Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
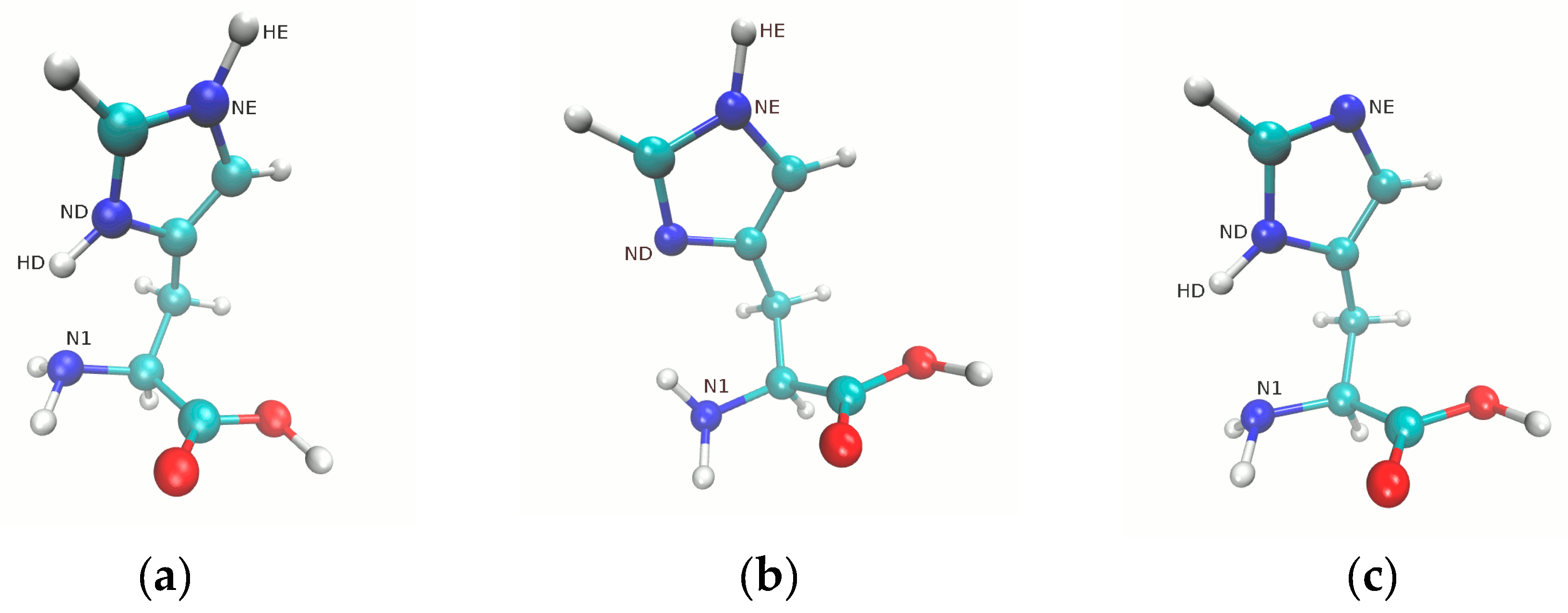
3.1. Metric Parameters Analysis
3.2. Atoms-In-Molecules (AIM) Analysis
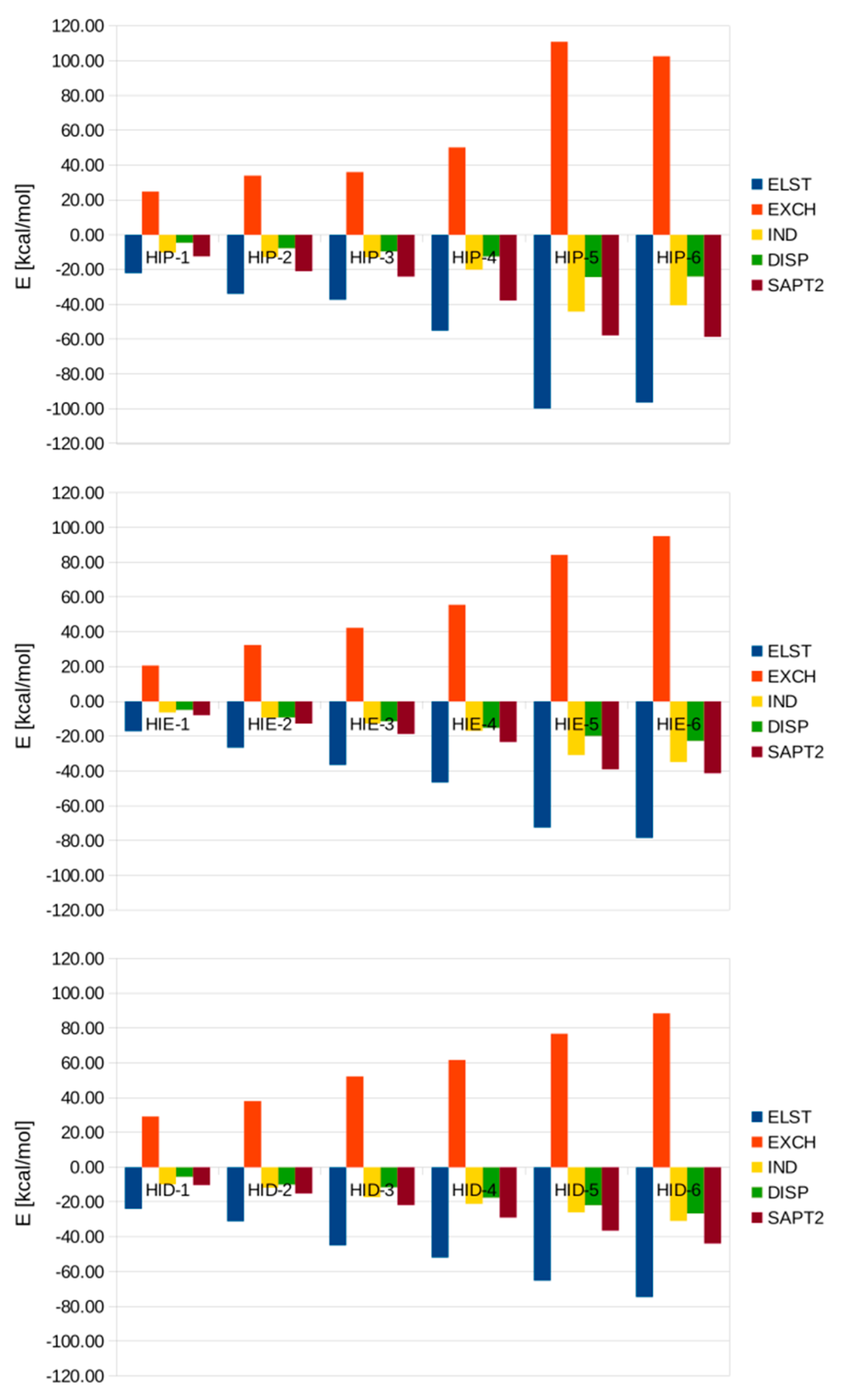
3.3. SAPT Analysis of Interaction Energies in the Microsolvated His-(H2O)n Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Braga, D.; Grepioni, F.; Desiraju, G.R. Crystal Engineering and Organometallic Architecture. Chem. Rev. 1998, 98, 1375–1405. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: New York, NY, USA, 1991. [Google Scholar]
- Wyttenbach, T.; Paizs, B.; Barran, P.; Breci, L.; Liu, D.; Suhai, S.; Wysocki, V.H.; Bowers, M.T. The Effect of the Initial Water of Hydration on the Energetics, Structures, and H/D Exchange Mechanism of a Family of Pentapeptides: An Experimental and Theoretical Study. J. Am. Chem. Soc. 2003, 125, 13768–13775. [Google Scholar] [CrossRef] [PubMed]
- Margenau, H.; Kestner, N. Theory of Inter-Molecular Forces, International Series of Monographs in Natural Philosophy; Pergamon Press: Oxford, UK, 1969. [Google Scholar]
- Chałasiński, G.; Gutowski, M. Weak interactions between small systems. Models for studying the nature of intermolecular forces and challenging problems for ab initio calculations. Chem. Rev. 1988, 88, 943–962. [Google Scholar] [CrossRef]
- Hobza, P.; Zahradnik, R. Intermolecular interactions between medium-sized systems. Nonempirical and empirical calculations of interaction energies. Successes and failures. Chem. Rev. 1988, 88, 871–897. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Grabowski, S.J. New type of halogen bond: Multivalent halogen interacting with π and σ-electrons. Molecules 2017, 22, 2150. [Google Scholar] [CrossRef]
- Jabłoński, M. Theoretical insight into the nature of the intermolecular charge-inverted hydrogen bond. Comput. Theor. Chem. 2012, 998, 39–45. [Google Scholar] [CrossRef]
- Jabłoński, M. Charge-inverted hydrogen bond vs. other interactions possessing a hydridic hydrogen atom. Chem. Phys. 2014, 433, 76–84. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2007. [Google Scholar]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B.O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed]
