Abstract
The homodimers of transiently chiral molecules offer physical insight into the process of molecular recognition, the preference for homo or heterochiral aggregation and the nature of the non-covalent interactions stabilizing the adducts. We report the observation of the benzyl mercaptan dimer in the isolation conditions of a supersonic jet expansion, using broadband (chirped-pulse) microwave spectroscopy. A single homochiral isomer was observed for the dimer, stabilized by a cooperative sequence of S-H···S and S-H···π hydrogen bonds. The structural data, stabilization energies and energy decomposition describe these non-covalent interactions as weak and dispersion-controlled. A comparison is also provided with the benzyl alcohol dimer.
1. Introduction
Molecular recognition is a subtle chemical process involving a combination of intermolecular interactions and intramolecular factors, largely dependent on the composition of the host and guest molecules. The consequences of molecular recognition, extending to large-scale chemical phenomena like supramolecular [1] and technological applications [2], justify the molecular investigation of the electronic and stereochemical features of the recognition process using high-resolution spectroscopy [3] and quantum mechanical methods [4,5]. In this context, the preparation of mass-selected intermolecular adducts in the gas phase is a well-known procedure for isolation and observation of specific weak interactions between target chemical groups, which otherwise would be blurred in the condensed media. This approach, when combined with rationalizing theoretical calculations has led, in the last decade, to an explosion of new weak intermolecular interactions previously unnoticed [6,7].
Molecular recognition is especially interesting between chiral species, as it may provide insight into biochemical docking, asymmetric synthesis, and chiral analysis. Among chiral molecules, those with very low (5–10 kJ mol−1) torsional stereomutation barriers display transient chirality. Transient enantiomers interconvert in nanosecond time scales that would be undetectable with conventional techniques. However, transient chirality can be revealed by the formation of diastereomeric dimers in the gas phase, simultaneously freezing stereomutation and providing information on the structural and energetic factors controlling homo- or heterochiral aggregation [8,9]. The process of chirality synchronization was first observed with electronic and vibrational spectroscopy [10,11,12], but these techniques require computational support for spectral interpretation. Alternatively, rotational spectroscopy [7,13] offers an unequivocal structural identification that complements molecular orbital calculations and may contribute to validating the computational models, which in turn are critical for the determination of the weak chirodiastaltic energies [14,15,16] between homo- and heterochiral aggregates.
Rotational experiments on homodimer synchronization have been mostly restricted to alcohols (ethanol [17,18], 2-propanol [19], 2-butanol [20], glycidol [21], cyclohexanol [22]) and fluoroalcohols (2-fluoroethanol [23], trifluoroethanol [24], hexafluoroisopropanol [25]), which benefit of the moderately strong O-H···O hydrogen bonds. Conversely, molecular studies of thiol aggregation are much scarcer [5] (pp. 15–45). The experiments available have mostly probed thiols as proton acceptors, in particular in O-H···S [26,27,28,29,30,31], N-H···S [32] and C-H···S [33,34] hydrogen bonds. Thiol dimerization studies particularly contribute to the description of thiol as proton donors in S-H···S [35,36,37] and other weak sulfur interactions (S-H···O [38], S-H···π [39], etc.), far less investigated [40,41] and consequently needed of molecular studies.
In this work, we selected benzyl mercaptan as a dimerization target. Benzyl mercaptan represents an extension of our previous investigation on thiophenol dimerization [36] and will establish if the two rings maintain the π-stacking thiophenol arrangement or tilted geometries similar to the benzyl alcohol dimer [42], recently revisited [43]. Additionally, it will discern whether the homochiral aggregation of benzyl alcohol is respected in the mercaptan and the structural and physical differences in the S-H···S hydrogen bond compared to the canonical O-H···O hydrogen bond.
2. Materials and Methods
The sample of benzyl mercaptan (>96%, bp 195 °C) was obtained commercially and required no further purification. The sample was vaporized inside the heating reservoir (45–55 °C) of a pulsed solenoid injector and expanded near adiabatically through a 0.8 mm nozzle to form a supersonic jet within an expansion chamber. Neon at stagnation pressures of 0.2 MPa was used as the carrier gas, with typical molecular pulses of 800–900 μs. The expanding jet was probed in the 2–8 GHz cm-wave region with a direct-digital chirped-pulse Fourier transform microwave (CP-FTMW) spectrometer, following Pate’s design [44]. The operation sequence is based on short (4 μs, 20 W) chirped pulses, which are broadcasted perpendicularly to the jet. The MW radiation produces a fast-passage broadband transient excitation [45,46], simultaneously covering the full spectral bandwidth. Following the excitation, the molecular ensemble emits a free-induction decay, which is detected in the time-domain (ca. 40 μs) and acquired using a (25 GSamples/s) digital oscilloscope. A Fourier transformation with a Kaiser–Bessel window results in FWHM linewidths of ca. 100 kHz. In this experiment, ca. 1 M averages were acquired at a repetition rate of 5 Hz. The uncertainty of the frequency measurements was estimated below 20 kHz.
Several computational calculations complemented the experimental study. Following an initial conformational screening with molecular mechanics (MMFFs [47]), all further calculations used density-functional theory (DFT) molecular orbital calculations. Two density-functional methods were selected here, including B3LYP [48] and the double hybrid B2PLYP [49] method, in combination with Ahlrichs’ polarized triple-zeta basis def2-TZVP [50]. Both methods were supplemented with D3 [51] dispersion corrections and Becke–Johnson damping [52]. Frequency calculations were performed at the same level of theory, using the harmonic approximation. The calculation of complexation energies considered the basis set superposition errors (BSSE) with the counterpoise approximation [53]. All DFT calculations were conducted with Gaussian 16 [54]. The physical contributions to the binding potential of the water clusters were estimated by energy decomposition analysis using second-order symmetry adapted perturbation theory [53,55] (SAPT), implemented in PSI4 [56]. Finally, the presence of non-covalent interactions was analyzed with the NCIPlot methodology, based on a reduced gradient of the electronic density [57].
