
Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State


Abstract
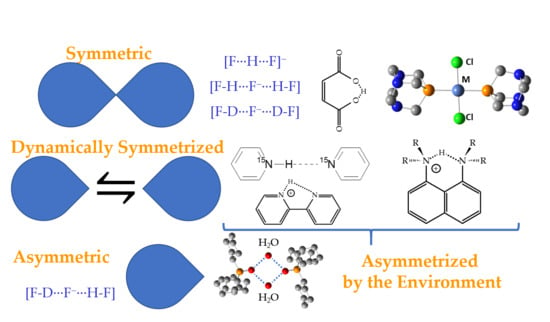
1. Introduction
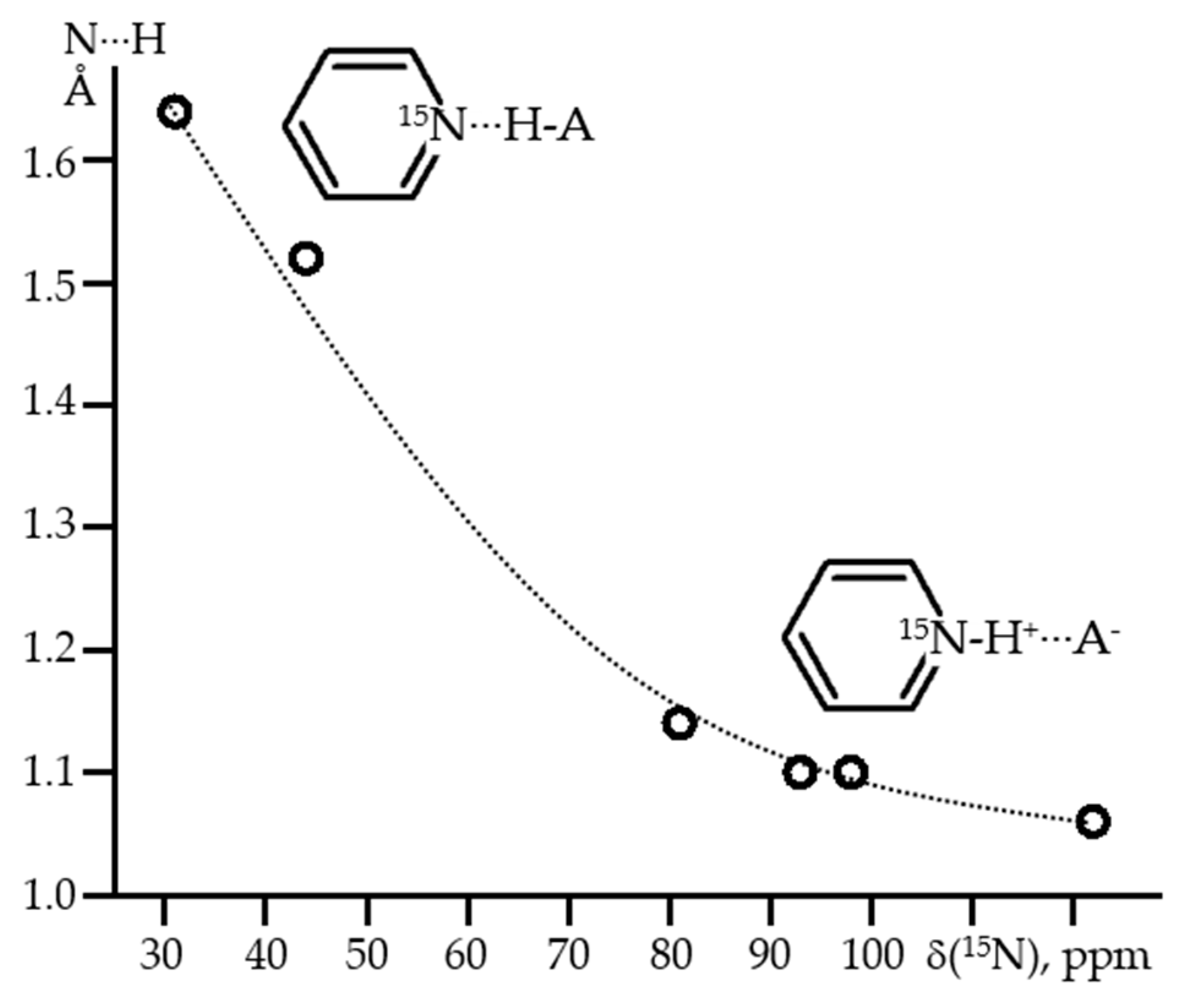
2. Hydrogen (Proton) as the Binding Unit
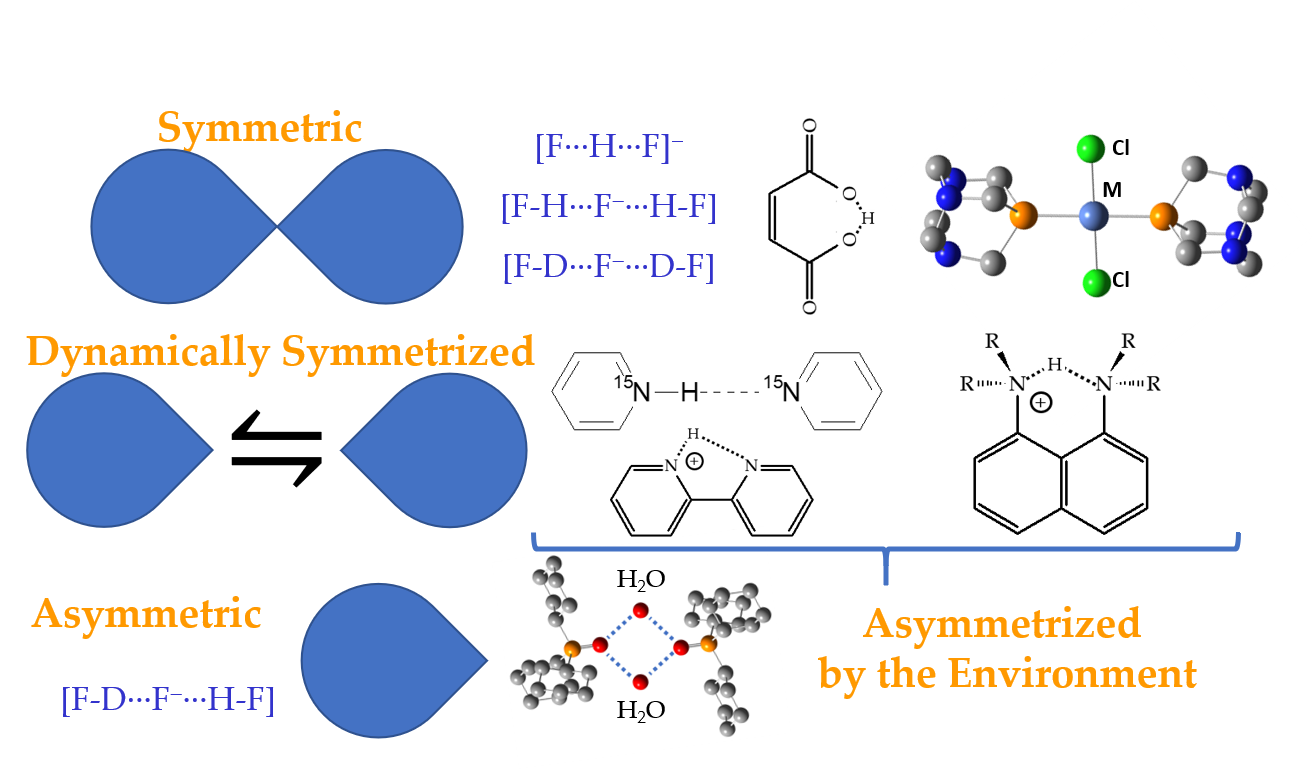
2.1. Intramolecular H-Bonds
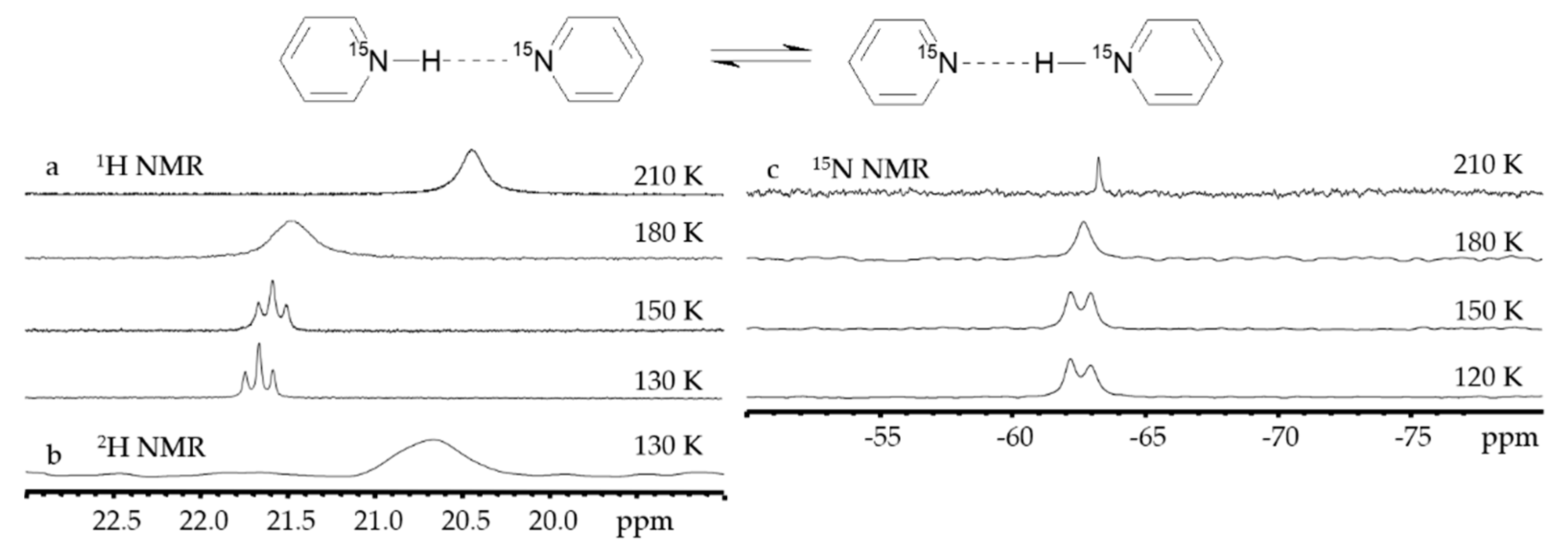
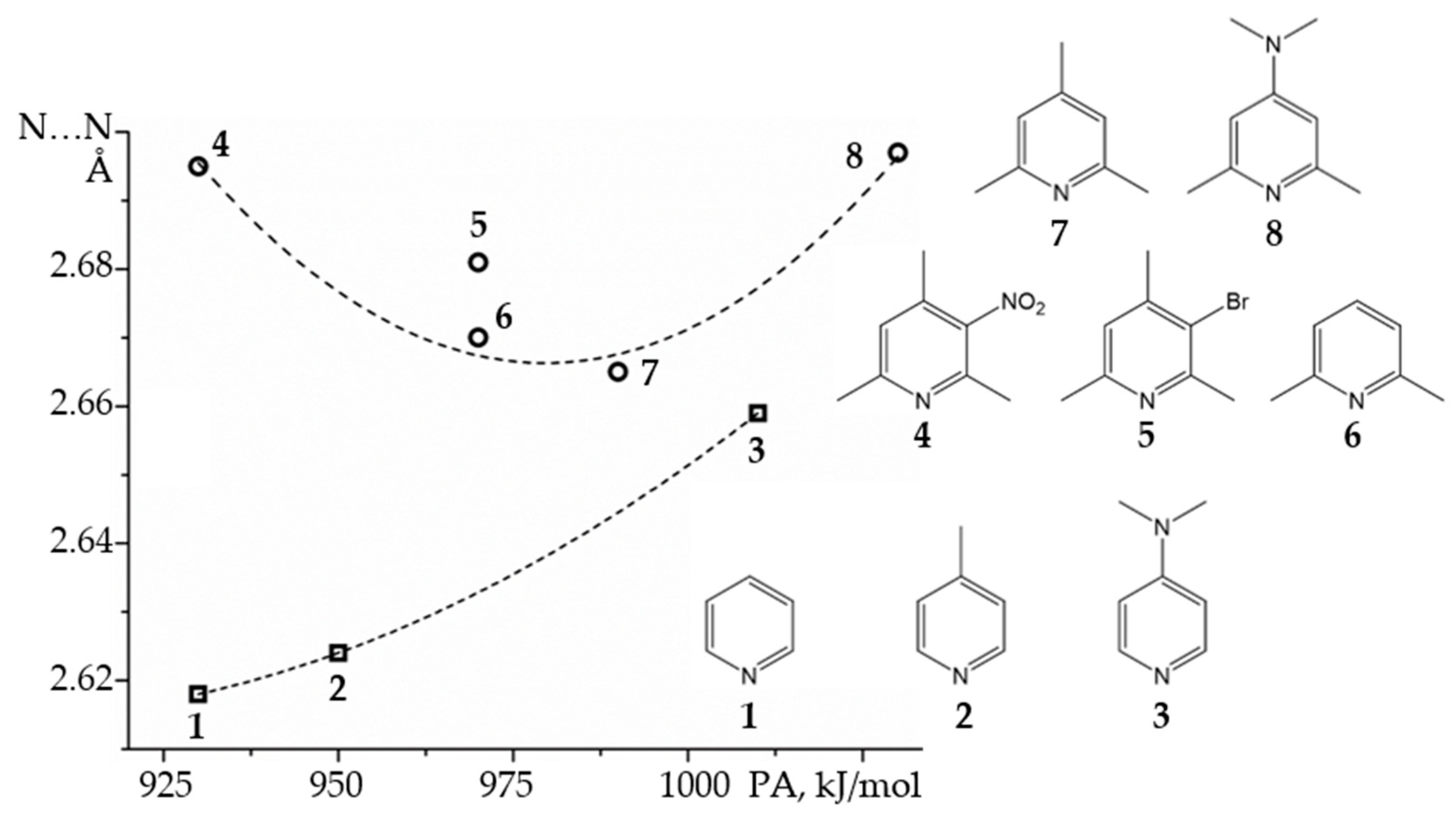
2.2. Proton-Bound Homodimers