- López, J.C.; Sánchez, R.; Blanco, S.; Alonso, J.L. Microsolvation of 2-azetidinone: A model for the peptide group-water interactions. Phys. Chem. Chem. Phys. 2015, 17, 2054–2066. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.O.; Mennucci, B.; Vreven, T. Combining microsolvation and polarizable continuum studies: New insights in the rotation mechanism of amides in water. J. Phys. Chem. A 2003, 107, 6630–6637. [Google Scholar] [CrossRef]
- Li, S.; Hong, M. Protonation, Tautomerization, and Rotameric Structure of Histidine: A Comprehensive Study by Magic-Angle-Spinning Solid-State NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. [Google Scholar] [CrossRef]
- Day, R.M.; Thalhauser, C.J.; Sudmeier, J.L.; Vincent, M.P.; Torchilin, E.V.; Sanford, D.G.; Bachovchin, C.W.; Bachovchin, W.W. Tautomerism, acid-base equilibria, and H bonding of the six histidines in subtilisin BPN′ by NMR. Protein Sci. 2003, 12, 794–810. [Google Scholar] [CrossRef]
- Krishna Deepak, R.N.V.; Sankararamakrishnan, R. N-H…N Hydrogen Bonds Involving Histidine Imidazole Nitrogen Atoms: A New Structural Role for Histidine Residues in Proteins. Biochemistry 2016, 55, 3774–3783. [Google Scholar] [CrossRef]
- Olivieri, F.A.; Burastero, O.; Drusin, S.I.; Defelipe, L.A.; Wetzler, D.E.; Turjanski, A.; Marti, M. Conformational and Reaction Dynamic Coupling in Histidine Kinases: Insights from Hybrid QM/MM Simulations. J. Chem. Inf. Model. 2020, 60, 833–842. [Google Scholar] [CrossRef]
- Kawamura, K.; Yamada, T.; Kurihara, K.; Tamada, T.; Kuroki, R.; Tanaka, I.; Takahashi, H.; Niimura, N. X-ray and neutron protein crystallographic analysis of the trypsin-BPTI complex. Acta Cryst. D 2011, 67, 140–148. [Google Scholar] [CrossRef]
- Saxena, A.K.; Singh, T.P.; Peters, K.; Fittkau, S.; Visanji, M.; Wilson, K.S.; Betzel, C. Structure of a ternary complex of proteinase K, mercury, and a substrate-analogue hexa-peptide at 2.2 Å resolution. Proteins 1996, 25, 195–201. [Google Scholar] [CrossRef]