3. Results
3.1. Benzyl Mercaptan Monomer
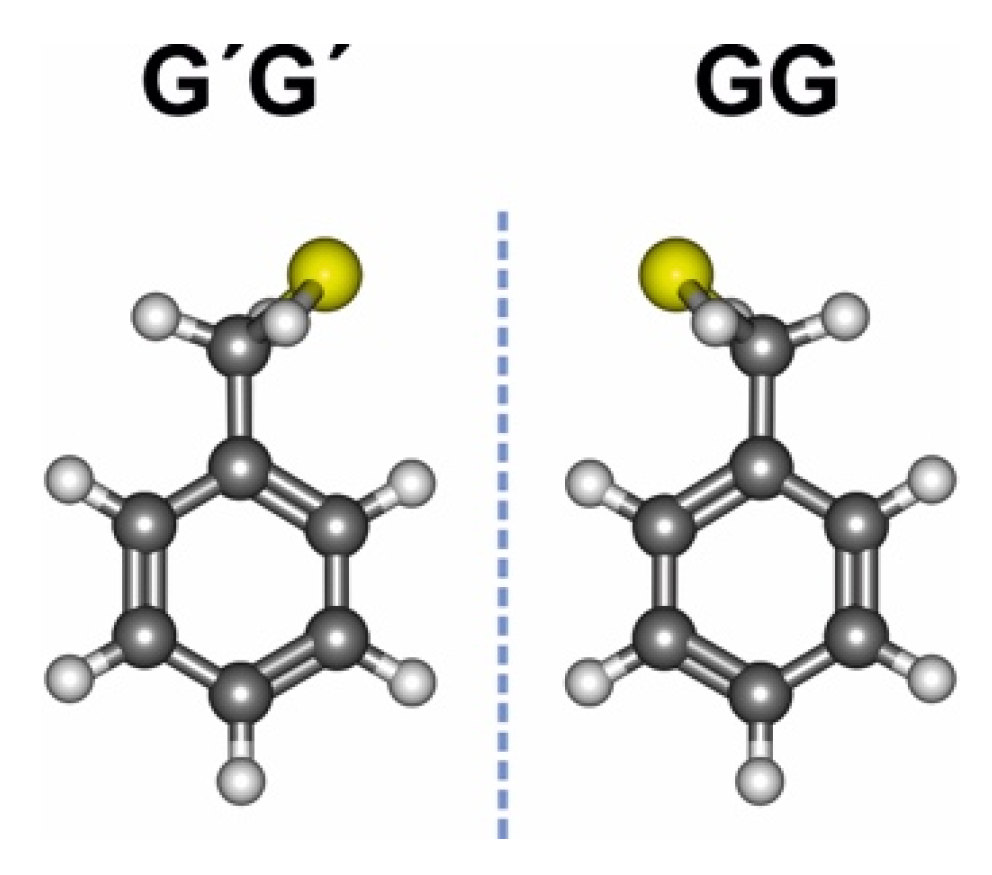
Benzyl mercaptan displays a bidimensional potential energy surface, qualitatively similar to benzyl alcohol [58]. The two torsional degrees of freedom are associated with the elevation and orientation of the terminal thiol group, given by dihedrals τ1(SCα-CipsoCortho) and τ2(HS-CαCipso). Our previous rotational investigation [59] of benzyl mercaptan confirmed that the monomer presents a single isomer in the gas phase, as in the alcohol. In this conformation the sulfur atom is synclinal to the ring plane (τ1~ ±74° vs. ±55° in benzyl alcohol) and the thiol hydrogen is synchronously oriented towards the π ring (τ2~ ±74° vs. ±53° in benzyl alcohol), denoted gauche-gauche or GG (Tables S1 and S2, Supporting Information, SI). The global minimum is four-fold degenerate, as the thiol group may tunnel between symmetry equivalent conformations either by reflection on the ring plane or in a perpendicular plane bisecting the phenyl ring through carbons Cipso and Cpara. Inversion through the perpendicular plane creates a detectable barrier (B2 = 248 cm−1 = 2.97 kJ mol−1), which produces characteristic tunneling doublings in the rotational spectrum of Figure S1 (SI) [59], strongly perturbed by Coriolis interactions [60]. The inversion barrier in benzyl mercaptan is ca. 11% lower than in the alcohol (B2 = 280 cm−1 = 3.35 kJ mol−1). The molecule thus changes chirality by inverting between the two gauche-gauche enantiomers GG and G′G′ of Figure 1 (primes denote negative dihedrals).
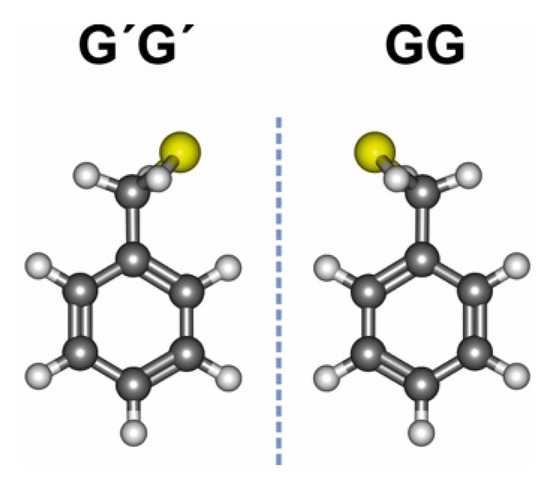
Figure 1.
The most stable GG conformer of benzyl mercaptan and its enantiomer G′G′.
3.2. Benzyl Mercaptan Homodimer
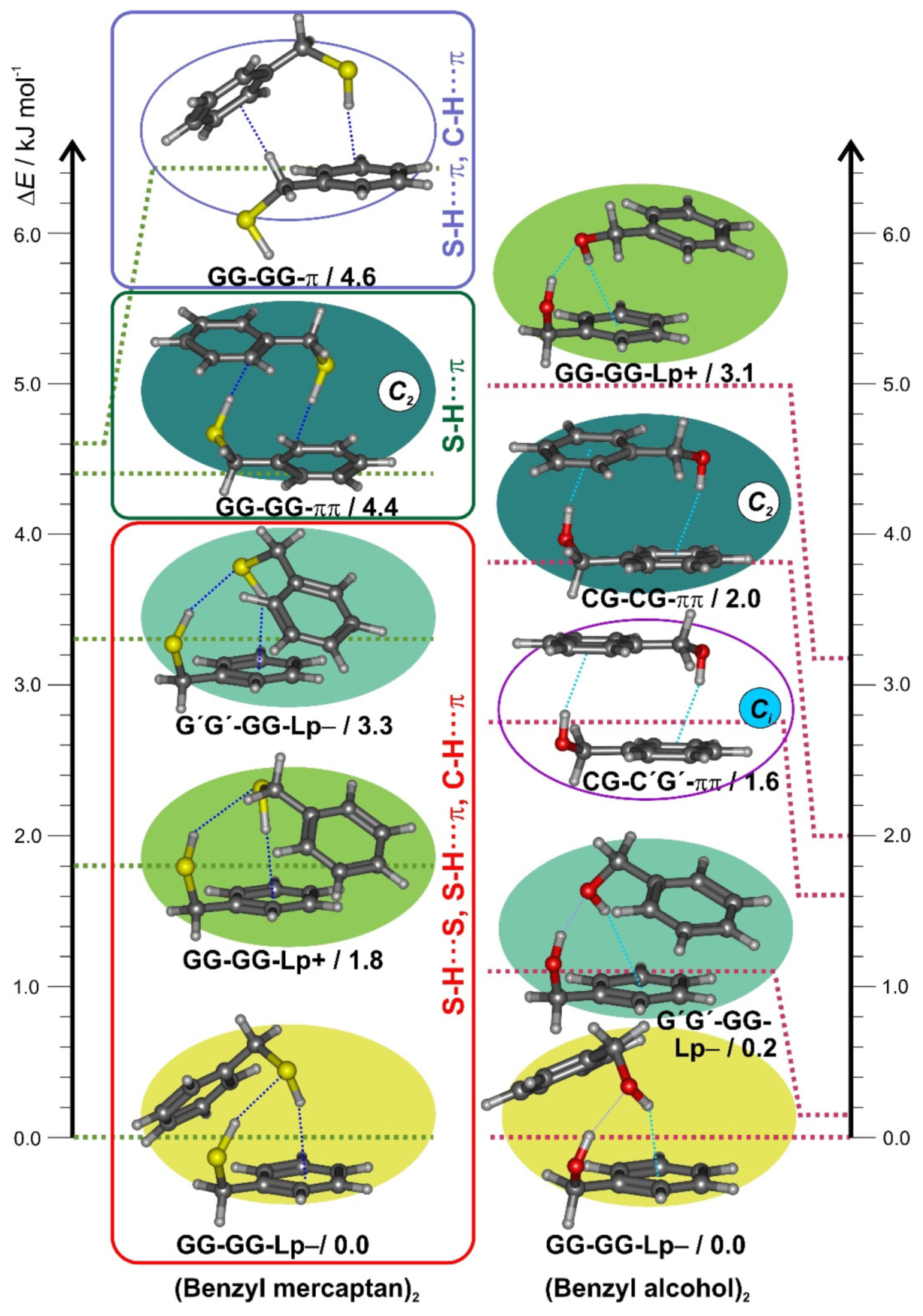
For benzyl mercaptan the dimerization from a single gauche-gauche conformation may in principle produce only homochiral (GG-GG = G′G′-G′G′) or heterochiral (GG-G′G′ = G′G′-GG) diastereoisomers (proton donor denoted first). The most stable isomers are expected to balance several intermolecular interactions, mainly involving the S-H and C-H groups as proton donors and the sulfur lone pairs and π electrons as acceptors. However, the two diastereotopic acceptor lone pairs (Lp+/−) and the multiple possibilities for interaction between the polar thiol groups and the two ring molecules are expected to produce multiple isomers. The results of a DFT (B3LYP-D3(BJ)) conformational search are summarized in Table S3 and Figure S2 (SI), suggesting 11 isomers within complexation energies below 10 kJ mol−1. The five most stable isomers are compared to the benzyl alcohol dimer [42] in Figure 2. The two most stable dimers 1 (= GG-GG-Lp−) and 2 (= GG-GG-Lp+) are homochiral and controlled by the two thiol groups (3D Figure S3, SI), which engage in two successive S-H···S and S-H···π interactions. The two rings adopt tilted (C1) unsymmetric orientations to permit further interaction via C(sp2)-H···π weak hydrogen bonds, with the alternative use of the two sulfur lone-pairs at the acceptor molecule producing two different