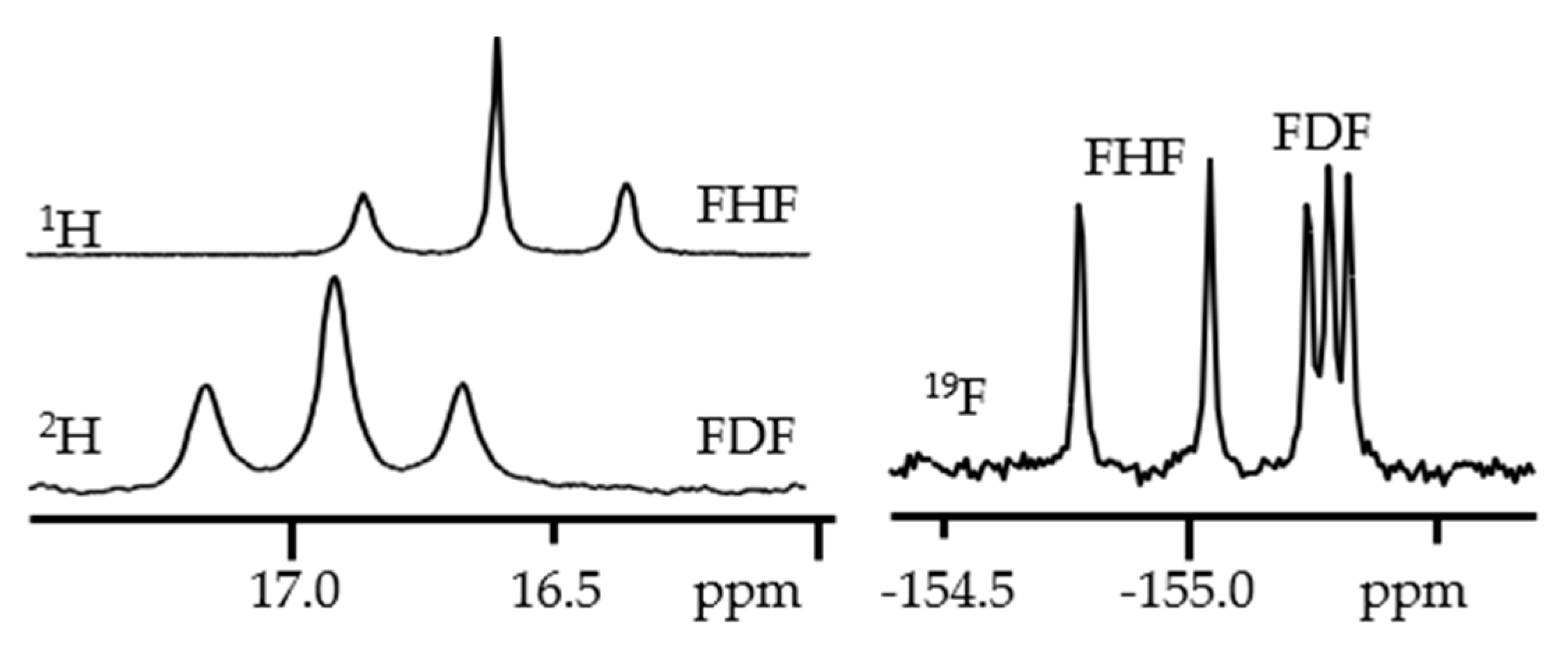
3. Halogen (Anion) as the Binding Unit
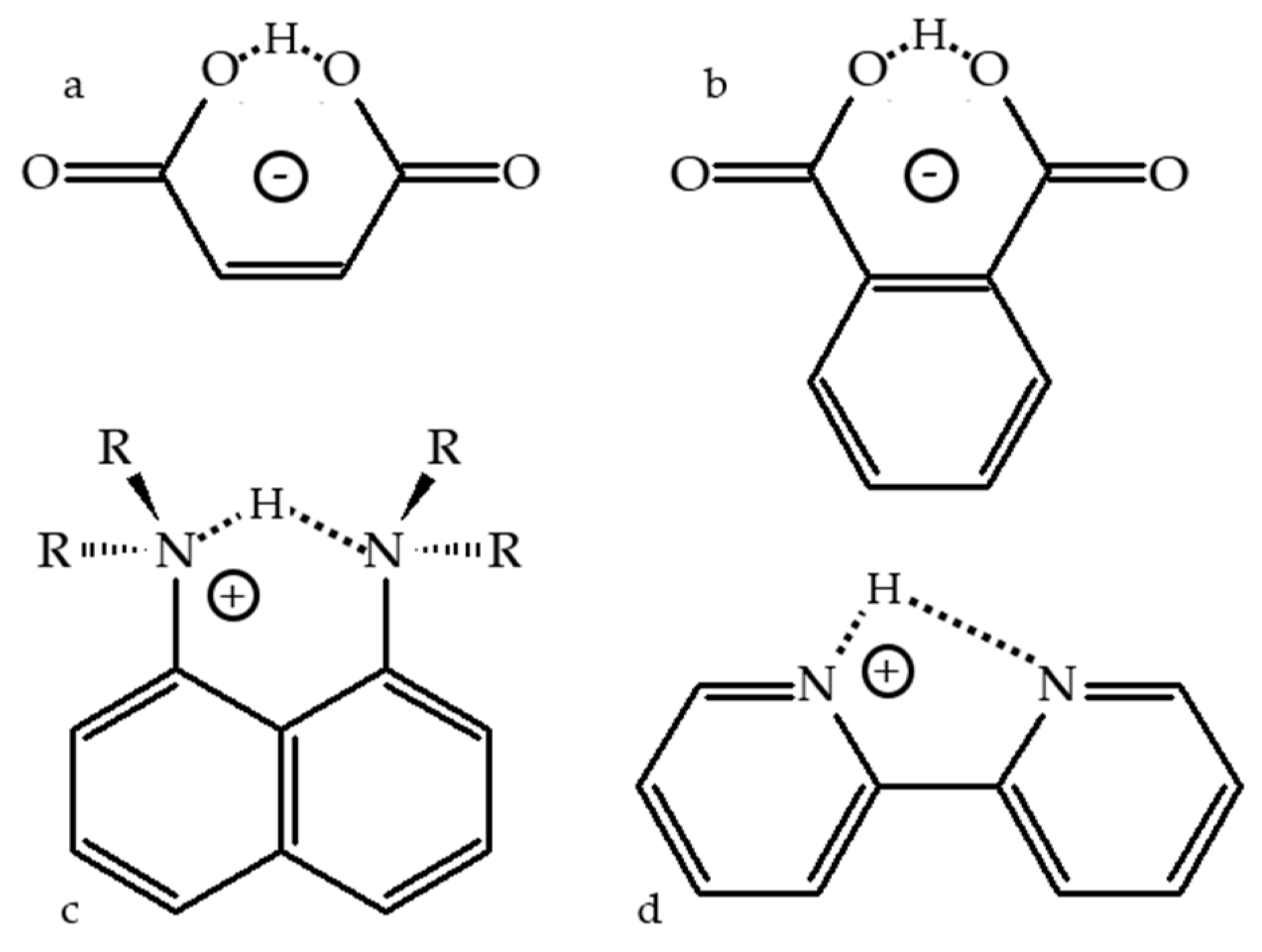
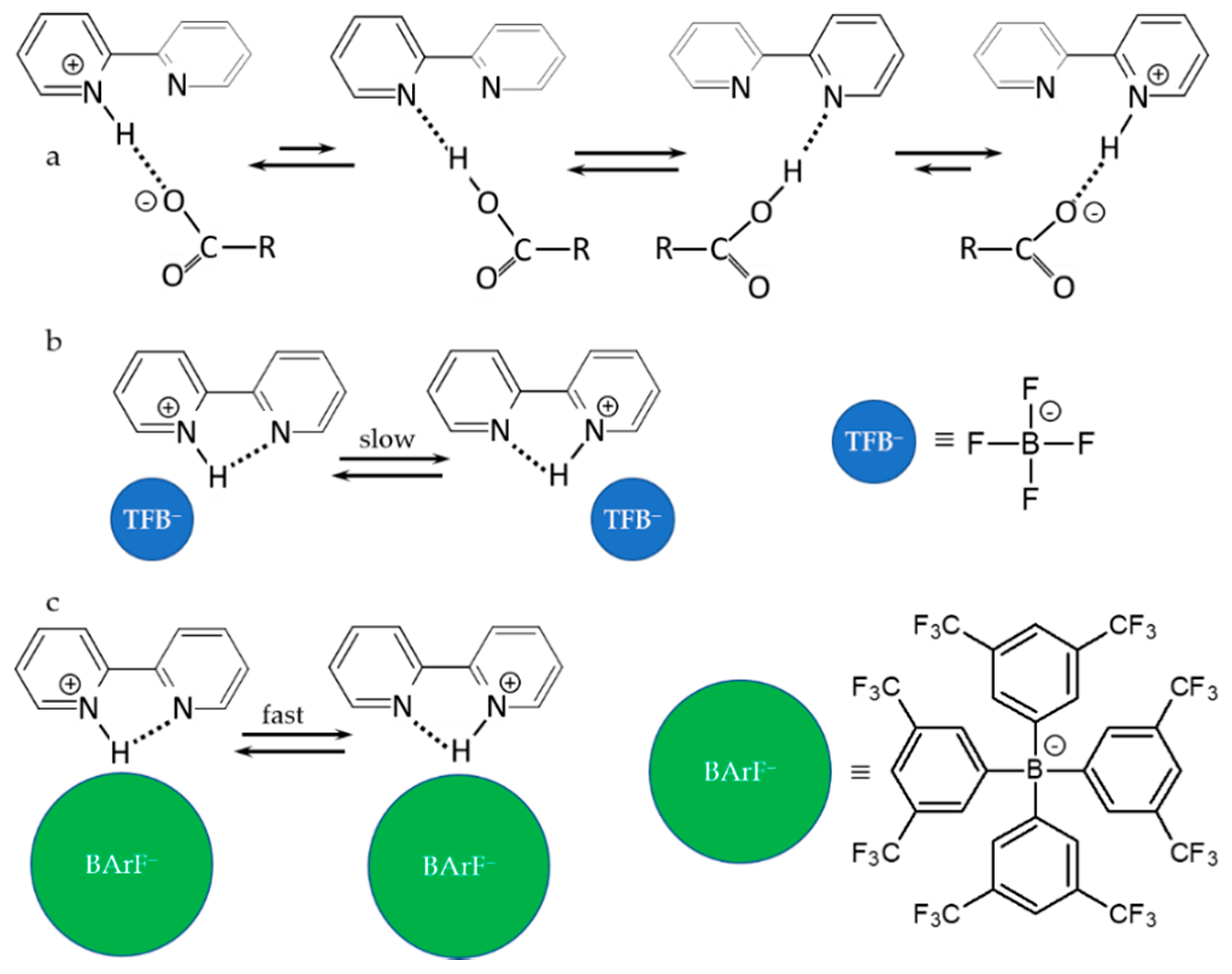
4. Metal (Cation) as the Binding Unit
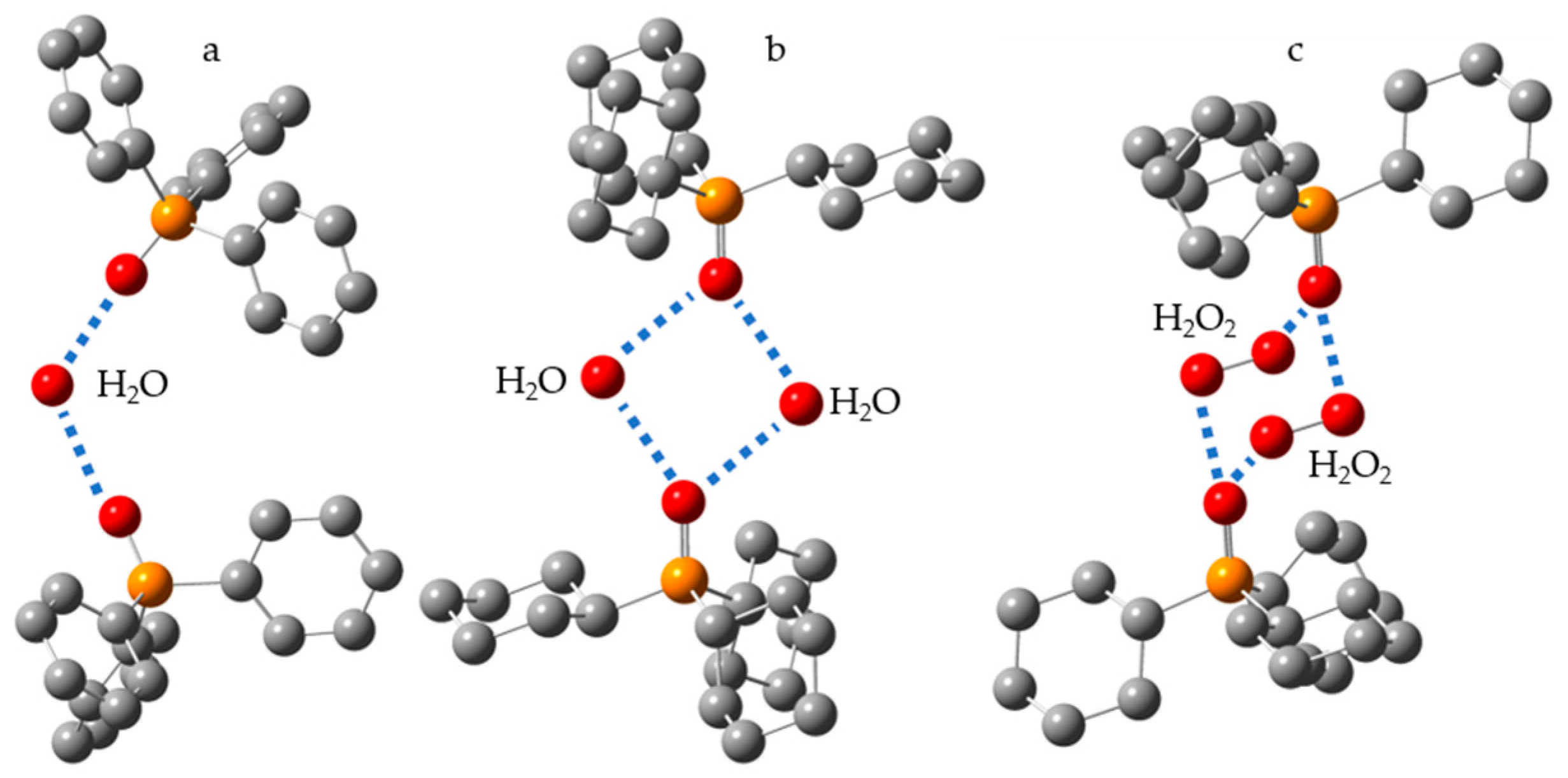
5. Water (Hydrogen Peroxide) as the Binding Unit
6. Conclusions
- (i)
- Steric hindrance and structural rigidity are not the only reasons why complexes can be asymmetric in the gas phase.
