeptide 6
3.2. Biological Tests
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blaskovich, M.A. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Rezhdo, A.; Islam, M.; Huang, M.; Deventer, J.V. Future prospects for noncanonical amino acids in biological therapeutics. Curr. Opin. Biotechnol. 2019, 60, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, J.; Izawa, K.; Sato, T.; White, S.; Meanwell, N.A.; Soloshonok, V.A. Cyclic tailor-made amino acids in the design of modern pharmaceuticals. Eur. J. Med. Chem. 2020, 208, 112736. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Konno, H.; Sato, T.; Soloshonok, V.A.; Izawa, K. Tailor-made amino acids in the design of small-molecule blockbuster drugs. Eur. J. Med. Chem. 2021, 220, 113448. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lyutenko, N.V.; Sorochinsky, A.E.; Okawara, A.; Konno, H.; White, S.; Soloshonok, V.A. Tailor-made amino acids in pharmaceutical industry: Synthetic approaches to aza-tryptophan derivatives. Chem. Eur. J. 2021, 27, 17510–17528. [Google Scholar] [CrossRef]
- Wang, Q.; Han, J.; Sorochinsky, A.; Landa, A.; Butler, G.; Soloshonok, V.A. The latest FDA-approved pharmaceuticals containing fragments of tailor-made amino acids and fluorine. Pharmaceuticals 2022, 15, 999. [Google Scholar] [CrossRef]
- Wang, N.; Mei, H.; Dhawan, G.; Zhang, W.; Han, J.; Soloshonok, V.A. New approved drugs appearing in the pharmaceutical market in 2022 featuring fragments of tailor-made amino acids and fluorine. Molecules 2023, 28, 3651. [Google Scholar] [CrossRef]
- Kazmaier, U. Amino acids–valuable organocatalysts in carbohydrate synthesis. Angew. Chem. Int. Ed. 2005, 44, 2186–2188. [Google Scholar] [CrossRef]
- Davie, E.A.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Supported proline and proline-derivatives as recyclable organocatalysts. Chem. Soc. Rev. 2008, 37, 1666–1688. [Google Scholar] [CrossRef]
- Honig, M.; Sondermann, P.; Turner, N.J.; Carreira, E. Enantioselective chemo- and biocatalysis: Partners in retrosynthesis. Angew. Chem. Int. Ed. 2017, 56, 8942–8973. [Google Scholar] [CrossRef] [PubMed]
- Saghyan, A.S.; Langer, P. Asymmetric Synthesis of Non-Proteinogenic Amino Acids; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2016. [Google Scholar]
- Weiner, B.; Szymanski, W.; Janssen, D.B.; Minnaard, A.J.; Feringa, B.L. Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem. Soc. Rev. 2010, 39, 1656–1691. [Google Scholar] [CrossRef] [PubMed]
- Belokon, Y.N.; Bulychev, A.G.; Vitt, S.V.; Struchkov, Y.T.; Batsanov, A.S.; Timofeeva, T.V.; Tsyryapkin, V.A.; Ryzhov, M.G.; Lysova, L.A.; Bakhmutov, V.I.; et al. General method of diastereo- and enantioselective synthesis of β-hydroxy-α-amino acids by condensation of aldehydes and ketones with glycine. J. Am. Chem. Soc. 1985, 107, 4252–4259. [Google Scholar] [CrossRef]
- Belokon, Y.N. Chiral complexes of Ni(II), Cu(II), and Cu(I) as reagents, catalysts and receptors for asymmetric synthesis and chiral recognition of amino acids. Pure. Appl. Chem. 1992, 64, 1917–1924. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases; Part 1: Alkyl halide alkylations. Amino Acids 2013, 45, 691–718. [Google Scholar] [CrossRef]
- Zou, Y.; Han, J.; Saghyan, A.S.; Mkrtchyan, A.F.; Konno, H.; Moriwaki, H.; Izawa, K.; Soloshonok, V.A. Asymmetric synthesis of tailor-made amino acids using chiral Ni(II) complexes of Schiff bases. Molecules 2020, 25, 2739. [Google Scholar] [CrossRef]
- Larionov, V.A.; Adonts, H.V.; Gugkaeva, Z.T.; Smol’yakov, A.F.; Saghyan, A.S.; Miftakhov, M.S.; Kuznetsova, S.A.; Maleev, V.I.; Belokon, Y.N. The elaboration of a general approach to the asymmetric synthesis of 1,4-substituted 1,2,3-triazole containing amino acids via Ni(II) complexes. ChemistrySelect 2018, 3, 3107–3110. [Google Scholar] [CrossRef]