ring orientations Lp+ and Lp− (Figure S4, SI), well separated in energy (ΔEZPE = 1.9 kJ mol−1, ΔEc = 3.1 kJ mol−1). The third isomer (=G′G′-GG-Lp−) is the heterochiral equivalent to isomer 1 (Figure S5, SI), predicted at ΔEZPE = 3.3 kJ mol−1 (ΔEc = 4.6 kJ mol−1) The fourth (=GG-GG-ππ) and fifth (=GG-GG-π) isomers reveal different homochiral S-H···π interactions, either alone or combined with a C(sp3)-H···π link, which is destabilized ca. 4–5 kJ mol−1 with respect to the thiol S-H···S hydrogen bonding. Isomer GG-GG-ππ uses two identical S-H···π interactions to produce the lowest-lying C2-symmetric dimer, characterized by two tilted rings which avoid π-stacking (Figure S6, SI). Only for the higher energy isomers (>10 kJ mol−1) near-parallel π···π or C-H···π interactions are predicted, as in the (C2) homochiral isomer 9 (=GG-GG) and the (Ci) heterochiral isomer 11 (=GG-G′G′). The preference for C2-symmetric heterochiral S-H···π dimers observed in the crystallographic structure [61] must thus be attributed to matrix effects and does not represent the isolated molecule nor the dimer. The three most stable isomers of the benzyl mercaptan dimer were reoptimized at B2PLYP-D3(BJ) level in Table 1, offering a prediction of the rotational parameters.
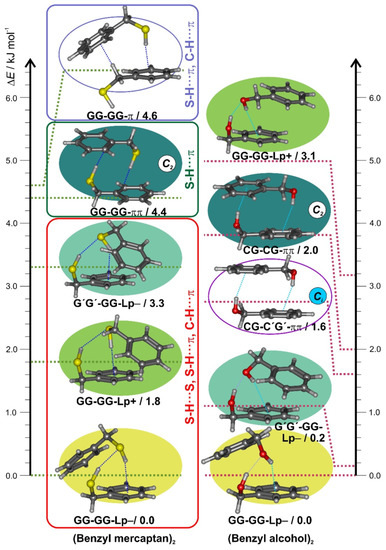
Figure 2.
Conformational search for the benzyl mercaptan dimer (left column), representing the B3LYP-D3(BJ) electronic energies (ΔE) of the five most stable isomers (see Figure S2 for a larger energy window). The right column represents the most stable isomers of the benzyl alcohol dimer.

Table 1.
Rotational parameters of the benzyl mercaptan dimer.
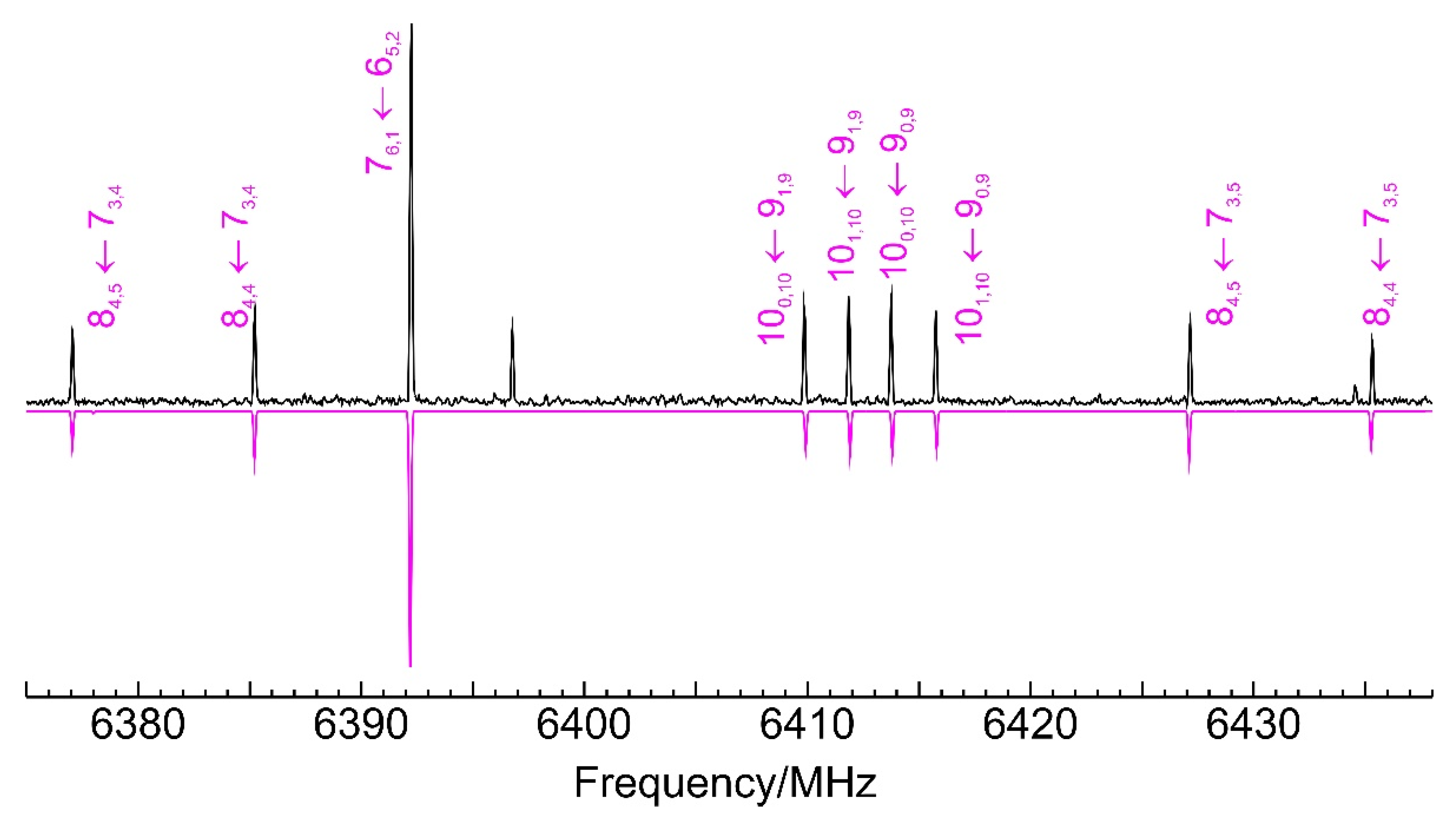
The jet-cooled microwave spectrum of Figure S1 provided experimental evidence on the nature of the dimerization adduct. Figure 3 shows a spectral section with typical rotational transitions. Noticeably, a single isomer was observed for the dimer of benzyl mercaptan. The spectrum was quite dense and more than 300 rotational transitions were measured experimentally, with all three (μa, μb, μc) selection rules active. The wide range of angular momentum quantum numbers (J = 3–15 and K−1 < 12) and the diversity of R-branch (J + 1←J) and some Q-branch (J←J) transitions assured a good determination of the rotational parameters. There was no indication of tunneling effects in the spectra, which were reproduced satisfactorily with a Watson’s (S-reduced) semirigid rotor Hamiltonian [62] and quartic centrifugal distortion terms. The experimental rotational parameters are compared with B2PLYP-D3(BJ) theoretical predictions in Table 1, with the full listing of observed transitions in Table S4 (SI). Since several low-lying isomers were predicted at small relative energies (<3–5 kJ mol−1), the observation of a single dimer species cannot be uniquely attributed to a pre-expansion thermal depopulation and probably suggests a conformational relaxation through low interconversion barriers, previously observed in jet experiments [63,64] and kinetically affecting the jet populations [65].