- (ii)
- At any given moment of time, the solvation shell is somewhat asymmetric.
- (iii)
- Dynamic processes in crystalline solids can be facilitated if the initial and final states are equivalent.
- (iv)
- The motion of the proton in a H-bond should always be treated as quantum.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mehlberg, H. Time, Causality, and the Quantum Theory. In Studies in the Philosophy of Science; Fawcett, C.R., Cohen, R.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar]
- Leifer, M.S.; Pusey, M.F. Is a time symmetric interpretation of quantum theory possible without retrocausality? Proc. R. Soc. A 2017, 473, 20160607. [Google Scholar] [CrossRef] [PubMed]
- Castilla, A.M.; Ramsay, W.J.; Nitschke, J.R. Stereochemistry in Subcomponent Self-Assembly. Acc. Chem. Res. 2014, 47, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Li, B.; Sarman, S.; Mocci, F.; Lu, Z.-Y.; Yuan, J.; Laaksonen, A.; Fayer, M.D. Microstructural and Dynamical Heterogeneities in Ionic Liquids. Chem. Rev. 2020, 120, 5798–5877. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Breed, D.; Manoharan, V.N.; Feng, L.; Hollingsworth, A.D.; Weck, M.; Pineet, D.J. Colloids with valence and specific directional bonding. Nature 2012, 491, 51–55. [Google Scholar] [CrossRef]
- Ungur, L.; Le Roy, J.J.; Korobkov, I.; Murugesu, M.; Chibotaru, L.F. Fine-tuning the Local Symmetry to Attain Record Blocking Temperature and Magnetic Remanence in a Single-Ion Magnet. Angew. Chem. Int. Ed. 2014, 53, 4413–4417. [Google Scholar] [CrossRef]
- Sun, W.-B.; Yan, P.-F.; Jiang, S.-D.; Wang, B.-W.; Zhang, Y.-Q.; Li, H.-F.; Chen, P.; Wang, Z.-M.; Gao, S. High symmetry or low symmetry, that is the question—high performance Dy(iii) single-ion magnets by electrostatic potential design. Chem. Sci. 2016, 7, 684–691. [Google Scholar] [CrossRef]
- Peng, J.; Cao, D.; He, Z.; Guo, J.; Hapala, P.; Ma, R.; Cheng, B.; Chen, J.; Xie, W.J.; Li, X.-Z.; et al. The effect of hydration number on the interfacial transport of sodium ions. Nature 2018, 557, 701–705. [Google Scholar] [CrossRef]
- Shenderovich, I.G. 1,3,5-Triaza-7-Phosphaadamantane (PTA) as a 31P NMR Probe for Organometallic Transition Metal Complexes in Solution. Molecules 2021, 26, 1390. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen Bond and Other Lewis Acid–Lewis Base Interactions as Preliminary Stages of Chemical Reactions. Molecules 2020, 25, 4668. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Ugalde, J.M.; Andrada, D.M.; Frenking, G. Comparison of Hydrogen and Gold Bonding in [XHX]-, [XAuX]-, and Isoelectronic [NgHNg]+, [NgAuNg]+ (X=Halogen, Ng=Noble Gas). Chem. Eur. J. 2016, 22, 11317–11328. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Review on DFT and ab initio Calculations of Scalar Coupling Constants. Int. J. Mol. Sci. 2003, 4, 64–92. [Google Scholar] [CrossRef]
- Kellman, M.E. Symmetry in chemistry from the hydrogen atom to proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14287–14294. [Google Scholar] [CrossRef] [PubMed]
- Zundel, G.; Metzger, H. Energiebänder der tunnelnden Überschuß-Protonen in flüssigen Säuren. Eine IR-spektroskopische Untersuchung der Natur der Gruppierungen H5O2+. Z. Phys. Chem. 1968, 58, 225–245. [Google Scholar] [CrossRef]
- Wicke, E.; Eigen, M.; Ackermann, T. Über den Zustand des Protons (Hydroniumions) in wäßriger Lösung. Z. Phys. Chem. 1954, 1, 340–364. [Google Scholar] [CrossRef]
- Carpenter, W.B.; Yu, Q.; Hack, J.H.; Dereka, B.; Bowman, J.M.; Tokmakoff, A. Decoding the 2D IR spectrum of the aqueous proton with high-level VSCF/VCI calculations. J. Chem. Phys. 2020, 153, 124506. [Google Scholar] [CrossRef]
- Wang, E.; Zhu, B.; Gao, Y. Discontinuous transition between Zundel and Eigen for H5O2+. Chin. Phys. B 2020, 29, 083101. [Google Scholar] [CrossRef]
- Li, C.; Swanson, J.M.J. Understanding and Tracking the Excess Proton in Ab Initio Simulations; Insights from IR Spectra. J. Phys. Chem. B 2020, 124, 5696–5708. [Google Scholar] [CrossRef]
- Vener, M.V.; Kong, S.; Levina, A.A.; Shenderovich, I.G. Spectroscopic Signatures of [H9O4]+ and [H13O6]+ Ions in a Polar Aprotic Environment Revealed under DFT-PCM Approximation. Acta Chim. Slov. 2011, 58, 402–410. [Google Scholar] [PubMed]
- Vener, M.V.; Librovich, N.B. The structure and vibrational spectra of proton hydrates: H5O2+ as a simplest stable ion. Int. Rev. Phys. Chem. 2009, 28, 407–434. [Google Scholar] [CrossRef]
- Shi, H.; Gong, L.-D.; Liu, C.; Lu, L.-N.; Yang, Z.-Z. ABEEM/MM OH- Models for OH-(H2O)n Clusters and Aqueous OH-: Structures, Charge Distributions, and Binding Energies. J. Phys. Chem. A 2020, 124, 5963–5978. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M. Fantasy versus reality in fragment-based quantum chemistry. J. Chem. Phys. 2019, 151, 170901. [Google Scholar] [CrossRef]
- Vener, M.V.; Shenderovich, I.G.; Rykounov, A.A. A qualitative study of the effect of a counterion and polar environment on the structure and spectroscopic signatures of a hydrated hydroxyl anion. Theor. Chem. Acc. 2013, 132, 1361. [Google Scholar] [CrossRef]
- Librovich, N.B.; Sakun, V.P.; Sokolov, N.D. H+ and OH- ions in aqueous solutions vibrational spectra of hydrates. Chem. Phys. 1979, 39, 351–366. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Winget, P.; Dolney, D.M.; Giesen, D.J.; Cramer, C.J.; Truhlar, D.G. Minnesota Solvent Descriptor Database. Available online: http://comp.chem.umn.edu/solvation/mnsddb.pdf (accessed on 26 April 2021).
- Onufriev, O. Implicit solvent models in molecular dynamics simulations: A brief overview. Annu. Rep. Comput. Chem. 2008, 4, 125–137. [Google Scholar] [CrossRef]
- Orozco, M.; Luque, F.J. Theoretical methods for the description of the solvent effect in biomolecular systems. Chem. Rev. 2000, 100, 4187–4225. [Google Scholar] [CrossRef]
- Pylaeva, S.; Allolio, C.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H.; Sebastiani, D.; Tolstoy, P.M. Proton transfer in a short hydrogen bond caused by solvation shell fluctuations: An ab initio MD and NMR/UV study of an (OHO)−bonded system. Phys. Chem. Chem. Phys. 2015, 17, 4634–4644. [Google Scholar] [CrossRef]
- Woo, H.-K.; Wang, X.-B.; Wang, L.-S.; Lau, K.-C. Probing the Low-Barrier Hydrogen Bond in Hydrogen Maleate in the Gas Phase: A Photoelectron Spectroscopy and ab Initio Study. J. Phys. Chem. A 2005, 109, 10633–10637. [Google Scholar] [CrossRef]
- Garcia-Viloca, M.; González-Lafont, A.; Lluch, J.M. Theoretical Study of the Low-Barrier Hydrogen Bond in the Hydrogen Maleate Anion in the Gas Phase. Comparison with Normal Hydrogen Bonds. J. Am. Chem. Soc. 1997, 119, 1081–1086. [Google Scholar] [CrossRef]
- Kiefer, P.M.; Hynes, J.T. Theoretical aspects of tunneling protontransfer reactions in a polar environment. J. Phys. Org. Chem. 2010, 23, 632–646. [Google Scholar] [CrossRef]
- Limbach, H.-H.; Tolstoy, P.M.; Pérez-Hernández, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO Hydrogen Bond Geometries and NMR Chemical Shifts: From Equilibrium Structures to Geometric H/D Isotope Effects, with Applications for Water, Protonated Water, and Compressed Ice. Isr. J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Vener, M.V. Model study of the primary H/D isotope effects on the NMR chemical shift in strong hydrogen-bonded systems. Chem. Phys. 1992, 166, 311–316. [Google Scholar] [CrossRef]
- Schah-Mohammedi, P.; Shenderovich, I.G.; Detering, C.; Limbach, H.-H.; Tolstoy, P.M.; Smirnov, S.N.; Denisov, G.S.; Golubev, N.S. H/D-Isotope Effects on NMR Chemical Shifts and Symmetry of Homoconjugated Hydrogen-Bonded Ions in Polar Solution. J. Am. Chem. Soc. 2000, 122, 12878–12879. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Burtsev, I.G.; Denisov, G.S.; Golubev, N.S.; Limbach, H.-H. Influence of the Temperature-Dependent Dielectric Constant on the H/D Isotope Effects on the NMR Chemical Shifts and the Hydrogen Bond Geometry of Collidine-HF Complex in CDF3/CDClF2 Solution. Magn. Reson. Chem. 2001, 39, S91–S99. [Google Scholar] [CrossRef]
- Gunnarsson, G.; Wennerström, H.; Egan, W.; Forsén, S. Proton and deuterium NMR of hydrogen bonds: Relationship between isotope effects and the hydrogen bond potential. Chem. Phys. Lett. 1976, 38, 96–99. [Google Scholar] [CrossRef]
- Perrin, C.L.; Nielson, J.B. Asymmetry of Hydrogen Bonds in Solutions of Monoanions of Dicarboxylic Acids. J. Am. Chem. Soc. 1997, 119, 12734–12741. [Google Scholar] [CrossRef]
- Perrin, C.L. Are Short, Low-Barrier Hydrogen Bonds Unusually Strong? Acc. Chem. Res. 2010, 43, 1550–1557. [Google Scholar] [CrossRef]
- Mavri, J.; Hodošček, M.; Hadži, D. Ab initio SCF and Møller-Plesset calculations on the hydrogen bond in hydrogen malonate: Effects of neighbour ions and polarizable medium. J. Mol. Struct. 1990, 209, 421–431. [Google Scholar] [CrossRef]
- Mavri, J.; Hadži, D. Influence of solvation on the hydrogen bond in hydrogen malonate an ab initio and semiempirical study. J. Mol. Struct. Theor. Chem. 1998, 432, 257–262. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Solid State NMR for Nonexperts: An overview of simple but general practical methods. Solids 2021, 2, 139–154. [Google Scholar] [CrossRef]
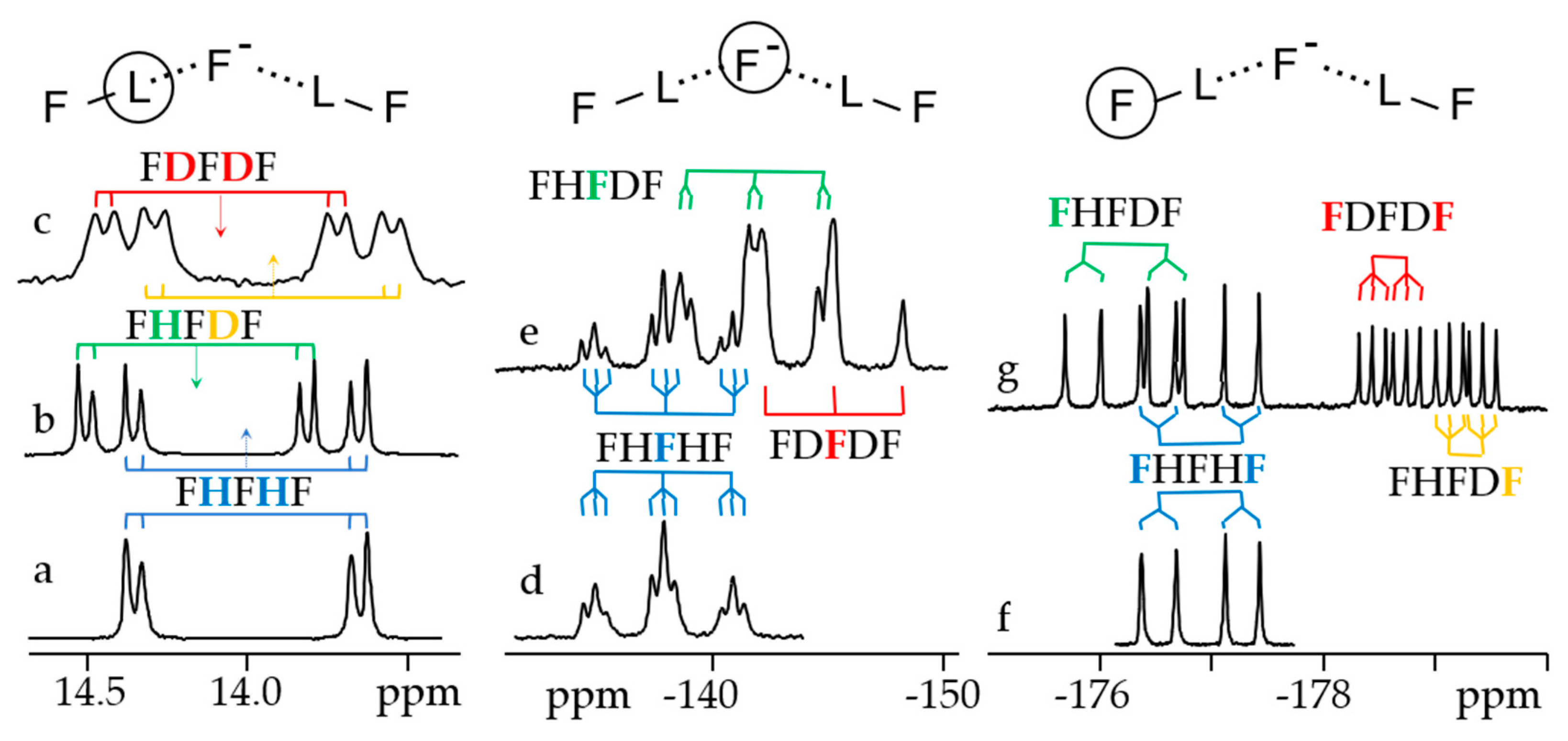
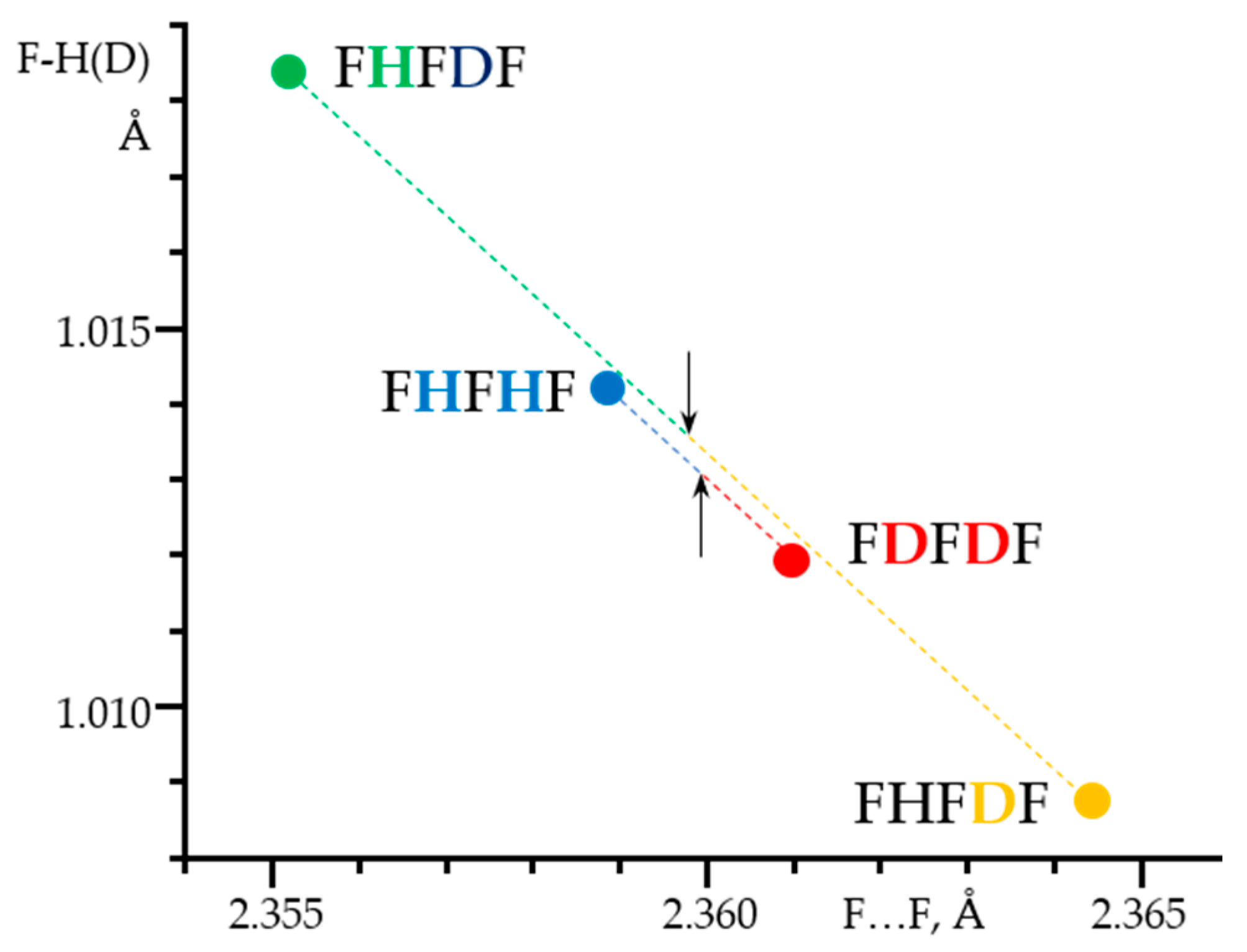
- Golubev, N.S.; Detering, C.; Smirnov, S.N.; Shenderovich, I.G.; Denisov, G.S.; Limbach, H.-H.; Tolstoy, P.M. H/D Isotope Effects on NMR Chemical Shifts of Nuclei Involved in a Hydrogen Bridge of Hydrogen Isocyanide Complexes with Fluoride Anion. Phys. Chem. Chem. Phys. 2009, 11, 5154–5159. [Google Scholar] [CrossRef] [PubMed]
- Golubev, N.S.; Melikova, S.M.; Shchepkin, D.N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S. Interpretation of H/D Isotope Effects on NMR Chemical Shifts of [FHF]- Ion Based on Calculations of Nuclear Magnetic Shielding Tensor Surface. Z. Phys. Chem. 2003, 217, 1549–1563. [Google Scholar] [CrossRef]