- Panek, J.J.; Mazzarello, R.; Novič, M.; Jezierska-Mazzarello, A. Impact of mercury(II) on proteinase K catalytic center: Investigations via classical and Born-Oppenheimer molecular dynamics. Mol. Divers. 2011, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Kuczer, M.; Błaszak, M.; Czarniewska, E.; Rosiński, G.; Kowalik-Jankowska, T. Mono- and Polynuclear Copper(II) Complexes of Alloferons 1 with Point Mutations (H6A) and (H12A): Stability Structure and Cytotoxicity. Inorg. Chem. 2013, 52, 5951–5961. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.S.; Mathews, A.J.; Rohlfs, R.J.; Springer, B.A.; Egeberg, K.D.; Sligar, S.G.; Tame, J.; Renaud, J.P.; Nagai, K. The role of the distal histidine in myoglobin and haemoglobin. Nature 1988, 336, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Vila, J.A.; Arnautova, Y.A.; Vorobjev, Y.; Scheraga, H.A. Assessing the fractions of tautomeric forms of the imidazole ring of histidine in proteins as a function of pH. Proc. Natl Acad. Sci. USA 2011, 108, 5602–5607. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Nichols, S.E.; Wang, Y.; McCammon, A. Effects of histidine protonation and rotameric states on virtual screening of M. tuberculosis RmlC. J. Comput. Aided Mol. Des. 2013, 27, 235–246. [Google Scholar] [CrossRef]
- Cysewski, P.; Szefler, B. Environment influences on the aromatic character of nucleobases and amino acids. J. Mol. Model. 2010, 16, 1709–1720. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Jeziorski, B.; Moszyński, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 7, 1887–1930. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. Cooperativity of hydrogen bonding network in microsolvated biotin, the ligand of avidin class proteins. J. Mol. Model. 2019, 25, 361. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Fei, W.; Lu, Z.; Lin, Z. Effects of microsolvation and aqueous solvation on the tautomers of histidine: A computational study on energy, structure and IR spectrum. Theor. Chem. Acc. 2009, 124, 37–47. [Google Scholar] [CrossRef]
- Lee, S.-S.; Kim, J.-Y.; Han, Y.; Shim, H.-J.; Lee, S. Thermodynamic and kinetic stability of zwitterionic histidine: Effects of gas phase hydration. Chem. Phys. Lett. 2015, 637, 42–50. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 1 July 2020).