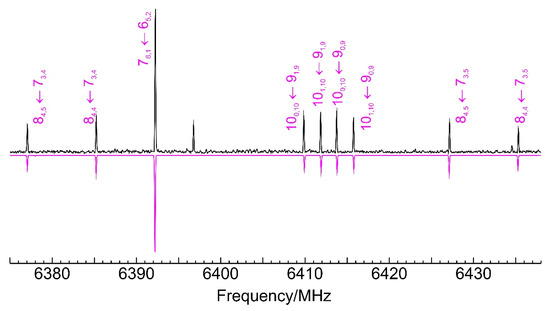
Figure 3.
A short 60 MHz section of the microwave spectrum of the benzyl mercaptan dimer, illustrating typical rotational transitions of the homochiral isomer 1 (GG-GG-Lp−). See Figure S1 (Supplementary Information) for the full 2–8 GHz spectrum. The positive trace is the experimental spectrum. The negative trace shows a simulation based on the fit of Table 1. The unmarked transitions belong to different species.
The identification of the benzyl mercaptan dimer was unequivocal. We confirmed the detection of the predicted global minimum, identified as the homochiral GG-GG-Lp−. The predictions for the equilibrium rotational constants slightly exceed the experimental ground-state values by 6–8 MHz, but the relative differences are below 1.6–2.8%, ensuring a positive identification. The agreement of the (harmonic) centrifugal distortion constants with the experiment values is consistent with the conformational assignment.
3.3. Non-Covalent Interactions
The intermolecular interactions associated to the benzyl mercaptan dimerization were explored using structural, energetic, and electronic density information. Unlike the parallel-displaced hydrogen-bonded dimer of thiophenol [36], the aggregation of benzyl mercaptan follows a pattern of non-stacked cooperative hydrogen bonding. A primary S-H···S hydrogen bond is formed in which the thiol acts as a proton donor to a second sulfur atom, followed by a secondary S-H···π hydrogen bond to the opposed π ring. Weak C-H···π or C-H···S hydrogen bonds are minor necessary contributors to the most stable isomers. The benzyl mercaptan dimers exhibit characteristically long S-H···S hydrogen bonds (B2PLYP: r(S-H···S) = 2.75 Å), relatively close to linearity (B2PLYP: ∠(S-H···S) = 163°). These values compare satisfactorily with the few rotational studies of thiol-thiol hydrogen bonding in the dimers of thiophenol [36] (B2PLYP: r(S-H···S) = 2.84 Å, ∠(S-H···S) = 135°) and hydrogen sulfide [35] (experiment: r(S-H···S) = 2.778(9) Å; B2PLYP: r(S-H···S) = 2.79 Å, ∠(S-H···S) = 172°). Crystallographic surveys of cysteine-methionine contacts have yielded shorter average values of r(S-H···S) = 2.55(47) Å [66] and r(S-H···S) = 2.5–2.7 Å [37]. The secondary hydrogen bond interactions between the thiol group and the π ring in Table 1 show distances to the ring centroid of r(S-H···centroid) = 2.52 Å (B2PLYP).
Binding energies are collected in Table 1 and Table 2, comparing B2PLYP (with BSSE corrections) and a second-order 2+(3) symmetry-adapted perturbation theory (SAPT) energy decomposition (at the optimized geometries of B3LYP-D3(BJ)/def2-TZVP). Not unexpectedly, the predicted binding energy of the benzyl mercaptan dimer (SAPT: −35.3 kJ mol−1) is larger than in thiophenol (−25.9 to −27 kJ mol−1) [36] but remains ca. 84% smaller than in the benzyl alcohol dimer (−42.1 kJ mol−1) [43], reflecting the weaker thiol H-bond. Additional insight into the nature of the thiol hydrogen bond is obtained from the attractive contributions to the energy decomposition [67,68]. The largest attractive contributor in the benzyl mercaptan dimer is dispersive (54%) but in smaller proportion than in the thiophenol dimer (60%) and with larger electrostatic participation (34% vs. 29–31% in thiophenol). This result reverts the calculation for the benzyl alcohol dimer, where the electrostatic component (45%) is larger but nearly matched by dispersion (41%). A comparison with the dispersive pyridine-methane dimer and the water and hydrogen sulfide dimers is also given in Table 2. Similar trends of decreasing strength and dispersion have been observed in other thiol clusters and monohydrates [30,31]. To our knowledge, there are no comparable experimental binding energies involving thiol dimers except for hydrogen sulfide clusters [26,27].