- Di Muzio, S.; Ramondo, F.; Gontrani, L.; Ferella, F.; Nardone, M.; Benassi, P. Choline Hydrogen Dicarboxylate Ionic Liquids by X-ray Scattering, Vibrational Spectroscopy and Molecular Dynamics: H-Fumarate and H-Maleate and Their Conformations. Molecules 2020, 25, 4990. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Simplified Calculation Approaches Designed to Reproduce the Geometry of Hydrogen Bonds in Molecular Complexes in Aprotic Solvents. J. Chem. Phys. 2018, 148, 124313. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H. NMR Study of Conformational Exchange and Double-Well Proton Potential in Intramolecular Hydrogen Bonds in Monoanions of Succinic Acid and Derivatives. J. Phys. Chem. A 2011, 115, 9828–9836. [Google Scholar] [CrossRef]
- Malaspina, L.A.; Edwards, A.J.; Woińska, M.; Jayatilaka, D.; Turner, M.J.; Price, J.R.; Herbst-Irmer, R.; Sugimoto, K.; Nishibori, E.; Grabowsky, S. Predicting the Position of the Hydrogen Atom in the Short Intramolecular Hydrogen Bond of the Hydrogen Maleate Anion from Geometric Correlations. Cryst. Growth Des. 2017, 17, 3812–3825. [Google Scholar] [CrossRef]
- Hussain, M.S.; Schlemper, E.; Fair, C. Neutron diffraction study of deuterated imidazolium hydrogen maleate: An evaluation of isotope effect on the hydrogen-bond length. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1980, 36, 1104–1108. [Google Scholar] [CrossRef]
- Hsu, B.; Schlemper, E. X−N deformation density studies of the hydrogen maleate ion and the imidazolium ion. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1980, 36, 3017–3023. [Google Scholar] [CrossRef]
- Olovsson, G.; Olovsson, I.; Lehmann, M.S. Neutron diffraction study of sodium hydrogen maleate trihydrate, NaH[C4H2O4].3H2O, at 120 K. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1984, 40, 1521–1526. [Google Scholar] [CrossRef]
- Vanhouteghem, F.; Lenstra, A.; Schweiss, P. Magnesium bis (hydrogen maleate) hexahydrate, [Mg(C4H3O4)2].6H2O, studied by elastic neutron diffraction and ab initio calculations. Acta Crystallogr. Sect. B Struct. Sci. 1987, 43, 523–528. [Google Scholar] [CrossRef]
- Sequeira, A.; Rajagopal, H.; Gupta, M.; Vanhouteghem, F.; Lenstra, A.; Geise, H. Tetraaquabis (hydrogen maleato) zinc (II) by neutron diffraction and tetraaquabis (hydrogen maleato) nickel (II) by high-order X-ray diffraction. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1992, 48, 1192–1197. [Google Scholar] [CrossRef]
- Madsen, D.; Flensburg, C.; Larsen, S. Properties of the experimental crystal charge density of methylammonium hydrogen maleate. A salt with a very short intramolecular O-H-O hydrogen bond. J. Phys. Chem. A 1998, 102, 2177–2188. [Google Scholar] [CrossRef]
- Malaspina, L.A.; Hoser, A.A.; Edwards, A.J.; Woińska, M.; Turner, M.J.; Price, J.R.; Sugimoto, K.; Nishibori, E.; Bürgi, H.-B.; Jayatilaka, D.; et al. Hydrogen atoms in bridging positions from quantum crystallographic refinements: Influence of hydrogen atom displacement parameters on geometry and electron density. CrystEngComm 2020, 22, 4778. [Google Scholar] [CrossRef]
- Poręba, T.; Macchi, P.; Casati, N. Structural Variety of Alkali Hydrogen Maleates at High Pressure. Cryst. Growth Des. 2020, 20, 4375–4386. [Google Scholar] [CrossRef]
- Garcia-Viloca, M.; González-Lafont, A.; Lluch, J.M. Asymmetry of the Hydrogen Bond of Hydrogen Phthalate Anion in Solution. A QM/MM Study. J. Am. Chem. Soc. 1999, 121, 9198–9207. [Google Scholar] [CrossRef]
- Perrin, C.L.; Lau, J.S. Hydrogen-Bond Symmetry in Zwitterionic Phthalate Anions: Symmetry Breaking by Solvation. J. Am. Chem. Soc. 2006, 128, 11820–11824. [Google Scholar] [CrossRef]
- Perrin, C.L. Symmetry of hydrogen bonds in solution. Pure Appl. Chem. 2009, 81, 571–583. [Google Scholar] [CrossRef]
- Mori, Y.; Masuda, Y. Effect of solvent on proton location and dynamic behavior in short intramolecular hydrogen bonds studied by molecular dynamics simulations and NMR experiments. Chem. Phys. 2015, 458, 18–29. [Google Scholar] [CrossRef]
- Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. Intramolecular Hydrogen Bond Patterns and Proton Dynamics in Nitrophthalic Acid Associates. Molecules 2020, 25, 4720. [Google Scholar] [CrossRef] [PubMed]
- Zhurov, V.V.; Pinkerton, A.A. Quantifying the Inter- and Intramolecular Interactions in Crystalline Phthalic Acid. Cryst. Growth Des. 2014, 14, 5685–5691. [Google Scholar] [CrossRef]
- Borissova, A.O.; Lyssenko, K.A.; Gurinov, A.A.; Shenderovich, I.G. Energy Analysis of Competing Non-Covalent Interaction in 1:1 and 1:2 Adducts of Collidine with Benzoic Acids by Means of X-Ray Diffraction. Z. Phys. Chem. 2013, 227, 775–790. [Google Scholar] [CrossRef]
- Küppers, H.; Takusagawa, F.; Koetzle, T.F. Neutron diffraction study of lithium hydrogen phthalate monohydrate: A material with two very short intramolecular O···H···O hydrogen bonds. J. Chem. Phys. 1985, 82, 5636. [Google Scholar] [CrossRef]
- Langkilde, A.; Madsen, D.; Larsen, S. Structures of three salts of phthalic acid; variation in crystal packing and geometry of the hydrogen phthalate ion. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Piękoś, P.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Pozharskii, A.F.; Antonov, A.S.; Tolstoy, P.M.; Filarowski, A. Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene. Symmetry 2020, 12, 1924. [Google Scholar] [CrossRef]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. Chem. Commun. 1968, 723–724. [Google Scholar] [CrossRef]
- Pozharskii, A.F. Naphthalene “Proton Sponges”. Russ. Chem. Rev. 1998, 67, 1–27. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Basicity and Proton Transfer in Proton Sponges and Related Compounds: An Ab Initio Study. Struct. Chem. 2000, 11, 335–340. [Google Scholar] [CrossRef]
- Perrin, C.L.; Ohta, B.K. Symmetry of N−H−N Hydrogen Bonds in 1,8-Bis(dimethylamino)naphthalene·H+ and 2,7-Dimethoxy-1,8-bis(dimethylamino)naphthalene·H+. J. Am. Chem. Soc. 2001, 123, 6520–6526. [Google Scholar] [CrossRef]
- Pietrzak, M.; Wehling, J.P.; Kong, S.; Tolstoy, P.M.; Shenderovich, I.G.; Lopez, C.; Claramunt, R.M.; Elguero, J.; Denisov, G.S.; Limbach, H.-H. Symmetrization of Cationic Hydrogen Bridges of Protonated Sponges Induced by Solvent and Counteranion Interactions as Revealed by NMR Spectroscopy. Chem. Eur. J. 2010, 16, 1679–1690. [Google Scholar] [CrossRef]
- Jones, A.O.F.; Kallay, A.A.; Lloyd, H.; McIntyre, G.J.; Wilson, C.C.; Thomas, L.H. The Effect of Local Crystalline Environment on Hydrogen Atom Behavior in Molecular Complexes of a Proton Sponge. Cryst. Growth Des. 2016, 16, 2123–2129. [Google Scholar] [CrossRef]
- Lopez, C.; Lorente, P.; Claramunt, R.M.; Marin, J.; Foces-Foces, C.; Llamas-Saiz, A.L.; Elguero, J.; Limbach, H.H. Localization of Hydrogen Bond Deuterons in Proton Sponges by Dipolar Solid State 15N NMR Spectroscopy. Ber. Bunsenges. Phys. Chem. Chem. Phys. 1998, 102, 414–418. [Google Scholar] [CrossRef]
- Woźniak, K.; Wilson, C.C.; Knight, K.S.; Jones, W.; Grech, E. Neutron Diffraction of a Complex of 1,8-Bis(dimethylamino)naphthalene with 1,2-Dichloromaleic Acid. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1996, 52, 691–696. [Google Scholar] [CrossRef]
- Llamas-Saiz, A.L.; Foces-Foces, C.; Elguero, J. Proton sponges. J. Mol. Struct. 1994, 328, 297–323. [Google Scholar] [CrossRef]
- Sobczyk, L. The specificity of the [NHN]+ hydrogen bonds in protonated naphthalene proton sponges. J. Mol. Struct. 2010, 972, 59–63. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, L. Symmetry and 1H NMR chemical shifts of short hydrogen bonds: Impact of electronic and nuclear quantum effects. Phys. Chem. Chem. Phys. 2020, 22, 4884–4895. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.; Ozeryanskii, V.A.; Sobczyk, L.; Pozharskii, A.F. Primary 1H/2H isotope effect in the NMR chemical shift of HClO4 salts of 1,8-bis(dimethylamino)naphthalene derivatives. J. Phys. Org. Chem. 2007, 20, 643–648. [Google Scholar] [CrossRef]
- White, P.B.; Hong, M. 15N and 1H Solid-State NMR Investigation of a Canonical Low-Barrier Hydrogen-Bond Compound: 1,8-Bis(dimethylamino)naphthalene. J. Phys. Chem. B 2015, 119, 11581–11589. [Google Scholar] [CrossRef]
- Gregorovic, A.; Apih, T.; Zagara, V.; Seliger, J. 14N NQR spectroscopy reveals the proton position in N–H N bonds: A case study with proton sponges. Phys. Chem. Chem. Phys. 2019, 21, 306–313. [Google Scholar] [CrossRef]
- Degtyarev, A.V.; Ryabtsova, O.V.; Pozharskii, A.F.; Ozeryanskii, V.A.; Starikova, Z.A.; Sobczyk, L.; Filarowski, A. 2,7-disubstituted proton sponges as borderline systems for investigating barrier-free intramolecular hydrogen bonds. Protonated 2,7-bis(trimethylsilyl)- and 2,7-di(hydroxymethyl)-1,8-bis(dimethylamino)naphthalenes. Tetrahedron 2008, 64, 6209–6214. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Spin-spin coupling across intramolecular N-H+-N hydrogen bonds in models for proton sponges: An ab initio investigation. Magn. Reson. Chem. 2008, 46, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Pawlukojć, A.; Natkaniec, I.; Grech, E.; Baran, J.; Malarski, Z.; Sobczyk, L. Incoherent inelastic neutron scattering, Raman and IR absorption studies on 1,8-bis(dimethylamino)naphthalene and its protonated forms. Spectrochim. Acta A 1998, 54, 439–448. [Google Scholar] [CrossRef]