- Skelton, B.W.; Piggott, M.J.; Walkey, M.C. CCDC 1840707: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2018. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Design 2000, 14, 123–134. Available online: http://cheminf.cmbi.ru.nl/molden/ (accessed on 4 June 2020). [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- PSI4 Open-Source Quantum Chemistry. Available online: http://www.psicode.org/psi4manual/master/dfmp2.html# (accessed on 4 June 2020).
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- AIMAll (Version 19.10.12) Software by T. A. Keith. Available online: aim.tkgristmill.com (accessed on 4 June 2020).
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The GIMP Development Team, GIMP. 2019. Available online: https://www.gimp.org (accessed on 1 July 2020).
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Grimme, S.; Mück-Lichtenfeld, C.; Erker, G.; Kehr, G.; Wang, H.; Beckers, H.; Willner, H. When Do Interacting Atoms Form a Chemical Bond? Spectroscopic Measurements and Theoretical Analyses of Dideuteriophenanthrene. Angew. Chem. Int. Ed. 2009, 48, 2592–2595. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomete, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric Parameters | Gas Phase | PCM |
|---|---|---|
| HIP | ||
| ND…N1 | 2.713 | 2.769 |
| ND-HD | 1.036 | 1.026 |
| HD…N1 | 1.917 | 2.030 |
| ∠NHN [°] | 131.2 | 126.9 |
| HIE | ||
| ND…N1 | 3.219 | 3.160 |
| N1-H | 1.012 | 1.017 |
| H…ND | 2.816 | 2.411 |
| ∠NHN [°] | 104.2 | 129.8 |
| HID | ||
| ND…N1 | 2.905 | 2.872 |
| ND-HD | 1.011 | 1.014 |
| HD…N1 | 2.247 | 2.197 |
| ∠NHN [°] | 121.4 | 122.5 |
| Metric Parameters | Histidine HIP Form | |
|---|---|---|
| HIP with 1–6 Water Molecules | HIP with 1–6 Water Molecules and with PCM | |
| One water molecule (HIP-1) | ||
| Intramolecular HB | ||
| ND...N1 | 2.706 | 2.763 |
| ND-HD | 1.038 | 1.027 |
| HD...N1 | 1.901 | 2.015 |
| ∠NHN [°] | 131.9 | 127.5 |
| Intermolecular HB | ||
| O1...O | 2.644 | 2.671 |
| O1-H1 | 0.992 | 0.992 |
| H1...O | 1.671 | 1.679 |
| ∠O1H1O [°] | 165.7 | 178.0 |
| Two water molecules (HIP-2) | ||
| Intramolecular HB | ||
| ND...N1 [A] | 2.668 | 2.737 |
| ND-HD | 1.048 | 1.030 |
| HD...N1 | 1.824 | 1.966 |
| ∠NHN [°] | 134.7 | 129.3 |
| Intermolecular HB | ||
| O1...O | 2.651 | 2.676 |
| O1-H1 | 0.991 | 0.991 |
| H1...O | 1.685 | 1.686 |
| ∠O1H1O [°] | 163.6 | 178.1 |
| N1...O | 2.996 | 3.005 |
| N1-H3 | 1.018 | 1.018 |
| H3...O | 2.067 | 2.052 |