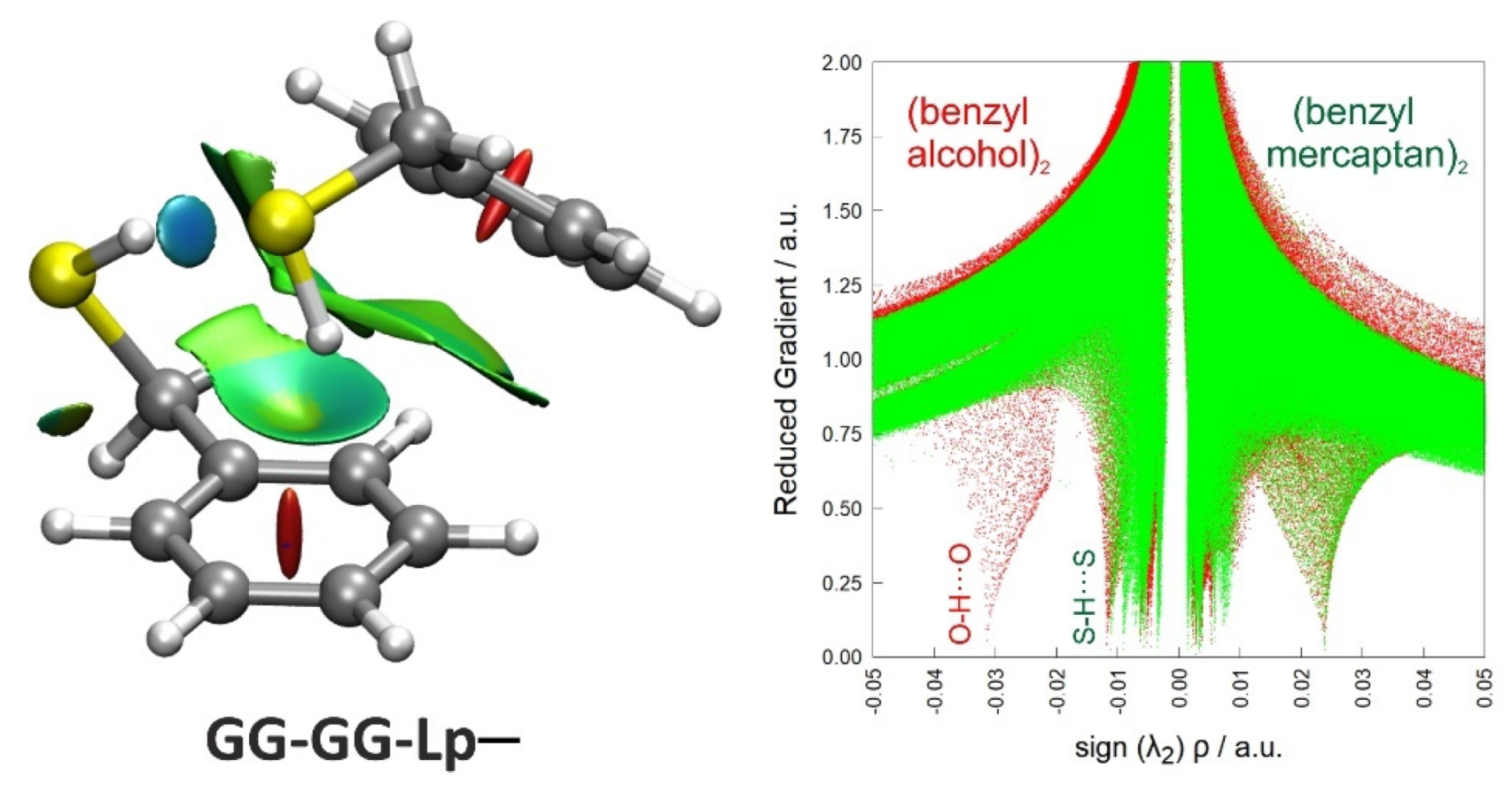
Non-covalent interactions can also be analyzed using the topological properties of the electron density () with the NCIPlot method [57]. Figure 4 represents a reduced electronic density gradient versus the signed electronic density using the second eigenvalue of the electron density Hessian, comparing the global minimum of the benzyl mercaptan and benzyl alcohol dimers (B3LYP-D3(BJ)/def2-TZVP geometries). This representation qualitatively identifies the stronger O-H···O hydrogen bond interactions in the alcohol (at more negative abscissas), simultaneously providing a 3D visualization of the most relevant interaction surfaces. The 3D plot clearly identifies the S-H···S hydrogen bond and broad interaction regions between the two rings mainly associated with the S-H···π contact. The reduced electronic density calculations thus confirm the cooperative scheme of intermolecular interactions present in the benzyl mercaptan dimer, complementing the structural and energetic descriptions in Table 1 and Table 2.
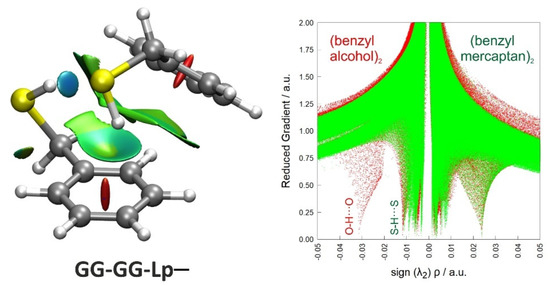
Figure 4.
NCI plot (left panel) and reduced electronic density gradient (right panel) for the observed dimer of benzyl mercaptan. In the NCI plot, blue shades indicate attractive interactions (associated to the S-H···S hydrogen bond), green colors indicate weak attractive interactions (like S-H···π), and red represents repulsive interactions (ring critical points). The reduced electronic density shows the attractive (negative minima) and repulsive (positive minima) interactions in the dimers of benzyl mercaptan (green) and benzyl alcohol (red), with a stronger O-H···O interaction.
Table 2.
Binding energy decomposition for the dimer of benzyl mercaptan, and comparison with several thiol and alcohol dimers and the van der Waals pyridine-methane dimer. The interaction energy is decomposed into electrostatic (ΔEelec), inductive (multipole interactions/charge transfer, ΔEind), exchange repulsion (ΔEexch) and dispersion (ΔEdisp) energy terms, using SAPT(2)+3/aug-cc-pVDZ//B3LYP-D3(BJ)/def2-TZVP (all values in kJ mol−1).
4. Discussion
The benzyl mercaptan dimer is a model cluster with a primary amphoteric thiol group and a combination of aliphatic proton donors and a π ring acceptor. The methylene pivot between the thiol and the aromatic ring permits some conformational flexibility and a variety of intermolecular forces, generally balancing two cooperative interactions. The rotational spectrum confirmed a single isomer in the gas phase, providing data for comparison between experiment and theory. The observed global minimum (GG-GG-Lp−) is homochiral and characterized by a leading thiol-thiol (S-H···S) hydrogen bond, in cooperation with a secondary thiol-aromatic (S-H···π) hydrogen bond. Characteristically, this interaction pattern and stereochemistry is coincident with the most stable (hom-Ogπ-I) benzyl alcohol dimer, stabilized by stronger O-H···O and O-H···π alcohol hydrogen bonds [42,43]. In consequence, the expected reduction in hydrogen bond strength of the thiol group does not affect the conformational equilibrium observed in the alcohol, and the π ring acceptor maintains its secondary role in both dimers. The smaller, more rigid phenol and thiophenol dimers, also observed in the gas phase [36,69], similarly depend on the S-H···S or O-H···O hydrogen bonds but adopt different hinged or stacked orientations which reflect a different balance with the ring interactions. Since the number of experimental observations is limited additional thiol-alcohol comparisons are presently possible only for a few other aromatic dimers like 2-phenylethanethiol [29,70], also showing similar thiol-thiol interactions in the global minimum.
The second isomer of the mercaptan dimer (GG-GG-Lp+), separated 1.7 kJ mol−1 (B2PLYP)–1.8 kJ mol−1 (B3LYP), differs in the use of a different sulfur lone-pair in the acceptor molecule but maintains its homochiral character. The equivalent isomer in the alcohol (hom-Otπ-V: 3.2 kJ mol−1) is much higher in energy. Consequently, the benzyl mercaptan dimer shows a reinforced preference for homochirality compared to the alcohol, where the heterochiral isomer (het-Ogπ-II) is nearly isoenergetic (0.2 kJ mol−1) with the (hom-Ogπ-I) global minimum. Conversely, in the benzyl mercaptan dimer the heterochiral isomer (G′G′-GG-Lp− ≡ het-Ogπ-II) is third in energy and more separated ((B2PLYP: 3.4 kJ mol−1; B3LYP: 3.3 kJ mol−1) from the homochiral partner.
The fourth and fifth isomers of the benzyl mercaptan dimer do not show thiol-thiol hydrogen bonds, which are replaced by separate thiol interactions with different proton acceptors. These isomers introduce the discussion on the strength of the thiol or alcohol groups as proton acceptors in presence of competing groups, which has been mostly studied for alcohol dimers with two acceptor groups. In those cases, the observed alcohol-alcohol hydrogen bonding of (benzyl alcohol)2 is confirmed with related aromatic acceptors, as in (indanol)2 [71] or (propargyl alcohol)2 [72]. However, ethers [21,73], carbonyl groups [74], or carboxylic acids [75] show preference as primary proton acceptors, more intense in amino [76] or cyano [77] groups. Additionally, when the two thiol/alcohol groups bind separately to secondary acceptors both homo and heterochiral symmetric geometries become possible. In the benzyl mercaptan dimer, the fourth isomer (GG-GG-ππ, 4.4 kJ mol−1) shows a homochiral C2 antiparallel geometry stabilized by two S-H···π hydrogen bonds, higher in energy than the equivalent C2 benzyl alcohol dimer (homππ-IV, 2.0 kJ mol−1) and reminiscent of the antiparallel aniline dimer [78]. However, the heterochiral Ci benzyl alcohol dimer (hetππ-III, 1.6 kJ mol−1) does not appear among the preferred geometries of the benzyl mercaptan dimer, where the fifth unsymmetric isomer (GG-GG-π, 4.6 kJ mol−1) instead shows a combination of S-H···π and C-H···π interactions.