- Mkrtchyan, A.F.; Hayriyan, L.A.; Karapetyan, A.J.; Tovmasyan, A.S.; Tsaturyan, A.H.; Khrustalev, V.N.; Maleev, V.I.; Saghyan, A.S. Using the Ni-[(benzylprolyl)amino]benzophenone complex in the Glaser reaction for the synthesis of bis α-amino acids. New J. Chem. 2020, 44, 11927–11932. [Google Scholar] [CrossRef]
- Larionov, V.A.; Stashneva, A.R.; Titov, A.A.; Lisov, A.A.; Medvedev, M.G.; Smol’yakov, A.F.; Tsedilin, A.M.; Shubina, E.S.; Maleev, V.I. Mechanistic study in azide-alkyne cycloaddition (CuAAC) catalyzed by bifunctional trinuclear copper(I) pyrazolate complex: Shift in rate-determining step. J. Catal. 2020, 390, 37–45. [Google Scholar] [CrossRef]
- Mkrtchyan, A.F.; Paloyan, A.M.; Hayriyan, L.A.; Sargsyan, A.S.; Tovmasyan, A.S.; Karapetyan, A.J.; Hambardzumyan, A.A.; Hovhannisyan, N.A.; Panosyan, H.A.; Khachatryan, H.N.; et al. Synthesis of enantiomerically enriched non-protein α-amino acids and their study as aldose reductase inhibitors. Synth. Commun. 2021, 51, 1433–1450. [Google Scholar] [CrossRef]
- Mkrtchyan, A.F.; Tovmasyan, A.S.; Paloyan, A.M.; Sargsyan, A.S.; Simonyan, H.M.; Sahakyan, L.Y.; Petrosyan, S.G.; Hayriyan, L.A.; Sargsyan, T.H. Asymmetric synthesis of derivatives of alanine via Michael addition reaction and their biological study. Synlett 2022, 33, 2013–2018. [Google Scholar] [CrossRef]
- Xu, M.; Tan, Z.; Zhu, C.; Zhuang, W.; Ying, H.; Ouyang, P. Recent advance of chemoenzymatic catalysis for the synthesis of chemicals: Scope and challenge. Chin. J. Chem. Eng. 2021, 30, 146–167. [Google Scholar] [CrossRef]
- Cativiela, C.; Ordóñez, M.; Viveros-Ceballos, J.L. Stereoselective synthesis of acyclic α,α-disubstituted α-amino acids derivatives from amino acids templates. Tetrahedron 2020, 76, 130875. [Google Scholar] [CrossRef]
- Ulijn, R.V.; Woolfson, D.N. Peptide and protein based materials in 2010: From design and structure to function and application. Chem. Soc. Rev. 2010, 39, 3349–3350. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future. J. Med. Chem. 2018, 61, 1382. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, R.; Chastel, A.; Previti, S.; Hindié, E.; Vimont, D.; Zanotti-Fregonara, P.; Fernandez, P.; Garrigue, P.; Lamare, F.; Schollhammer, R.; et al. Silicon-containing neurotensin analogues as radiopharmaceuticals for NTS1-positive tumors imaging. Bioconjugate Chem. 2020, 31, 2339–2349. [Google Scholar] [CrossRef]
- Uhlig, T.; Kyprianou, T.; Martinelli, F.G.; Oppici, C.A.; Heiligers, D.; Hills, D.; Calvo, X.R.; Verhaert, P. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPA Open Proteomics 2004, 4, 58–69. [Google Scholar] [CrossRef]
- Sun, L. Peptide-based drug development. Mod. Chem. Appl. 2013, 1, e103. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability challenges in peptide synthesis and purification: From R&D to production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar]
- Muramatsu, W.; Hattori, T.; Yamamoto, H. Amide bond formation: Beyond the dilemma between activation and racemization. Chem. Commun. 2021, 57, 6346–6359. [Google Scholar] [CrossRef]
- Nakashima, E.; Yamamoto, H. Biomimetic peptide catalytic bond-forming utilizing a mild Brønsted acid. Chem. Eur. J. 2022, 28, e202103989. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Albericio, F.; Dawson, P.E. Methods, setup and safe handling for anhydrous hydrogen fluoride cleavage in Boc solid-phase peptide synthesis. Nat. Protoc. 2015, 10, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Verlander, M. Industrial applications of solid-phase peptide synthesis—A status report. Int. J. Pept. Res. Ther. 2007, 13, 75–82. [Google Scholar] [CrossRef]