- Antonov, A.S.; Pozharskii, A.F.; Tolstoy, P.M.; Filarowski, A.; Khoroshilova, O.V. 1,8-Bis(dimethylamino) naphthyl-2-ketimines: Inside vs. outside protonation. Beilstein J. Org. Chem. 2018, 14, 2940–2948. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, A.; Panek, J.J. “Zwitterionic proton sponge” hydrogen bonding investigations on the basis of Car-Parrinello molecular dynamics. J. Chem. Inf. Model. 2015, 55, 1148–1157. [Google Scholar] [CrossRef]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Antonov, A.S.; Filarowski, A. Out-Basicity of 1,8-bis(dimethylamino) naphthalene: The experimental and theoretical challenge. Org. Biomol. Chem. 2014, 12, 2360–2369. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Degtyarev, A.V.; Ryabtsova, O.V.; Ozeryanskii, V.A.; Kletskii, M.E.; Starikova, Z.A.; Sobczyk, L.; Filarowski, A. 2-α-hydroxyalkyl- and 2,7-di(α-hydroxyalkyl)-1,8-bis(dimethylamino)naphthalenes: Stabilization of nonconventional in/out conformers of “proton sponges” via N···H-O intramolecular hydrogen bonding. A remarkable kind of tandem nitrogen inversion. J. Org. Chem. 2007, 72, 3006–3019. [Google Scholar] [CrossRef]
- Howard, S.T. Conformers, Energetics, and Basicity of 2,2′-Bipyridine. J. Am. Chem. Soc. 1996, 118, 10269–10274. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Kamdem, N.; Mauder, D.; Denisov, G.S.; Shenderovich, I.G. Studies of Adsorption of 2,2′-Bipyridyl on the Surface of Highly Regulated Silica Matrix of the MCM-41 Type by Means of 15N NMR Spectroscopy. Russ. J. Gen. Chem. 2010, 80, 2027–2031. [Google Scholar] [CrossRef]
- Kühn, F.E.; Groarke, M.; Bencze, E.; Herdtweck, E.; Prazeres, A.; Santos, A.M.; Calhorda, M.J.; Romao, C.C.; Goncalves, I.S.; Lopes, A.D.; et al. Octahedral Bipyridine and Bipyrimidine Dioxomolybdenum(VI) Complexes: Characterization, Application in Catalytic Epoxidation, and Density Functional Mechanistic Study. Chem. Eur. J. 2002, 8, 2370–2383. [Google Scholar] [CrossRef]
- Lavender, E.S.; Glidewell, C.; Ferguson, G. 1,3,5-Trihydroxybenzene-2,2′-Bipyridyl (1/2): A Hydrogen-Bonded Structure Based on a Stem-and-Leaves Motif. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1637–1639. [Google Scholar] [CrossRef]
- Vogler, A.; Shenderovich, I.G. Photochemistry of deprotonated rhenium(I) (3,3’-dihydroxy-2,2’-bipyridine) tricarbonyl chloride. Photoisomerization at the chelate in basic solution. Inorg. Chim. Acta 2014, 421, 496–499. [Google Scholar] [CrossRef]
- Morancho, R.; Pouvreau, P.; Constant, G.; Jaud, J.; Galy, J. Synthese et etude structurale d’un nouveau compose du silicium: Di-2,2′-bipyridine silicium. J. Organomet. Chem. 1979, 166, 329–338. [Google Scholar] [CrossRef]
- Barker, D.J.; Summers, L.A.; Cooney, R.P. Conformations of 2,2′-bipyridine in acidic media. J. Mol. Struct. 1987, 159, 249–254. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Tolstoy, P.M.; Limbach, H.-H.; Shenderovich, I.G. Counteranion-Dependent Mechanisms of Intramolecular Proton Transfer in Aprotic Solution. Phys. Chem. Chem. Phys. 2010, 12, 10373–10379. [Google Scholar] [CrossRef]
- Chan, B.; Del Bene, J.E.; Radom, L. What factors determine whether a proton-bound homodimer has a symmetric or an asymmetric hydrogen bond? Mol. Phys. 2009, 107, 1095–1105. [Google Scholar] [CrossRef]
- Chan, B.; Del Bene, J.E.; Radom, L. Proton-Bound Homodimers: How Are the Binding Energies Related to Proton Affinities? J. Am. Chem. Soc. 2007, 129, 12197–12199. [Google Scholar] [CrossRef]
- Chan, B.; Del Bene, J.E.; Radom, L. Proton-bound homodimers involving second-row atoms. Theor. Chem. Acc. 2012, 131, 1088. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Ugalde, J.M. High-level ab initio calculations on low barrier hydrogen bonds and protonbound homodimers. Chem. Phys. Lett. 2010, 493, 37–44. [Google Scholar] [CrossRef]
- Cotton, C.E.; Francisco, J.S.; Klemperer, W. Computational study of the linear proton bound ion–molecule complexes of HCNH+ with HCN and HNC. J. Chem. Phys. 2013, 139, 014304. [Google Scholar] [CrossRef]
- Denisov, G.S.; Melikova, S.M.; Rutkovskii, K.S.; Tokhadze, K.G. Spectral Diagnostics of the Dynamics of the Formation of a Homoconjugated Complex [HCN.H.NCH]+. Opt. Spectrosc. 2020, 128, 467–469. [Google Scholar] [CrossRef]
- Terrill, K.; Nesbitt, D.J. Ab initio anharmonic vibrational frequency predictions for linear proton-bound complexes OC–H+–CO and N2–H+–N2. Phys. Chem. Chem. Phys. 2010, 12, 8311–8322. [Google Scholar] [CrossRef] [PubMed]
- Fridgen, T.D.; Parnis, J.M. Electron bombardment matrix isolation of Rg/Rg′/methanol mixtures (Rg=Ar, Kr, Xe): Fourier-transform infrared characterization of the proton-bound dimers Kr2H+, Xe2H+, (ArHKr)+ and (ArHXe)+ in Ar matrices and (KrHXe)+ and Xe2H+ in Kr matrices. J. Chem. Phys. 1998, 109, 2155. [Google Scholar] [CrossRef]
- Kunttu, H.; Seetula, J.; Räsänen, M.; Apkarian, V.A. Photogeneration of ions via delocalized charge transfer states. I. Xe2H+ and Xe2D+ in solid Xe. J. Chem. Phys. 1992, 96, 5630. [Google Scholar] [CrossRef]
- Grabowski, S.J. [FHF]-—The Strongest Hydrogen Bond under the Influence of External Interactions. Crystals 2016, 6, 3. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Gindin, V.A.; Golubev, N.S.; Dunger, A.; Reibke, R.; Kirpekar, S.; Malkina, O.L.; Limbach, H.-H. Nuclear Magnetic Resonance of Hydrogen Bonded Clusters between F- and (HF)n: Experiment and Theory. Ber. Bunsenges. Phys. Chem. Chem. Phys. 1998, 102, 422–428. [Google Scholar] [CrossRef]
- Panich, A.M. NMR study of the F-H···F hydrogen bond. Relation between hydrogen atom position and F-H···F bond length. Chem. Phys. 1995, 196, 511–519. [Google Scholar] [CrossRef]
- Fujiwara, F.Y.; Martin, J.S. Heterobihalide ions. Nuclear magnetic resonance spectroscopy of strong hydrogen bonds. J. Am. Chem. Soc. 1974, 96, 7625–7631. [Google Scholar] [CrossRef]
- Wenthold, P.G.; Squires, R.R. Bond Dissociation Energies of F2 and HF2-. A Gas-Phase Experimental and G2 Theoretical Study. J. Phys. Chem. 1995, 99, 2002–2005. [Google Scholar] [CrossRef]
- Yamdagni, R.; Kebarle, P. The Hydrogen Bond Energies in ClHCl- and Cl-(HCl)n. Can. J. Chem. 1974, 52, 2449–2453. [Google Scholar] [CrossRef]
- Cho, H.-G.; Andrews, L. Matrix Infrared Spectra, Photochemistry and Density Functional Calculations of Cl-−HCCl2, ClHCl-, Cl−ClCCl, and Cl-−HCHCl Produced from CHCl3 and CH2Cl2 Exposed to Irradiation from Laser Ablation. J. Phys. Chem. A 2019, 123, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Maiorov, V.D.; Sysoeva, S.G.; Temkin, O.N.; Kislina, L.S. IR Study of ion-molecular interactions in a DMF-HCl system. Russ. Chem. Bull. 1993, 42, 1511–1516. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Tolstoy, P.M.; Golubev, N.S.; Smirnov, S.N.; Denisov, G.S.; Limbach, H.-H. Low-Temperature NMR Studies of the Structure and Dynamics of a Novel Series of Acid-Base Complexes of HF with Collidine Exhibiting Scalar Couplings Across Hydrogen Bonds. J. Am. Chem. Soc. 2003, 125, 11710–11720. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, S.A.; Elgabarty, H.; Sebastiani, D.; Tolstoy, P.M. Symmetry and dynamics of FHF- anion in vacuum, in CD2Cl2 and in CCl4. Ab initio MD study of fluctuating solvent–solute hydrogen and halogen bonds. Phys. Chem. Chem. Phys. 2017, 19, 26107–26120. [Google Scholar] [CrossRef]
- Golubev, N.S.; Shenderovich, I.G.; Tolstoy, P.M.; Shchepkin, D.N. Solvent Induced Temperature Dependencies of NMR Parameters of Hydrogen Bonded Anionic Clusters. J. Molec. Struct. 2004, 697, 9–15. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Tolstoy, P.M.; Titova, A.A.; Kostin, M.A.; Denisov, G.S. Estimations of FH···X hydrogen bond energies from IR intensities: Iogansen’s rule revisited. J. Comput. Chem. 2021, 42, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Tupikina, E.Y.; Tokhadze, K.G.; Karpov, V.V.; Denisov, G.S.; Tolstoy, P.M. Stretching force constants as descriptors of energy and geometry of F···HF hydrogen bonds. Spectrochim. Acta A 2020, 241, 118677. [Google Scholar] [CrossRef] [PubMed]
- Tupikina, E.Y.; Denisov, G.S.; Melikova, S.M.; Kucherov, S.Y.; Tolstoy, P.M. New look at the Badger-Bauer rule: Correlations of spectroscopic IR and NMR parameters with hydrogen bond energy and geometry. FHF complexes. J. Mol. Struct. 2018, 1164, 129–136. [Google Scholar] [CrossRef]
- Denisov, G.S.; Bureiko, S.F.; Kucherov, S.Y.; Tolstoy, P.M. Correlation relationships between the energy and spectroscopic parameters of complexes with F···HF hydrogen bond. Dokl. Phys. Chem. 2017, 475, 115–118. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Jordan, M.J.T.; Perera, S.A.; Bartlett, R.J. Vibrational effects on the F-F spin-spin coupling constant (2hJF-F) in FHF- and FDF-. J. Phys. Chem. A 2001, 105, 8399–8402. [Google Scholar] [CrossRef]
- Limbach, H.-H.; Pietrzak, M.; Sharif, S.; Tolstoy, P.M.; Shenderovich, I.G.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S. NMR-Parameters and Geometries of OHN and ODN Hydrogen Bonds of Pyridine-Acid Complexes. Chem. Eur J. 2004, 10, 5195–5204. [Google Scholar] [CrossRef] [PubMed]
- Akcakayiran, D.; Mauder, D.; Hess, C.; Sievers, T.K.; Kurth, D.G.; Shenderovich, I.; Limbach, H.-H.; Findenegg, G.H. Carboxylic Acid-Doped SBA-15 Silica as a Host for Metallo-supramolecular Coordination Polymers. J. Phys. Chem. B 2008, 112, 14637–14647. [Google Scholar] [CrossRef]
- Ip, B.C.K.; Andreeva, D.V.; Buntkowsky, G.; Akcakayiran, D.; Findenegg, G.H.; Shenderovich, I.G. NMR Study of Proton Transfer to Strong Bases on Inner Surfaces of MCM-41. Micropor. Mesopor. Mater. 2010, 134, 22–28. [Google Scholar] [CrossRef]
- Andreeva, D.V.; Ip, B.; Gurinov, A.A.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G.; Limbach, H.-H. Geometrical Features of Hydrogen Bonded Complexes Involving Sterically Hindered Pyridines. J. Phys. Chem. A 2006, 110, 10872–10879. [Google Scholar] [CrossRef]