Differences in the physical forces operating in the benzyl mercaptan and alcohol dimers are notorious in the topological analysis of the electronic density and the SAPT energy decomposition of Table 2 and Figure 3. The weaker, dispersive character of the thiol dimer, thus, contributes to a better description of non-covalent forces involving low-electronegativity atoms.
Future advances in the understanding of intermolecular forces are expected to extend to different chemical groups, incorporating a synergic combination of empirical data and computational models. In this work, rotational spectroscopy has an increasingly important role thanks to the development of chirped-pulsed broadband techniques.
5. Conclusions
The combination of rotational data and molecular orbital calculations constitutes a very effective way of interrogating weak non-covalent interactions, simultaneously extending the knowledge base on intermolecular forces and the benchmark capacity of the experiments. The availability of empirical data is expected to contribute to the development of improved density-functional methods, which will be essential for the molecular modelling of large chemical systems.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/sym13112022/s1, Figure S1: Rotational spectrum of benzyl mercaptan in the 3–8 GHz frequency region. The positive trace shows the experimental spectrum; the negative trace is the simulation of fitted rotational constants of the monomer and the dimer (isomer 1, GG-GG-Lp−), Figure S2: The most stable isomers of the benzyl mercaptan dimer, classified by their intermolecular interactions and ordered by complexation energy (ΔEc, kJ/mol). Isomers in the dashed rectangle exhibit a sulfur S-H···S hydrogen bond. The only symmetric isomers are 5 (C2), 9 (C2) and 11 (Ci), Figure S3: A rotatable 3D figure of the two most stable isomers of the benzyl mercaptan dimer, Figure S4: A comparison of the three asymmetric (C1) most stable isomers of the benzyl mercaptan dimer. Isomers 1 (G′G′-G′G′-Lp+ = GG-GG-Lp−, left) and 2 (G′G′-G′G′-Lp− = GG-GG-Lp+, right), are both homochiral but differ in the acceptor lone pair position of sulfur (the dihedral formed by the lone pair or τ(LpS-CαCipso) have different signs for isomer 1 and isomer 2), producing a change of relative orientation between the rings, Figure S5: A comparison of the three asymmetric (C1) most stable isomers of the benzyl mercaptan dimer. Isomers 1 (G′G′-G′G′-Lp+ = GG-GG-Lp−, left) and 3 (GG-G′G′-Lp+ = G′G′-GG-Lp−, right) differ in the stereochemistry of the donor group, either homochiral in isomer 1 or heterochiral in isomer 3, Figure S6: The most stable symmetric dimer of benzyl mercaptan (isomer 5 GG-GG-ππ) has C2 symmetry but avoids the parallel arrangement of the two phenyl rings, unlike in the symmetric benzyl alcohol dimer. The Figure shows the (a, b, c) principal inertial axes, with a vector pointing in the C2 (≡b) symmetry axis direction, Figure S7: A comparison of the NCI Plots for isomers 1 (G′G′-G′G′-Lp+ = GG-GG-Lp−, upper panel) and 2 (G′G′-G′G′-Lp− = GG-GG-Lp+, lower panel) of the benzyl mercaptan dimer, Table S1: Rotational parameters of benzyl mercaptan, Table S2: Atomic coordinates of benzyl mercaptan gauche GG, Table S3: Conformational search for the benzyl mercaptan dimer (B3LYP-D3(BJ)) and comparison with the experimental rotational parameters, Table S4: List of observed rotational transitions of the benzyl mercaptan dimer GG-GG-Lp− and (observed-calculated, o-c) residuals for the fit of Table 1 (all units in MHz).
Author Contributions
Conceptualization, A.L.; methodology, A.L. and J.A.F.; software, R.P. and J.E.R.; validation, R.T.S. and M.J.; formal analysis, R.T.S.; investigation, R.T.S.; resources, A.L.; data curation, A.L.; writing—original draft preparation, A.L.; writing—review and editing, R.T.S., M.J., R.P., J.E.R., J.A.F. and A.L.; visualization, R.T.S.; supervision, A.L.; project administration, A.L.; funding acquisition, A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministerio de Ciencia e Innovación MICINN-FEDER, grants numbers PGC2018-098561-B-C21 and PGC2018-098561-B-C22. The APC were funded by PGC2018-098561-B-C22.
Data Availability Statement
The data presented in this study are available in the supplementary material.
Acknowledgments
M.J. and R.T.S. thank predoctoral contracts from the MECD and UVa, respectively.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-Assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.R.; Salerno, F.; Fuchter, M.J. The added value of small-molecule chirality in technological applications. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Schermann, J.-P. Spectroscopy and Modeling of Biomolecular Building Blocks; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780444527080. [Google Scholar]
- Hobza, P.; Muller-Dethlefs, K. Non-Covalent Interactions; Hobza, P., Muller-Dethlefs, K., Eds.; Theoretical and Computational Chemistry Series; Royal Society of Chemistry: Cambridge, UK, 2009; ISBN 9781847558534. [Google Scholar]
- Scheiner, S. (Ed.) Challenges and Advances in Computational Chemistry and Physics. In Noncovalent Forces; Springer International Publishing: Cham, Switzerland, 2015; Volume 19, ISBN 9783319141626. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Juanes, M.; Saragi, R.T.; Caminati, W.; Lesarri, A. The Hydrogen Bond and Beyond: Perspectives for Rotational Investigations of Non-Covalent Interactions. Chem. A Eur. J. 2019, 25, 11402–11411. [Google Scholar] [CrossRef]
- Zehnacker, A.; Suhm, M.A. Chirality Recognition between Neutral Molecules in the Gas Phase. Angew. Chem. Int. Ed. 2008, 47, 6970–6992. [Google Scholar] [CrossRef]
- Zehnacker, A. (Ed.) Chiral Recognition in the Gas Phase; CRC Press: Boca Raton, FL, USA, 2010; ISBN 9781420082272. [Google Scholar]
- Pierini, M.; Troiani, A.; Speranza, M.; Piccirillo, S.; Bosman, C.; Toja, D.; Giardini-Guidoni, A. Gas-Phase Enantiodifferentiation of Chiral Molecules: Chiral Recognition of 1-Phenyl-1-propanol/2-Butanol Clusters by Resonance Enhanced Multiphoton Ionization Spectroscopy. Angew. Chem. Int. Ed. 1997, 36, 1729–1731. [Google Scholar] [CrossRef]
- Le Barbu, K.; Brenner, V.; Millié, P.; Lahmani, F.; Zehnacker-Rentien, A. An Experimental and Theoretical Study of Jet-Cooled Complexes of Chiral Molecules: The Role of Dispersive Forces in Chiral Discrimination. J. Phys. Chem. A 1998, 102, 128–137. [Google Scholar] [CrossRef]
- Borho, N.; Häber, T.; Suhm, M.A. Chiral self-recognition in the gas phase: The case of glycidol dimers. Phys. Chem. Chem. Phys. 2001, 3, 1945–1948. [Google Scholar] [CrossRef]