- Winkler, D.F.H.; Tian, K. Investigation of the automated solid-phase synthesis of a 38mer peptide with difficult sequence pattern under different synthesis strategies. Amino Acids 2015, 47, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Masuda, K.; Ooyama, H.; Shikano, K.; Kondo, K.; Furumitsu, M.; Iwakoshi-Ukena, E.; Ukena, K. Microwave-assisted solid-phase peptide synthesis of neurosecretory protein GL composed of 80 amino acid residues. J. Pept. Sci. 2015, 21, 454–460. [Google Scholar] [CrossRef]
- Hattori, T.; Yamamoto, H. Synthesis of silacyclic dipeptides: Peptide elongation at both N- and C-termini of dipeptide. J. Am. Chem. Soc. 2022, 144, 1758–1765. [Google Scholar] [CrossRef]
- Okada, Y.; Hosoya, S.; Suzuki, H.; Chiba, K. Total synthesis of elastin peptide using high pressure−liquid phase synthesis assisted by a soluble tag strategy. Org. Lett. 2014, 16, 6448–6451. [Google Scholar] [CrossRef]
- Takahashi, D.; Inomata, T.; Fukui, T. AJIPHASE®: A highly efficient synthetic method for one-pot peptide elongation in the solution phase by an Fmoc strategy. Angew. Chem. Int. Ed. 2017, 56, 7803–7807. [Google Scholar] [CrossRef]
- Condron, M.M.; Monien, B.H.; Bitan, G. Synthesis and purification of highly hydrophobic peptides derived from the C-terminus of amyloid β-protein. Open Biotechnol. J. 2008, 2, 87–93. [Google Scholar] [CrossRef]
- Tickler, A.K.; Wade, J.D. Current Protocols in Protein Science; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Mäde, V.; Els-Heindl, S.; Beck-Sickinger, A.G. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J. Org. Chem. 2014, 10, 1197–1212. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, A.S.; Babayan, B.G.; Avetisyan, N.S.; Mkrtchyan, A.G.; Hovhannisyan, A.M.; Hovhannisyan, N.A. Heterocyclic substituted non-protein amino acids as inhibitors of clostridium histolyticum collagenase. Arm. Biolog. J. 2018, 1, 12–15. [Google Scholar]
- Sargsyan, T.H.; Jamgaryan, S.M.; Gyulumyan, E.A.; Sargsyan, A.S.; Hakobyan, H.I.; Mardiyan, Z.Z. Targeted synthesis of N-tert-butyloxycarbonylglycyl-(S)-alanine tripeptide and study of its effect on collagenase activity. Chem. J. Arm. 2021, 74, 58–65. [Google Scholar]
- Belokon, Y.N.; Tararov, V.I.; Maleev, V.I.; Savel’eva, T.F.; Ryzhov, M.G. Improved procedures for the synthesis of (S)-2-[N-(N′-benzylprolyl)amino]benzophenone (BPB) and Ni(II) complexes of Schiff’s bases derived from BPB and amino acids. Tetrahedron Asymmetry 1998, 9, 4249–4252. [Google Scholar] [CrossRef]
- Arsenov, M.A.; Stoletova, N.V.; Savel’yeva, T.F.; Smol’yakov, A.F.; Maleev, V.I.; Loginov, D.A.; Larionov, V.A. Asymmetric metal-templated route to amino acids with an isoquinolone core via a Rh(III)-catalyzed coupling of aryl hydroxamates with chiral propargylglycine Ni(II) complexes. Org. Biomol. Chem. 2022, 20, 9385–9391. [Google Scholar] [CrossRef] [PubMed]
- Parpart, S.; Petrosyan, A.; Ali Shah, S.J.; Adewale, R.A.; Ehlers, P.; Grigoryan, T.; Mkrtchyan, A.F.; Mardiyan, Z.Z.; Karapetyan, A.J.; Tsaturyan, A.H.; et al. Synthesis of optically pure (S)-2-amino-5-arylpent-4-ynoic acids by Sonogashira reactions and their potential use as highly selective potent inhibitors of aldose reductase. RSC Adv. 2015, 5, 107400–107412. [Google Scholar] [CrossRef]
- Sargsyan, A.; Hakobyan, H.; Mardiyan, Z.; Jamharyan, S.; Dadayan, A.; Sargsyan, T.; Hovhannisyan, N. Modeling, synthesis and in vitro screening of unusual amino acids and peptides as protease inhibitors. J. Chem. Technol. Metallurgy 2023, 58, 615–620. [Google Scholar]