- Lorente, P.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.-H. 1H/15N NMR Chemical Shielding, Dipolar 15N,2H Coupling and Hydrogen Bond Geometry Correlations in a Novel Serious of Hydrogen-Bonded Acid-Base Complexes of Collidine with Carboxylic Acids. Magn. Reson. Chem. 2001, 39, S18–S29. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Shenderovich, I.G.; Muljati, T.; Limbach, H.-H.; Silverman, D. Intrinsic Proton Donating Power of Zinc-bound Water in a Carbonic Anhydrase Active Site Model Estimated by NMR. J. Am. Chem. Soc. 2011, 133, 11331–11338. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Smirnov, S.N.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. NMR Studies of Solid State—Solvent and H/D Isotope Effects on Hydrogen Bond Geometries of 1:1 Complexes of Collidine with Carboxylic Acids. J. Molec. Struct. 2004, 700, 19–27. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Rozhkova, Y.A.; Zukal, A.; Čejka, J.; Shenderovich, I.G. Mutable Lewis and Brønsted Acidity of Aluminated SBA-15 as Revealed by NMR of Adsorbed Pyridine-15N. Langmuir 2011, 27, 12115–12123. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Buntkowsky, G.; Schreiber, A.; Gedat, E.; Sharif, S.; Albrecht, J.; Golubev, N.S.; Findenegg, G.H.; Limbach, H.-H. Pyridine-15N—a Mobile NMR Sensor for Surface Acidity and Surface Defects of Mesoporous Silica. J. Phys. Chem. B 2003, 107, 11924–11939. [Google Scholar] [CrossRef]
- Chan-Huot, M.; Dos, A.; Zander, R.; Sharif, S.; Tolstoy, P.M.; Compton, S.; Fogle, E.; Toney, M.D.; Shenderovich, I.; Denisov, G.S.; et al. NMR Studies of Protonation and Hydrogen Bond States of Internal Aldimines of Pyridoxal 5′-Phosphate Acid−Base in Alanine Racemase, Aspartate Aminotransferase, and Poly-L-lysine. J. Am. Chem. Soc. 2013, 135, 18160–18175. [Google Scholar] [CrossRef] [PubMed]
- Limbach, H.-H.; Chan-Huot, M.; Sharif, S.; Tolstoy, P.M.; Shenderovich, I.G.; Denisov, G.S. Critical Hydrogen Bonds and Protonation States of Pyridoxal 5′-phosphate Revealed by NMR. Biochim. Biophys. Acta 2011, 1814, 1426–1437. [Google Scholar] [CrossRef]
- Ip, B.C.K.; Shenderovich, I.G.; Tolstoy, P.M.; Frydel, J.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.-H. NMR Studies of Solid Pentachlorophenol-4-Methylpyridine Complexes Exhibiting Strong OHN Hydrogen Bonds: Geometric H/D Isotope Effects and Hydrogen Bond Coupling Cause Isotopic Polymorphism. J. Phys. Chem. A 2012, 116, 11370–11387. [Google Scholar] [CrossRef] [PubMed]
- Bismarck, A.; Aranberri-Askargorta, I.; Springer, J.; Lampke, T.; Wielage, B.; Stamboulis, A.; Shenderovich, I.; Limbach, H.-H. Surface Characterization of Flax, Hemp and Cellulose Fibers; Surface Properties and the Water Uptake Behavior. Polym. Compos. 2002, 23, 872–894. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.-H.; Shenderovich, I.G. NMR Study of Solvation Effect on the Geometry of Proton-Bound Homodimers of Increasing Size. J. Phys. Chem. A 2017, 121, 8697–8705. [Google Scholar] [CrossRef] [PubMed]
- Attah, I.K.; Platt, S.P.; Meot-Ner, M.; El-Shall, M.S.; Aziz, S.G.; Alyoubi, A.O. Proton-bound dimers of nitrogen heterocyclic molecules: Substituent effects on the structures and binding energies of homodimers of diazine, triazine, and fluoropyridine. J. Chem. Phys. 2014, 140, 114313. [Google Scholar] [CrossRef]
- Pollice, R.; Bot, M.; Kobylianskii, I.J.; Shenderovich, I.; Chen, P. Attenuation of London Dispersion in Dichloromethane Solutions. J. Am. Chem. Soc. 2017, 139, 13126–13140. [Google Scholar] [CrossRef] [PubMed]
- Gurinov, A.A.; Lesnichin, S.B.; Limbach, H.-H.; Shenderovich, I.G. How Short is the Strongest Hydrogen Bond in the Proton-Bound Homodimers of Pyridine Derivatives? J. Phys. Chem. A 2014, 118, 10804–10812. [Google Scholar] [CrossRef]
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and Spectral Properties of the Protonated Homodimer of Pyridine in the Liquid and Solid States. A Combined NMR, X-ray Diffraction and Inelastic Neutron Scattering Study. J. Phys. Chem. A 2011, 115, 8041–8048. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Qualitative analysis of the geometry of the hydrogen bond in the homoconjugated pyridine ion. Russ. J. Gen. Chem. 2007, 77, 620. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Modeling of Solute–Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides. Molecules 2021, 26, 1283. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Adduct under Field—A Qualitative Approach to Account for Solvent Effect on Hydrogen Bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Solvent effects on acid-base complexes. What is more important: A macroscopic reaction field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef]
- Mori, Y.; Takano, K. Location of protons in N-H···N hydrogen-bonded systems: A theoretical study on intramolecular pyridine-dihydropyridine and pyridine-pyridinium pairs. Phys. Chem. Chem. Phys. 2012, 14, 11090–11098. [Google Scholar] [CrossRef]
- Rozhkova, Y.A.; Gurinov, A.A.; Orlova, A.O.; Maslov, V.G.; Shenderovich, I.G.; Korotkov, V.I. Spectrophotometric investigations of protonated forms of heterocyclic compounds. Opt. Spectrosc. 2012, 113, 275–278. [Google Scholar] [CrossRef]
- Melikova, S.M.; Rutkowski, K.S.; Gurinov, A.A.; Denisov, G.S.; Rospenk, M.; Shenderovich, I.G. FTIR study of the hydrogen bond symmetry in protonated homodimers of pyridine and collidine in solution. J. Mol. Struct. 2012, 1018, 39–44. [Google Scholar] [CrossRef]
- Guzei, I.A.; Roberts, J.; Saulys, D.A. Pseudosymmetry in pyridinium-tetrachloro(oxo)pyridineniobate(V) pyridine solvate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2002, 58, m141–m143. [Google Scholar] [CrossRef] [PubMed]
- Katrusiak, A. Stereochemistry and transformations of NH---N hydrogen bonds Part I. Structural preferences for the hydrogen site. J. Mol. Struct. 1999, 474, 125–133. [Google Scholar] [CrossRef]
- Brencic, J.V.; Ceh, B.; Leban, I. Pyridinium tetrabromobis (pyridine)tungstate (III), [(py)2H][WBr4(py)2]. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1979, 35, 3028–3030. [Google Scholar] [CrossRef]
- Minshall, P.C.; Sheldrick, G.M. Pyridinium 2,2,5,5-tetrathio-cyclo-di(phosphadithianate), [(pyridine)2H]22+[P2S8]2−. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1978, 34, 1378–1380. [Google Scholar] [CrossRef]
- Drew, M.G.B.; McKee, V.; Nelson, S.M. Crystal and molecular structure and some properties of pyridinium µ-oxo-bis[trichioroferrate(III)]–pyridine. J. Chem. Soc. Dalton Trans. 1978, 80–84. [Google Scholar] [CrossRef]
- Villarreal-Salinas, B.E.; Schlemper, E.O. Crystal structure of a salt of the pyridinium-pyridine ion by X-ray and neutron diffraction. J. Cryst. Mol. Struct. 1978, 8, 217–237. [Google Scholar] [CrossRef]
- Pollice, R.; Fleckenstein, F.; Shenderovich, I.; Chen, P. Compensation of London Dispersion in the Gas Phase and in Aprotic Solvents. Angew. Chem. Int. Ed. 2019, 58, 14281–14288. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.W.; Bordwell, F.G. Structural and solvent effects evaluated from acidities measured in dimethyl sulfoxide and in the gas phase. Acc. Chem. Res. 1988, 21, 463–469. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. QTAIM Characteristics of Halogen Bond and Related Interactions. J. Phys. Chem. A 2012, 116, 1838–1845. [Google Scholar] [CrossRef]
- Nejad, A.; Suhm, M.A. Concerted Pair Motion Due to Double Hydrogen Bonding: The Formic Acid Dimer Case. J. Indian I. Sci. 2020, 100, 5–19. [Google Scholar] [CrossRef]
- Tautermann, C.S.; Loferer, M.J.; Voegele, A.F.; Liedl, K.R. Double hydrogen tunneling revisited: The breakdown of experimental tunneling criteria. J. Chem. Phys. 2004, 120, 11650. [Google Scholar] [CrossRef]
- Emmeluth, C.; Suhm, M.A.; Luckhaus, D. A monomers-in-dimers model for carboxylic acid dimers. J. Chem. Phys. 2003, 118, 2242–2255. [Google Scholar] [CrossRef]
- Vener, M.V.; Kühn, O.; Bowman, J.M. Vibrational spectrum of the formic acid dimer in the OH stretch region. A model 3D study. Chem. Phys. Lett. 2001, 349, 562–570. [Google Scholar] [CrossRef]
- Elsaesser, T.; Huse, N.; Dreyer, J.; Dwyer, J.R.; Heyne, K.; Nibbering, E.T.J. Ultrafast vibrational dynamics and anharmonic couplings of hydrogen-bonded dimers in solution. Chem. Phys. 2007, 341, 175–188. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Schah-Mohammedi, P.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. Characterization of Fluxional Hydrogen-Bonded Complexes of Acetic Acid and Acetate by NMR: Geometries and Isotope and Solvent Effects. J. Am. Chem. Soc. 2004, 126, 5621–5634. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Hung, I.; Gan, Z.; Terskikh, V.; Kong, X. Solid-State 17O NMR Study of Carboxylic Acid Dimers: Simultaneously Accessing Spectral Properties of Low- and High-Energy Tautomers. J. Phys. Chem. A 2019, 123, 8243–8253. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Davies, J.A.; Hanson-Heine, M.W.D.; Besley, N.A.; Shirley, A.; Trowers, J.; Yang, S.; Ellis, A.M. Dimers of acetic acid in helium nanodroplets. Phys. Chem. Chem. Phys. 2019, 21, 13950–13958. [Google Scholar] [CrossRef] [PubMed]
- Manriquez, R.; Lopez-Dellamary, F.A.; Frydel, J.; Emmler, T.; Breitzke, H.; Buntkowsky, G.; Limbach, H.-H.; Shenderovich, I.G. Solid-State NMR Studies of Aminocarboxylic Salt Bridges in L-Lysine Modified Cellulose. J. Phys. Chem. B 2009, 113, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Golubev, N.S.; Smirnov, S.N.; Schah-Mohammedi, P.; Shenderovich, I.; Denisov, G.S.; Gindin, V.; Limbach, H.-H. Study of Acid-Base Interaction by Means of Low-Temperature NMR Spectra. Structure of Salicilic Acid Complexes. Russ. J. Gen. Chem. 1997, 67, 1082–1087. [Google Scholar]
- Jing Guo, J.; Tolstoy, P.M.; Koeppe, B.; Golubev, N.S.; Denisov, G.S.; Smirnov, S.N.; Limbach, H.-H. Hydrogen Bond Geometries and Proton Tautomerism of Homoconjugated Anions of Carboxylic Acids Studied via H/D Isotope Effects on 13C NMR Chemical Shifts. J. Phys. Chem. A 2012, 116, 11180–11188. [Google Scholar] [CrossRef]
- Reiersølmoen, A.C.; Battaglia, S.; Øien-Ødegaard, S.; Gupta, A.K.; Fiksdahl, A.; Lindh, R.; Erdélyi, M. Symmetry of three-center, four-electron bonds. Chem. Sci. 2020, 11, 7979. [Google Scholar] [CrossRef]