- Caminati, W.; Grabow, J.-U. Advancements in Microwave Spectroscopy. In Frontiers and Advances in Molecular Spectroscopy; Laane, J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 569–598. ISBN 9780128112212. [Google Scholar]
- Portmann, S.; Inauen, A.; Lüthi, H.P.; Leutwyler, S. Chiral discrimination in hydrogen-bonded complexes. J. Chem. Phys. 2000, 113, 9577–9585. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Discrimination of hydrogen-bonded complexes with axial chirality. J. Chem. Phys. 2002, 117, 6463–6468. [Google Scholar] [CrossRef] [Green Version]
- Borho, N.; Xu, Y. Tailoring the key in a molecular lock-and-key model system: The propylene oxide⋯2-fluoroethanol complex. J. Am. Chem. Soc. 2008, 130, 5916–5921. [Google Scholar] [CrossRef] [PubMed]
- Hearn, J.P.I.; Cobley, R.V.; Howard, B.J. High-resolution spectroscopy of induced chiral dimers: A study of the dimers of ethanol by Fourier transform microwave spectroscopy. J. Chem. Phys. 2005, 123, 134324. [Google Scholar] [CrossRef] [PubMed]
- Loru, D.; Peña, I.; Sanz, M.E. Ethanol dimer: Observation of three new conformers by broadband rotational spectroscopy. J. Mol. Spectrosc. 2017, 335, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Snow, M.S.; Howard, B.J.; Evangelisti, L.; Caminati, W. From Transient to Induced Permanent Chirality in 2-Propanol upon Dimerization: A Rotational Study. J. Phys. Chem. A 2011, 115, 47–51. [Google Scholar] [CrossRef] [PubMed]
- King, A.K.; Howard, B.J. A microwave study of the hetero-chiral dimer of butan-2-ol. Chem. Phys. Lett. 2001, 348, 343–349. [Google Scholar] [CrossRef]
- Maris, A.; Giuliano, B.M.; Bonazzi, D.; Caminati, W. Molecular recognition of chiral conformers: A rotational study of the dimers of glycidol. J. Am. Chem. Soc. 2008, 130, 13860–13861. [Google Scholar] [CrossRef]
- Juanes, M.; Usabiaga, I.; León, I.; Evangelisti, L.; Fernández, J.A.; Lesarri, A. The Six Isomers of the Cyclohexanol Dimer: A Delicate Test for Dispersion Models. Angew. Chem. Int. Ed. 2020, 59, 14081–14085. [Google Scholar] [CrossRef]
- Liu, X.; Borho, N.; Xu, Y. Molecular self-recognition: Rotational spectra of the dimeric 2-fluoroethanol conformers. Chem. A Eur. J. 2009, 15, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Xu, Y. Chirality synchronization in trifluoroethanol dimer revisited: The missing heterochiral dimer. J. Phys. Chem. Lett. 2014, 5, 1850–1855. [Google Scholar] [CrossRef]
- Oswald, S.; Seifert, N.A.; Bohle, F.; Gawrilow, M.; Grimme, S.; Jäger, W.; Xu, Y.; Suhm, M.A. The Chiral Trimer and a Metastable Chiral Dimer of Achiral Hexafluoroisopropanol: A Multi-Messenger Study. Angew. Chem. Int. Ed. 2019, 58, 5080–5084. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wategaonkar, S. ZEKE photoelectron spectroscopy of p-fluorophenol H2S/H2O complexes and dissociation energy measurement using the Birge-Sponer extrapolation method. J. Phys. Chem. A 2014, 118, 9386–9396. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Bhattacharyya, S.; Wategaonkar, S. Dissociation Energies of Sulfur-Centered Hydrogen-Bonded Complexes. J. Phys. Chem. A 2015, 119, 10863–10870. [Google Scholar] [CrossRef]
- Martin, D.E.; Robertson, E.G.; Thompson, C.D.; Morrison, R.J.S. Resonant 2-photon ionization study of the conformation and the binding of water molecules to 2-phenylethanethiol (PhC H2 C H2 SH). J. Chem. Phys. 2008, 128, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Lobo, I.A.; Robertson, P.A.; Villani, L.; Wilson, D.J.D.; Robertson, E.G. Thiols as Hydrogen Bond Acceptors and Donors: Spectroscopy of 2-Phenylethanethiol Complexes. J. Phys. Chem. A 2018, 122, 7171–7180. [Google Scholar] [CrossRef]
- Juanes, M.; Lesarri, A.; Pinacho, R.; Charro, E.; Rubio, J.E.; Enríquez, L.; Jaraíz, M. Sulfur Hydrogen Bonding in Isolated Monohydrates: Furfuryl Mercaptan versus Furfuryl Alcohol. Chem. A Eur. J. 2018, 24, 6564–6571. [Google Scholar] [CrossRef]
- Juanes, M.; Saragi, R.T.; Pinacho, R.; Rubio, J.E.; Lesarri, A. Sulfur hydrogen bonding and internal dynamics in the monohydrates of thenyl mercaptan and thenyl alcohol. Phys. Chem. Chem. Phys. 2020, 22, 12412–12421. [Google Scholar] [CrossRef] [PubMed]
- Wategaonkar, S.; Bhattacherjee, A. N-H···S Interaction Continues to Be an Enigma: Experimental and Computational Investigations of Hydrogen-Bonded Complexes of Benzimidazole with Thioethers. J. Phys. Chem. A 2018, 122, 4313–4321. [Google Scholar] [CrossRef] [PubMed]
- Cocinero, E.J.; Sánchez, R.; Blanco, S.; Lesarri, A.; López, J.C.; Alonso, J.L. Weak hydrogen bonds C–H⋯S and C–H⋯F–C in the thiirane–trifluoromethane dimer. Chem. Phys. Lett. 2005, 402, 4–10. [Google Scholar] [CrossRef]
- Ghosh, S.; Chopra, P.; Wategaonkar, S. C-H⋯S interaction exhibits all the characteristics of conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2020, 22, 17482–17493. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Mandal, P.K.; Lovas, F.J.; Medcraft, C.; Walker, N.R.; Arunan, E. The H2S Dimer is Hydrogen-Bonded: Direct Confirmation from Microwave Spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 15199–15203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saragi, R.T.; Juanes, M.; Pérez, C.; Pinacho, P.; Tikhonov, D.S.; Caminati, W.; Schnell, M.; Lesarri, A. Switching Hydrogen Bonding to π-Stacking: The Thiophenol Dimer and Trimer. J. Phys. Chem. Lett. 2021, 12, 1367–1373. [Google Scholar] [CrossRef]
- Mishra, K.K.; Borish, K.; Singh, G.; Panwaria, P.; Metya, S.; Madhusudhan, M.S.; Das, A. Observation of an Unusually Large IR Red-Shift in an Unconventional S–H···S Hydrogen-Bond. J. Phys. Chem. Lett. 2021, 12, 1228–1235. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Matsuda, Y.; Fujii, A.; Wategaonkar, S. Acid-base formalism in dispersion-stabilized S–H···Y (Y=O, S) hydrogen-bonding interactions. J. Phys. Chem. A 2015, 119, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chopra, P.; Wategaonkar, S.; Fujii, A. Electronic and Infrared Spectroscopy of Benzene-(H2S)n (n = 1 and 2): The Prototype of the SH-πInteraction. J. Phys. Chem. A 2019, 123, 7255–7260. [Google Scholar] [CrossRef] [PubMed]
- Biswal, H.S.; Bhattacharyya, S.; Bhattacherjee, A.; Wategaonkar, S. Nature and strength of sulfur-centred hydrogen bonds: Laser spectroscopic investigations in the gas phase and quantum-chemical calculations. Int. Rev. Phys. Chem. 2015, 34, 99–160. [Google Scholar] [CrossRef]