| ∠N1H3O [°] | 150.5 | 154.9 |
| Three water molecules (HIP-3) | ||
| Intramolecular HB | ||
| ND...N1 | 2.644 | 2.739 |
| ND-HD | 1.055 | 1.030 |
| HD...N1 | 1.780 | 1.968 |
| ∠NDHDN [°] | 136.2 | 129.3 |
| Intermolecular HB | ||
| O1...O | 2.661 | 2.677 |
| O1-H1 | 0.989 | 0.991 |
| H1...O | 1.703 | 1.686 |
| ∠O1H1O [°] | 161.7 | 178.2 |
| N1...O | 3.225 | 3.081 |
| N1-H3 | 1.017 | 1.018 |
| H3...O | 2.300 | 2.091 |
| ∠N1H3O [°] | 150.6 | 163.6 |
| Four water molecules (HIP-4) | ||
| Intramolecular HB | ||
| ND...N1 [A] | 2.699 | 2.750 |
| ND-HD | 1.039 | 1.028 |
| HD...N1 | 1.881 | 1.990 |
| ∠NDHDN1 [°] | 133.0 | 128.5 |
| Intermolecular HB | ||
| O1...O | 2.664 | 2.676 |
| O1-H1 | 0.989 | 0.991 |
| H1...O | 1.707 | 1.686 |
| ∠O1H1O [°] | 161.6 | 177.3 |
| N1...O | 3.281 | 3.092 |
| N1-H3 | 1.016 | 1.018 |
| H3...O | 2.291 | 2.074 |
| ∠N1H3O [°] | 164.6 | 180.0 |
| NE...O | 2.781 | 2.773 |
| NE-HE | 1.029 | 1.033 |
| HE...O | 1.753 | 1.741 |
| ∠NEHEO [°] | 178.2 | 177.8 |
| Five water molecules (HIP-5) | ||
| No intramolecular HB | ||
| Intermolecular HB | ||
| O1...O | 2.664 | 2.655 |
| O1-H1 | 0.990 | 0.995 |
| H1...O | 1.687 | 1.661 |
| ∠O1H1O [°] | 168.1 | 177.7 |
| N1...O | 2.751 | 2.764 |
| O-H | 0.988 | 0.991 |
| H...N1 | 1.789 | 1.785 |
| ∠N1HO [°] | 163.5 | 169.0 |
| NE...O | 2.785 | 2.769 |
| NE-HE | 1.029 | 1.033 |
| HE...O | 1.756 | 1.736 |
| ∠NEHEO [°] | 178.1 | 178.1 |
| ND...O | 2.676 | 2.742 |
| ND-HD | 1.051 | 1.039 |
| HD...O | 1.640 | 1.721 |
| ∠NDHDO [°] | 167.6 | 166.7 |
| O...O2 | 2.869 | 2.816 |
| O-H | 0.970 | 0.971 |
| H...O2 | 1.910 | 1.851 |
| ∠OHO2 [°] | 169.5 | 171.7 |
| Six water molecules (HIP-6) | ||
| No intramolecular HB | ||
| Intermolecular HB | ||
| O1...O | 2.696 | 2.643 |
| O1-H1 | 0.986 | 0.998 |
| H1...O | 1.741 | 1.659 |
| ∠O1H1O [°] | 161.7 | 168.1 |
| N1...O | 2.754 | 2.780 |
| O-H | 0.990 | 0.990 |
| H...N1 | 1.789 | 1.801 |
| ∠N1HO [°] | 163.8 | 169.3 |
| NE...O | 2.811 | 2.773 |
| NE-HE | 1.023 | 1.022 |
| HE...O | 1.884 | 1.910 |
| ∠NEHEO [°] | 149.2 | 140.1 |
| ND...O | 2.673 | 2.743 |
| ND-HD | 1.050 | 1.037 |
| HD...O | 1.652 | 1.746 |
| ∠NDHDO [°] | 162.6 | 160.0 |
| O...O2 | 2.837 | 2.819 |
| O-H | 0.971 | 0.971 |
| H...O2 | 1.880 | 1.855 |
| ∠OHO2 [°] | 168.0 | 171.7 |
| HIP-(H2O)n | Simulation Environment | ND | HD | NE | HE | O2 | Mol. Charge |
|---|---|---|---|---|---|---|---|
| HIP-1 n = 1 | Gas phase | −1.246 | 0.537 | −1.206 | 0.476 | −1.179 | 0.958 |