- Hakkert, S.B.; Erdélyi, M. Halogen bond symmetry: The N–X–N bond. J. Phys. Org. Chem. 2015, 28, 226–233. [Google Scholar] [CrossRef]
- Karim, A.; Reitti, M.; Carlsson, A.-C.C.; Gräfenstein, J.; Erdélyi, M. The nature of [N–Cl–N]+ and [N–F–N]+ halogen bonds in solution. Chem. Sci. 2014, 5, 3226–3233. [Google Scholar] [CrossRef]
- Carlsson, A.-C.C.; Gräfenstein, J.; Budnjo, A.; Laurila, J.L.; Bergquist, J.; Karim, A.; Kleinmaier, R.; Brath, U.; Erdélyi, M. Symmetric Halogen Bonding Is Preferred in Solution. J. Am. Chem. Soc. 2012, 134, 5706–5715. [Google Scholar] [CrossRef]
- Troyanov, S.I.; Morozov, I.V.; Kemnitz, E. Crystal Structure of Cesium Dihydrotrifluoride, CsH2F3. Refinement of the Crystal Structures of NMe4HF2 and NMe4H2F3. Z. Anorg. Allg. Chem. 2005, 631, 1651–1654. [Google Scholar] [CrossRef]
- Tian, C.; Nie, W.; Chen, Q.; Sun, G.; Borzov, M.V. Modified approach to the Arduengo carbene complexes of silver. Russ. Chem. Bull. 2014, 63, 2675–2680. [Google Scholar] [CrossRef]
- Mootz, D.; Boenigk, D. Poly(hydrogen Fluorides) with the Tetramethylammonium Cation: Preparation, Stability Ranges, Crystal Structures, [HnFn+1]- Anion Homology, Hydrogen Bonding F-H…F. Z. Anorg. Allg. Chem. 1987, 544, 159–166. [Google Scholar] [CrossRef]
- Fernández, F.J.; Alfonso, M.; Schmalle, H.W.; Berke, H. Utilization of Redox and Acid/Base Chemistry for the Deprotection of a Mn(dmpe)2(C⋮CSiMe3)2 Complex. Organometallics 2001, 20, 3122–3131. [Google Scholar] [CrossRef]
- Forrester, J.D.; Senko, M.E.; Zalkin, A.; Templeton, D. Crystal structure of KH2F3 and geometry of the H2F3- ion. Acta Cryst. 1963, 16, 58–62. [Google Scholar] [CrossRef]
- Wilson, W.W.; Chxiste, K.O.; Feng, J.; Bau, R. Tetramethylammonium bifluoride, crystal structure and vibrational spectra. Can. J. Chem. 1989, 67, 1898–1901. [Google Scholar] [CrossRef]
- Freza, S.; Skurski, P. Enormously large (approaching 14 eV!) electron binding energies of [HnFn+1]- (n = 1–5, 7, 9, 12) anions. Chem. Phys. Lett. 2010, 487, 19–23. [Google Scholar] [CrossRef]
- Schneider, H.-J. Hydrogen bonds with fluorine. Studies in solution, in gas phase and by computations, conflicting conclusions from crystallographic analyses. Chem. Sci. 2012, 3, 1381–1394. [Google Scholar] [CrossRef]
- Bulychev, V.P.; Buturlimova, M.V. Anharmonic calculation of structural and vibrational properties of the isolated complexes [F(HF)2]-, [F(DF)2]-, and [F(TF)2]-. J. Mol. Struct. 2009, 928, 32–39. [Google Scholar] [CrossRef]
- Bulychev, V.P.; Denisov, G.S.; Limbach, H.-H.; Shukailov, R.M. Calculation of vibrations of the H-bonds and electrooptical parameters of the [F(HF)2]- complex. Opt. Spectrosc. 2001, 90, 356–361. [Google Scholar] [CrossRef]
- de la Vega, G.J.M.; Fabian, S.J. Assessment of DFT functionals with fluorine-fluorine coupling constants. Mol. Phys. 2015, 113, 1924–1936. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Limbach, H.-H.; Shenderovich, I.G.; Winkler, T. A DFT and AIM Analysis of the Spin-Spin Couplings Across the Hydrogen Bond in the 2-fluorobenzamide and Related Compounds. Magn. Reson. Chem. 2009, 47, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. A Systematic Comparison of Second-Order Polarization Propagator Approximation (SOPPA) and Equation-of-Motion Coupled Cluster Singles and Doubles (EOM-CCSD) Spin-Spin Coupling Constants for Selected Singly Bonded Molecules, and the Hydrides NH3, H2O, and HF and Their Protonated and Deprotonated Ions and Hydrogen-Bonded Complexes. J. Chem. Theory Comput. 2008, 4, 967–973. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Elguero, J. 19F–19F and 19F–1H spin–spin coupling constants in cyclic FH polymers (FH)n, n = 2–6. Solid State Nucl. Magn. Reson. 2008, 34, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Elguero, J.; Alkorta, I.; Yanez, M.; Mo, O. 19F-19F spin-spin coupling constant surfaces for (HF)2 clusters: The orientation and distance dependence of the sign and magnitude of J(F-F). J. Chem. Phys. 2004, 120, 3237–3243. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Computed EOM-CCSD 19F-19F spin-spin coupling constants in small organic molecules. Z. Phys. Chem. 2003, 217, 1565–1575. [Google Scholar] [CrossRef]
- Perera, S.A.; Bartlett, R.J. NMR spin-spin coupling constants for hydrogen bonds of [F(HF)n]-, n=1-4, clusters. J. Am. Chem. Soc. 2000, 122, 1231–1232. [Google Scholar] [CrossRef]
- Golubev, N.S.; Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Limbach, H.-H. Nuclear Scalar Spin-Spin Coupling Reveals Novel Properties of Low-Barrier Hydrogen Bonds in a Polar Environment. Chem. Eur. J. 1999, 5, 492–497. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Denisov, G.S.; Tolstoy, P.M. Anticooperativity of FH···Cl- hydrogen bonds in [FH)nCl]- clusters (n = 1…6). J. Comput. Chem. 2019, 40, 2858–2867. [Google Scholar] [CrossRef] [PubMed]
- Kucherov, S.Y.; Bureiko, S.F.; Denisov, G.S. Anticooperativity of FHF hydrogen bonds in clusters of the type F-×(HF)n, RF×(HF)n and XF×(HF)n, R = alkyl and X = H, Br, Cl, F. J. Mol. Struct. 2016, 1105, 246–255. [Google Scholar] [CrossRef]
- Grabowski, S.J. Cooperativity of hydrogen and halogen bond interactions. Theor. Chem. Acc. 2013, 132, 1347. [Google Scholar] [CrossRef]
- Ziółkowski, M.; Grabowski, S.J.; Leszczynski, J. Cooperativity in Hydrogen-Bonded Interactions: Ab Initio and “Atoms in Molecules” Analyses. J. Phys. Chem. A 2006, 110, 6514–6521. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Lesnichin, S.B.; Tu, C.; Silverman, D.N.; Tolstoy, P.M.; Denisov, G.S.; Limbach, H.-H. NMR Studies of Active-Site Properties of Human Carbonic Anhydrase II by using 15N-Labeled 4-Methylimidazole as a Local Probe and Histidine Hydrogen-Bond Correlations. Chem. Eur. J. 2015, 21, 2915–2929. [Google Scholar] [CrossRef]
- Sharif, S.; Fogle, E.; Toney, M.D.; Denisov, G.S.; Shenderovich, I.G.; Buntkowsky, G.; Tolstoy, P.M.; Chan Huot, M.; Limbach, H.-H. NMR Localization of Protons in Critical Enzyme Hydrogen Bonds. J. Am. Chem. Soc. 2007, 129, 9558–9559. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Giba, I.S.; Kostin, M.A.; Denisov, G.S.; Shenderovich, I.G.; Tolstoy, P.M. Cyclic Trimers of Phosphinic Acids in Polar Aprotic Solvent: Symmetry, Chirality and H/D Isotope Effects on NMR Chemical Shifts. Phys. Chem. Chem. Phys. 2018, 20, 4901–4910. [Google Scholar] [CrossRef]
- Shenderovich, I.G. The Partner Does Matter: The Structure of Heteroaggregates of Acridine Orange in Water. Molecules 2019, 24, 2816. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Mauder, D.; Akcakayiran, D.; Findenegg, G.H.; Shenderovich, I.G. Does Water Affect the Acidity of Surfaces? The Proton-Donating Ability of Silanol and Carboxylic Acid Groups at Mesoporous Silica. ChemPhysChem 2012, 13, 2282–2285. [Google Scholar] [CrossRef]
- Mauder, D.; Akcakayiran, D.; Lesnichin, S.B.; Findenegg, G.H.; Shenderovich, I.G. Acidity of Sulfonic and Phosphonic Acid-Functionalized SBA-15 under Almost Water-Free Conditions. J. Phys. Chem. C 2009, 113, 19185–19192. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Limbach, H.-H.; Smirnov, S.N.; Tolstoy, P.M.; Denisov, G.S.; Golubev, N.S. H/D Isotope Effects on the Low-Temperature NMR Parameters and Hydrogen Bond Geometries of (FH)2F- and (FH)3F- Dissolved in CDF3/CDF2Cl. Phys. Chem. Chem. Phys. 2002, 4, 5488–5497. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Ustimchuk, D.O.; Filarowski, A.; Tolstoy, P.M. H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules 2020, 25, 1907. [Google Scholar] [CrossRef]
- Machida, S.; Sohmiya, M.; Ide, Y.; Sugahara, Y. Solid-State 31P Nuclear Magnetic Resonance Study of Interlayer Hydroxide Surfaces of Kaolinite Probed with an Interlayer Triethylphosphine Oxide Monolayer. Langmuir 2018, 34, 12694–12701. [Google Scholar] [CrossRef]
- Seitz, A.E.; Hippauf, F.; Kremer, W.; Kaskel, S.; Scheer, M. Facile storage and release of white phosphorus and yellow arsenic. Nat. Commun. 2018, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Moussa, M.E.; Shelyganov, P.A.; Wegley, B.; Seidl, M.; Scheer, M. The Potential of the Diphosphorus Complex [Cp2W2(CO)4( η2-P2)] as an Organometallic Connecter in Supramolecular Chemistry. Eur. J. Inorg. Chem. 2019, 2019, 4241–4248. [Google Scholar] [CrossRef]
- Guenther, J.; Reibenspies, J.; Blümel, J. Synthesis and characterization of tridentate phosphine ligands incorporating long methylene chains and ethoxysilane groups for immobilizing molecular rhodium catalysts. Mol. Catal. 2019, 479, 110629. [Google Scholar] [CrossRef]
- Cluff, K.J.; Bhuvanesh, N.; Blümel, J. Monometallic Ni-0 and Heterobimetallic Ni-0/Au-I Complexes of Tripodal Phosphine Ligands: Characterization in Solution and in the Solid State and Catalysis. Chem. Eur. J. 2015, 21, 10138–10148. [Google Scholar] [CrossRef]
- Pazderski, L. 15N and 31P NMR Coordination Shifts in Transition Metal Complexes with Nitrogen- and Phosphorus-Containing Heterocycles. Annu. Rep. NMR Spectrosc. 2013, 80, 33–179. [Google Scholar] [CrossRef]
- Hubbard, P.J.; Benzie, J.W.; Bakhmutov, V.I.; Blümel, J. Disentangling Different Modes of Mobility of Triphenylphosphine Oxide Adsorbed on Alumina. J. Chem. Phys. 2020, 152, 054718. [Google Scholar] [CrossRef]
- Kharel, S.; Cluff, K.J.; Bhuvanesh, N.; Gladysz, J.A.; Blümel, J. Structures and Dynamics of Secondary and Tertiary Alkylphosphine Oxides Adsorbed on Silica. Chem. Asian J. 2019, 14, 2704–2711. [Google Scholar] [CrossRef]
- Hilliard, C.R.; Kharel, S.; Cluff, K.; Bhuvanesh, N.; Gladysz, J.; Bluemel, J. Structures and Unexpected Dynamic Properties of Phosphine Oxides Adsorbed on Silica Surfaces. Chem. Eur. J. 2014, 20, 17292–17295. [Google Scholar] [CrossRef]