- Chand, A.; Sahoo, D.K.; Rana, A.; Jena, S.; Biswal, H.S. The Prodigious Hydrogen Bonds with Sulfur and Selenium in Molecular Assemblies, Structural Biology, and Functional Materials. Acc. Chem. Res. 2020, 53, 1580–1592. [Google Scholar] [CrossRef] [PubMed]
- Medel, R.; Suhm, M.A. Understanding benzyl alcohol aggregation by chiral modification: The pairing step. Phys. Chem. Chem. Phys. 2020, 22, 25538–25551. [Google Scholar] [CrossRef]
- Medel, R.; Camiruaga, A.; Saragi, R.T.; Pinacho, P.; Pérez, C.; Schnell, M.; Lesarri, A.; Suhm, M.A.; Fernandez, J.A. Rovibronic Signatures of Molecular Aggregation in the Gas Phase: Subtle Homochirality Trends in the Dimer, Trimer and Tetramer of Benzyl Alcohol. Phys. Chem. Chem. Phys. 2021, in press. [Google Scholar] [CrossRef]
- Neill, J.L.; Shipman, S.T.; Alvarez-Valtierra, L.; Lesarri, A.; Kisiel, Z.; Pate, B.H. Rotational spectroscopy of iodobenzene and iodobenzene-neon with a direct digital 2-8 GHz chirped-pulse Fourier transform microwave spectrometer. J. Mol. Spectrosc. 2011, 269, 21–29. [Google Scholar] [CrossRef]
- Shipman, S.T.; Pate, B.H. New Techniques in Microwave Spectroscopy. In Handbook of High-Resolution Spectroscopy; Major Reference Works; Merkt, F., Quack, M., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2011; pp. 801–828. ISBN 9780470749593. [Google Scholar]
- Grabow, J.-U. Fourier Transform Microwave Spectroscopy Measurement and Instrumentation. In Handbook of High-Resolution Spectroscopy; Merkt, F., Quack, M., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2011; pp. 723–799. ISBN 9780470749593. [Google Scholar]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press on Demand: Oxford, UK, 1997; ISBN 019509011X. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utzat, K.A.; Bohn, R.K.; Montgomery, J.A.; Michels, H.H.; Caminati, W. Rotational spectrum, tunneling motions, and potential barriers of benzyl alcohol. J. Phys. Chem. A 2010, 114, 6913–6916. [Google Scholar] [CrossRef] [PubMed]
- Saragi, R.T.; Juanes, M.; Caminati, W.; Lesarri, A.; Enríquez, L.; Jaraíz, M. Rotational Spectrum, Tunneling Motions, and Intramolecular Potential Barriers in Benzyl Mercaptan. J. Phys. Chem. A 2019, 123, 8435–8440. [Google Scholar] [CrossRef] [PubMed]
- Papousek, D.; Aliev, M.R. Molecular Vibrational-Rotational Spectra Studies in Physical and Theoretical Chemistry 17; Elsevier: Amsterdam, The Netherlands, 1982; ISBN 0444997377. [Google Scholar]
- Nayak, S.K.; Sathishkumar, R.; Row, T.N.G. Directing role of functional groups in selective generation of C–H⋯π interactions: In situ cryo-crystallographic studies on benzyl derivatives. CrystEngComm 2010, 12, 3112–3118. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure; Durig, J.R., Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Godfrey, P.D.; Brown, R.D. Proportions of species observed in jet spectroscopy-vibrational-energy effects: Histamine tautomers and conformers. J. Am. Chem. Soc. 1998, 120, 10724–10732. [Google Scholar] [CrossRef]
- Florio, G.M.; Christie, R.A.; Jordan, K.D.; Zwier, T.S. Conformational preferences of jet-cooled melatonin: Probing trans- and cis-amide regions of the potential energy surface. J. Am. Chem. Soc. 2002, 124, 10236–10247. [Google Scholar] [CrossRef]
- Lesarri, A.; Pinacho, R.; Enríquez, L.; Rubio, J.E.; Jaraíz, M.; Abad, J.L.; Gigosos, M.A. Rotational spectra of tetracyclic quinolizidine alkaloids: Does a water molecule flip sparteine? Phys. Chem. Chem. Phys. 2017, 19, 17553–17559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Tian, F.; Lv, F.; Shang, Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins Struct. Funct. Bioinform. 2009, 76, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Cukras, J.; Skóra, G.; Jankowska, J.; Lundell, J. Computational structures and SAPT interaction energies of HXeSH···H2Y (Y=O or S) complexes. Inorganics 2018, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Cukras, J.; Sadlej, J. Structure and energetics of weakly bound water-sulfur dioxide complexes. J. Mol. Struct. THEOCHEM 2007, 819, 41–51. [Google Scholar] [CrossRef]
- Seifert, N.A.; Steber, A.L.; Neill, J.L.; Pérez, C.; Zaleski, D.P.; Pate, B.H.; Lesarri, A. The interplay of hydrogen bonding and dispersion in phenol dimer and trimer: Structures from broadband rotational spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 11468–11477. [Google Scholar] [CrossRef]
- Mons, M.; Robertson, E.G.; Simons, J.P. Intra- and Intermolecular π-Type Hydrogen Bonding in Aryl Alcohols: UV and IR−UV Ion Dip Spectroscopy. J. Phys. Chem. A 2000, 104, 1430–1437. [Google Scholar] [CrossRef]
- Altnöder, J.; Bouchet, A.; Lee, J.J.; Otto, K.E.; Suhm, M.A.; Zehnacker-Rentien, A. Chirality-dependent balance between hydrogen bonding and London dispersion in isolated (±)-1-indanol clusters. Phys. Chem. Chem. Phys. 2013, 15, 10167–10180. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. Rotational spectra of propargyl alcohol dimer: A dimer bound with three different types of hydrogen bonds. J. Chem. Phys. 2014, 141, 164311. [Google Scholar] [CrossRef]
- Longarte, A.; Redondo, C.; Fernández, J.A.; Castaño, F. IR/UV and UV/UV double-resonance study of guaiacol and eugenol dimers. J. Chem. Phys. 2005, 122, 164304. [Google Scholar] [CrossRef] [PubMed]
- Altnöder, J.; Lee, J.J.; Otto, K.E.; Suhm, M.A. Molecular Recognition in Glycolaldehyde, the Simplest Sugar: Two Isolated Hydrogen Bonds Win Over One Cooperative Pair. ChemistryOpen 2012, 1, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, L.; Spada, L.; Li, W.; Vazart, F.; Barone, V.; Caminati, W. The Borderline between Reactivity and Pre-reactivity of Binary Mixtures of Gaseous Carboxylic Acids and Alcohols. Angew. Chem. Int. Ed. 2017, 56, 3872–3875. [Google Scholar] [CrossRef] [Green Version]
- Asselin, P.; Madebène, B.; Soulard, P.; Georges, R.; Goubet, M.; Huet, T.R.; Pirali, O.; Zehnacker-Rentien, A. Competition between inter- and intra-molecular hydrogen bonding: An infrared spectroscopic study of jet-cooled amino-ethanol and its dimer. J. Chem. Phys. 2016, 145, 224313. [Google Scholar] [CrossRef] [Green Version]
- Seurre, N.; Le Barbu-Debus, K.; Lahmani, F.; Zehnacker-Rentien, A.; Sepioł, J. Electronic and vibrational spectroscopy of jet-cooled m-cyanophenol and its dimer: Laser-induced fluorescence and fluorescence-dip IR spectra in the S0 and S1 states. Chem. Phys. 2003, 295, 21–33. [Google Scholar] [CrossRef]
- Pérez, C.; León, I.; Lesarri, A.; Pate, B.H.; Martínez, R.; Millán, J.; Fernández, J.A. Isomerism of the Aniline Trimer. Angew. Chem. Int. Ed. 2018, 57, 15112–15116. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).