- Hakobyan, H.; Mardiyan, Z.; Khachaturyan, N.; Gevorgyan, S.; Jamgaryan, S.; Gyulumyan, E.; Danghyan, Y.; Sargsyan, T.; Saghyan, A. Synthesis of (S)-alanyl-(S)-β-(thiazol-2-yl-carbamoyl)-α-alanine, dipeptide containing it and in vitro investigation of the antifungal activity. Farmacia 2022, 70, 1148–1154. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Ran, L.Y.; Li, C.Y.; Chen, X.L. Diversity, structures, and collagen-degrading mechanisms of bacterial collagenolytic proteases. Appl. Environ. Microbiol. 2015, 81, 6098–6107. [Google Scholar] [CrossRef]
- Schönauer, E.; Kany, A.M.; Haupenthal, J.; Hüsecken, K.; Hoppe, I.J.; Voos, K.; Yahiaoui, S.; Elsässer, B.; Ducho, C.; Brandstetter, H.; et al. Discovery of a potent inhibitor class with high selectivity toward clostridial collagenases. J. Am. Chem. Soc. 2017, 139, 12696–12703. [Google Scholar] [CrossRef]
- Hartmann, A.; Gostner, J.; Fuchs, J.E.; Chaita, E.; Aligiannis, N.; Skaltsounis, L.; Ganzera, M. Inhibition of collagenase by mycosporine-like amino acids from Marine sources. Planta Med. 2015, 81, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Eckhard, U.; Schönauer, E.; Nüss, D.; Brandstetter, H. Structure of collagenase G reveals a chew-and-digest mechanism of bacterial collagenolysis. Nat. Struct. Mol. Biol. 2011, 18, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.; Goodsell, D.; Halliday, R.; Huey, R.; Hart, W.; Belew, R.; Olson, A. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Matsushita, O.; Koide, T.; Kobayashi, R.; Nagata, K.; Okabe, A. Substrate recognition by the collagen-binding domain of Clostridium histolyticum class I collagenase. J. Biol. Chem. 2001, 276, 8761–8770. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
 | ||
|---|---|---|
| Aryl Bromide | Product | Yield, % b |
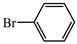 | complex 2a | 75 |
| AA 3a | 94 | |
 | complex 2b | 70 |
| AA 3b | 91 | |
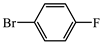 | complex 2c | 75 |
| AA 3c | 96 | |
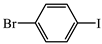 | complex 2d | 68 |
| AA 3d | 71 | |
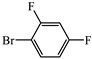 | complex 2e | 69 |
| AA 3e | 95 | |
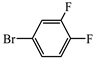 | complex 2f | 63 |
| AA 3f | 72 | |
| Run | Compound | ΔG (kcal/mol) | IC50 (mM) |
|---|---|---|---|
| 1 | 3a | −6.2 | 1.25 |
| 2 | 3b | −5.9 | 0.93 |
| 3 | 3c | −6.0 | 1.45 |
| 4 | 3d | −6.6 | 1.14 |
| 5 | 3e | −6.7 | 2.57 |
| 6 | 3f | −6.1 | 0.59 |
| 7 | 6 | −9.2 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakobyan, H.I.; Jamgaryan, S.M.; Sargsyan, A.S.; Danghyan, Y.M.; Larionov, V.A.; Maleev, V.I.; Saghyan, A.S.; Mardiyan, Z.Z. A Stereoselective Entry to Enantiopure (S)-2-Amino-2-methyl-5-arylpent-4-ynoic Acids and Evaluation of Their Inhibitory Activity against Bacterial Collagenase G. Symmetry 2023, 15, 1924. https://doi.org/10.3390/sym15101924
Hakobyan HI, Jamgaryan SM, Sargsyan AS, Danghyan YM, Larionov VA, Maleev VI, Saghyan AS, Mardiyan ZZ. A Stereoselective Entry to Enantiopure (S)-2-Amino-2-methyl-5-arylpent-4-ynoic Acids and Evaluation of Their Inhibitory Activity against Bacterial Collagenase G. Symmetry. 2023; 15(10):1924. https://doi.org/10.3390/sym15101924
Chicago/Turabian StyleHakobyan, Hegine I., Silva M. Jamgaryan, Armen S. Sargsyan, Yuri M. Danghyan, Vladimir A. Larionov, Victor I. Maleev, Ashot S. Saghyan, and Zorayr Z. Mardiyan. 2023. "A Stereoselective Entry to Enantiopure (S)-2-Amino-2-methyl-5-arylpent-4-ynoic Acids and Evaluation of Their Inhibitory Activity against Bacterial Collagenase G" Symmetry 15, no. 10: 1924. https://doi.org/10.3390/sym15101924
APA StyleHakobyan, H. I., Jamgaryan, S. M., Sargsyan, A. S., Danghyan, Y. M., Larionov, V. A., Maleev, V. I., Saghyan, A. S., & Mardiyan, Z. Z. (2023). A Stereoselective Entry to Enantiopure (S)-2-Amino-2-methyl-5-arylpent-4-ynoic Acids and Evaluation of Their Inhibitory Activity against Bacterial Collagenase G. Symmetry, 15(10), 1924. https://doi.org/10.3390/sym15101924