- Shenderovich, I.G. For Whom a Puddle Is the Sea? Adsorption of Organic Guests on Hydrated MCM-41 Silica. Langmuir 2020, 36, 11383–11392. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G. Effect of Noncovalent Interactions on the 31P Chemical Shift Tensor of Phosphine Oxides, Phosphinic, Phosphonic, and Phosphoric Acids, and Their Complexes with Lead(II). J. Phys. Chem. C 2013, 117, 26689–26702. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Electric field effect on 31P NMR magnetic shielding. J. Chem. Phys. 2020, 153, 184501. [Google Scholar] [CrossRef] [PubMed]
- Chernyshov, I.Y.; Vener, M.V.; Shenderovich, I.G. Local-structure effects on 31P NMR chemical shift tensors in solid state. J. Chem. Phys. 2019, 150, 144706. [Google Scholar] [CrossRef]
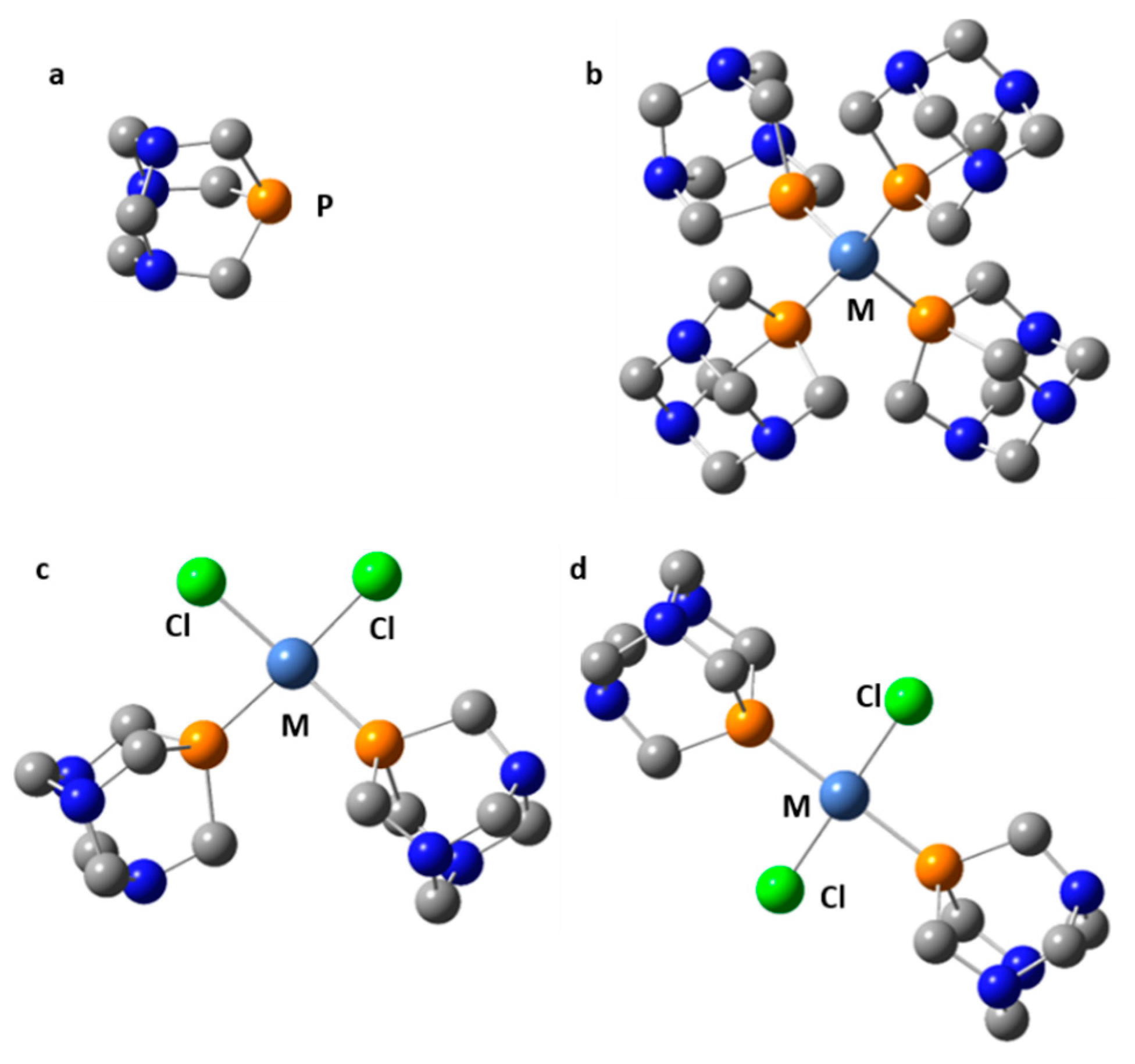
- Fisher, K.J.; Alyea, E.C.; Shehnazarian, N. A 31P NMR Study of the Water Soluble Derivatives of 1,3,5-triaza-7-phosphaadamantane (PTA). Phosphorus Sulfur Silicon 1990, 48, 37–40. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA) Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Battistin, F.; Balducci, G.; Milani, B.; Alessio, E. Water-Soluble Ruthenium(II) Carbonyls with 1,3,5-Triaza-7-phosphoadamantane. Inorg. Chem. 2018, 57, 6991–7005. [Google Scholar] [CrossRef]
- Battistin, F.; Balducci, G.; Iengo, E.; Demitri, N.; Alessio, E. Neutral 1,3,5-Triaza-7-phosphaadamantane-Ruthenium(II) Complexes as Precursors for the Preparation of Highly Water-Soluble Derivatives. Eur. J. Inorg. Chem. 2016, 2016, 2850–2860. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Alyea, E.C.; Ferguson, G.; Kannan, S. Intermolecular hydrogen···metal interactions. The crystal structure of {cis-[PdCl2(TPA)2]}2·H2O, a water-soluble palladium (II) tertiary phosphine complex. Chem. Commun. 1998, 345–346. [Google Scholar] [CrossRef]
- Otto, S.; Roodt, A.; Purcell, W. Synthesis and characterisation of water soluble Pt(II) complexes of 1,3,5-triaza-7-phosphaadamantane (PTA). Crystal and molecular structure of {cis-[PtCl2(PTA)2]}2·H2O. Inorg. Chem. Commun. 1998, 1, 415–417. [Google Scholar] [CrossRef]
- Braddock-Wilking, J.; Acharya, S.; Rath, N.P. Synthesis and characterization of Pt(II) and Pd(II) PTA and DAPTA complexes. Polyhedron 2014, 79, 16–28. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Experimentally Established Benchmark Calculations of 31P NMR Quantities. Chem. Methods 2021, 1, 61–70. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Robertson, J.B.; Larkins, D.L.; Reibenspies, J.H. Water-Soluble Organometallic Compounds. 7.1 Further Studies of 1,3,5-Triaza-7-Phosphaadamantane Derivatives of Group 10 Metals, Including Metal Carbonyls and Hydrides. Inorg. Chem. 1999, 38, 2473–2481. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Decuir, T.J.; Stafford, N.W.; Robertson, J.B.; Draper, J.D.; Reibenspies, J.H. Water-Soluble Organometallic Compounds. 6.1 Synthesis, Spectral Properties, and Crystal Structures of Complexes of 1,3,5-Triaza-7-phosphaadamantane with Group 10 Metals. Inorg. Chem. 1997, 36, 4218–4226. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Smoleński, P.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. The First Copper Complexes Bearing the 1,3,5-Triaza-7- phosphaadamantane (PTA) Ligand. Eur. J. Inorg. Chem. 2007, 2007, 2686–2692. [Google Scholar] [CrossRef]
- Alyea, E.C.; Ferguson, G.; Kannan, S. Some water-soluble organometallic complexes of group 10 transition metal(II) ions with 1,3,5-triaza-7-phosphaadamantane (TPA). Syntheses, characterization and reactivity. The crystal and molecular structure of [Ni(CN)2(TPA)3]·4.3H2O. Polyhedron 1998, 17, 2727–2732. [Google Scholar] [CrossRef]
- Grünberg, B.; Emmler, T.; Gedat, E.; Shenderovich, I.; Findenegg, G.H.; Limbach, H.-H.; Buntkowsky, G. Hydrogen Bonding of Water Confined in Mesoporous Silica MCM-41 and SBA-15 Studied by 1H Solid-State NMR. Chem. Eur. J. 2004, 10, 5689–5696. [Google Scholar] [CrossRef]
- Buntkowsky, G.; Breitzke, H.; Adamczyk, A.; Roelofs, F.; Emmler, T.; Gedat, E.; Grünberg, B.; Xu, Y.; Limbach, H.-H.; Shenderovich, I.; et al. Structural and Dynamical Properties of Guest Molecules Confined in Mesoporous Silica Materials Revealed by NMR. Phys. Chem. Chem. Phys. 2007, 9, 4843–4853. [Google Scholar] [CrossRef]
- Gedat, E.; Schreiber, A.; Findenegg, G.H.; Shenderovich, I.; Limbach, H.-H.; Buntkowsky, G. Stray Field Gradient NMR Reveals Effects of Hydrogen Bonding on Diffusion Coefficients of Pyridine in Mesoporous Silica. Magn. Reson. Chem. 2001, 39, S149–S157. [Google Scholar] [CrossRef]
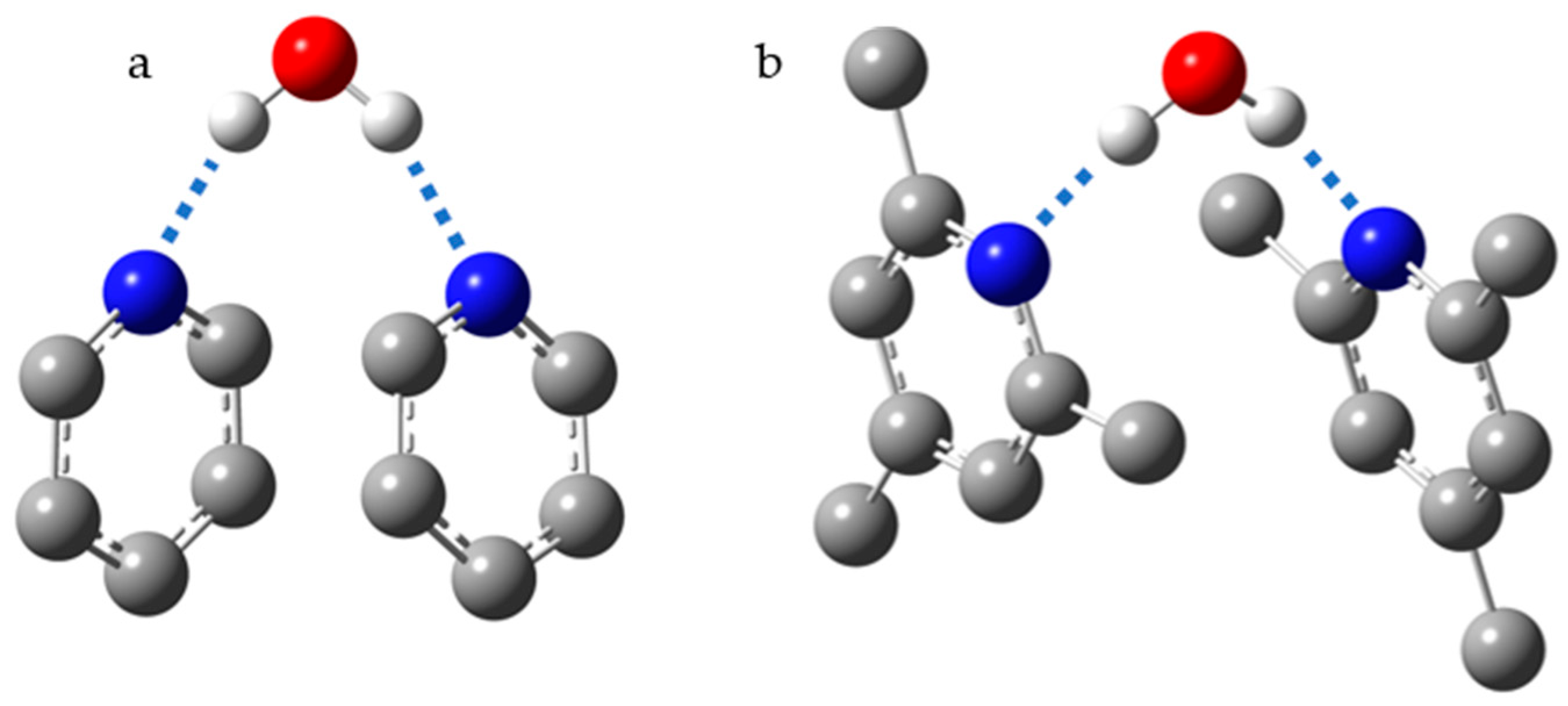
- Masaru, N.; Chihiro, W. Monomeric and Cluster States of Water Molecules in Organic Solvent. Chem. Lett. 1992, 21, 809–812. [Google Scholar] [CrossRef]
- Sharif, S.; Shenderovich, I.G.; González, L.; Denisov, G.S.; Silverman, D.N.; Limbach, H.-H. NMR and Ab initio Studies of Small Complexes Formed between Water and Pyridine Derivatives in Solid and Liquid Phase. J. Phys. Chem. A 2007, 111, 6084–6093. [Google Scholar] [CrossRef]
- Baures, P.W. Monoclinic Triphenylphosphine Oxide Hemihydrate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1991, 47, 2715–2716. [Google Scholar] [CrossRef]
- Ng, S.W. A Second Monoclinic Modification of Triphenylphosphine Oxide Hemihydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o1431. [Google Scholar] [CrossRef]
- Begimova, G.U.; Tupikina, E.Y.; Yu, V.K.; Denisov, G.S.; Bodensteiner, M.; Shenderovich, I.G. Effect of Hydrogen Bonding to Water on the 31P Chemical Shift Tensor of Phenyl- and Trialkylphosphine Oxides and a-Amino Phosphonates. J. Phys. Chem. C 2016, 120, 8717–8729. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P=O Moiety as an Ambidextrous Hydrogen Bond Acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Hilliard, C.R.; Bhuvanesh, N.; Gladysz, J.A.; Blümel, J. Synthesis, purification, and characterization of phosphine oxides and their hydrogen peroxide adducts. Dalton Trans. 2012, 41, 1742–1754. [Google Scholar] [CrossRef]
- Ahn, S.H.; Cluff, K.J.; Bhuvanesh, N.; Blümel, J. Hydrogen Peroxide and Di(hydroperoxy)propane Adducts of Phosphine Oxides as Stoichiometric and Soluble Oxidizing Agents. Angew. Chem. Int. Ed. 2015, 54, 13341–13345. [Google Scholar] [CrossRef]
- Arp, F.F.; Bhuvanesh, N.; Blümel, J. Hydrogen peroxide adducts of triarylphosphine oxides. Dalton Trans. 2019, 48, 14312–14325. [Google Scholar] [CrossRef]
- Forlani, L. Hydrogen bonds of anilines. In The Chemistry Anilines; Rappoport, Z., Ed.; The Atrium, Southern Gate, West Sussex PO19 8SQ, Part 1; John Wiley & Sons Ltd.: Chichester, UK, 2007; pp. 407–454. [Google Scholar]
- Szatyłowicz, H.; Krygowski, T.M.; Hobza, P. How the Shape of the NH2 Group Depends on the Substituent Effect and H-Bond Formation in Derivatives of Aniline. J. Phys. Chem. A 2007, 111, 170–175. [Google Scholar] [CrossRef]
- Borisenko, V.E.; Filarovski, A.I. The electrooptical parameters of aniline and its halogen derivatives in hydrogen bonded complexes. J. Mol. Struct. 1989, 196, 353–370. [Google Scholar] [CrossRef]
- Grabowski, S.J. Pnicogen and tetrel bonds—tetrahedral Lewis acid centres. Struct. Chem. 2019, 30, 1141–1152. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel bond and coordination of triel centres—Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171. [Google Scholar] [CrossRef]
- Pelties, S.; Maier, T.; Herrmann, D.; de Bruin, B.; Rebreyend, C.; Gärtner, S.; Shenderovich, I.G.; Wolf, R. Selective P4 Activation by a Highly Reduced Cobaltate: Synthesis of Dicobalt Tetraphosphido Complexes. Chem. Eur. J. 2017, 23, 6094–6102. [Google Scholar] [CrossRef]
- Maier, T.M.; Sandl, S.; Shenderovich, I.G.; von Wangelin, A.J.; Weigand, J.J.; Wolf, R. Amine-Borane Dehydrogenation and Transfer Hydrogenation Catalyzed by alpha-Diimine Cobaltates. Chem. Eur. J. 2019, 25, 238–245. [Google Scholar] [CrossRef]
- Medvedev, A.G.; Churakov, A.V.; Prikhodchenko, P.V.; Lev, O.; Vener, M.V. Crystalline Peroxosolvates: Nature of the Coformer, Hydrogen-Bonded Networks and Clusters, Intermolecular Interactions. Molecules 2021, 26, 26. [Google Scholar] [CrossRef] [PubMed]
- Giba, I.S.; Tolstoy, P.M. Self-Assembly of Hydrogen-Bonded Cage Tetramers of Phosphonic Acid. Symmetry 2021, 13, 258. [Google Scholar] [CrossRef]
- Kizior, B.; Panek, J.J.; Szyja, B.M.; Jezierska, A. Structure-Property Relationship in Selected Naphtho- and Anthra-Quinone Derivatives on the Basis of Density Functional Theory and Car–Parrinello Molecular Dynamics. Symmetry 2021, 13, 564. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Experimental δiso(31P), ppm | Calculated δiso(31P), ppm 1 |
|---|---|---|
| PTA | −104.3 [228] | −104 |
| Ni(PTA)4 | −44.8 [229],−45.7 [230] | −46; −46; −47; −47 |
| Pd(PTA)4 | −56.5 [229], −58.7 [230] | −53; −56; −57; −57 |
| Pt(PTA)4 | −74.5 [230] | −34; −39; −39; −39 |
| [NO3]-Cu+(PTA)4 | −78.2 [231] | −84; −84; −84; −85 |
| Cl2Ni(PTA)2 | −1.2 [232] | − |
| cis-Cl2Ni(PTA)2 | − | −13; −21 |
| trans-Cl2Ni(PTA)2 | − | −36; −36 |
| Cl2Pd(PTA)2 | −21 [230], −18 [232] | − |
| cis-Cl2Pd(PTA)2 | − | −13; −14 |
| trans-Cl2Pd(PTA)2 | − | −37; −37 |
| Cl2Pt(PTA)2 | −51 [230], −47.5 [232] | − |
| cis-Cl2Pt(PTA)2 | − | −17; −18 |
| trans-Cl2Pt(PTA)2 | − | −46; −46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shenderovich, I.G. Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State. Symmetry 2021, 13, 756. https://doi.org/10.3390/sym13050756
Shenderovich IG. Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State. Symmetry. 2021; 13(5):756. https://doi.org/10.3390/sym13050756
Chicago/Turabian StyleShenderovich, Ilya G. 2021. "Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State" Symmetry 13, no. 5: 756. https://doi.org/10.3390/sym13050756
APA StyleShenderovich, I. G. (2021). Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State. Symmetry, 13(5), 756. https://doi.org/10.3390/sym13050756