| PCM H2O | −1.240 | 0.528 | −1.209 | 0.496 | −1.203 | 0.952 | |
| Δ(PCM-gas) | 0.006 | −0.009 | −0.003 | 0.020 | −0.024 | ||
| HIP-2 n = 2 | Gas phase | −1.251 | 0.544 | −1.207 | 0.473 | −1.182 | 0.946 |
| PCM H2O | −1.244 | 0.531 | −1.209 | 0.495 | −1.205 | 0.939 | |
| Δ(PCM-gas) | 0.007 | −0.013 | −0.002 | 0.022 | −0.023 | ||
| HIP-3 n = 3 | Gas phase | −1.252 | 0.549 | −1.207 | 0.472 | −1.184 | 0.953 |
| PCM H2O | −1.244 | 0.531 | −1.209 | 0.495 | −1.206 | 0.933 | |
| Δ(PCM-gas) | 0.008 | −0.018 | −0.002 | 0.024 | −0.022 | ||
| HIP-4 n = 4 | Gas phase | −1.254 | 0.533 | −1.249 | 0.539 | −1.190 | 0.916 |
| PCM H2O | −1.246 | 0.526 | −1.249 | 0.545 | −1.206 | 0.893 | |
| Δ(PCM-gas) | 0.008 | −0.008 | 0.000 | 0.006 | −0.016 | ||
| HIP-5 n = 5 | Gas phase | −1.253 | 0.553 | −1.256 | 0.538 | −1.207 | 0.927 |
| PCM H2O | −1.252 | 0.541 | −1.255 | 0.547 | −1.212 | 0.943 | |
| Δ(PCM-gas) | 0.001 | −0.012 | 0.001 | 0.008 | −0.004 | ||
| HIP-6 n = 6 | Gas phase | −1.251 | 0.552 | −1.246 | 0.517 | −1.201 | 0.938 |
| PCM H2O | −1.248 | 0.540 | −1.239 | 0.529 | −1.211 | 0.956 | |
| Δ(PCM-gas) | 0.003 | −0.012 | 0.007 | 0.012 | −0.010 |
| HIE-(H2O)n | Simulation Environment | ND | NE | HE | O2 | Mol. Charge |
|---|---|---|---|---|---|---|
| HIE-1 n = 1 | Gas phase | −1.150 | −1.227 | 0.424 | −1.183 | 0.047 |
| PCM H2O | −1.185 | −1.231 | 0.459 | −1.207 | 0.059 | |
| Δ(PCM-gas) | −0.036 | −0.004 | 0.035 | −0.024 | ||
| HIE-2 n = 2 | Gas phase | −1.156 | −1.228 | 0.424 | −1.174 | 0.058 |
| PCM H2O | −1.188 | −1.232 | 0.458 | −1.198 | 0.067 | |
| Δ(PCM-gas) | −0.031 | −0.003 | 0.035 | −0.024 | ||
| HIE-3 n = 3 | Gas phase | −1.164 | −1.263 | 0.486 | −1.176 | 0.041 |
| PCM H2O | −1.194 | −1.269 | 0.508 | −1.199 | 0.039 | |
| Δ(PCM-gas) | −0.030 | −0.005 | 0.023 | −0.023 | ||
| HIE-4 n = 4 | Gas phase | −1.188 | −1.263 | 0.492 | −1.176 | 0.062 |
| PCM H2O | −1.203 | −1.267 | 0.513 | −1.199 | 0.089 | |
| Δ(PCM-gas) | −0.015 | −0.003 | 0.021 | −0.023 | ||
| HIE-5 n = 5 | Gas phase | −1.180 | −1.255 | 0.493 | −1.222 | 0.051 |
| PCM H2O | −1.198 | −1.259 | 0.505 | −1.225 | 0.058 | |
| Δ(PCM-gas) | −0.017 | −0.004 | 0.012 | −0.003 | ||
| HIE-6 n = 6 | Gas phase | −1.176 | −1.253 | 0.471 | −1.232 | 0.021 |
| PCM H2O | −1.196 | −1.256 | 0.498 | −1.229 | 0.052 | |
| Δ(PCM-gas) | −0.020 | −0.003 | 0.027 | 0.003 |
| HID-(H2O)n | Simulation Environment | ND | HD | NE | O2 | Mol. Charge |
|---|---|---|---|---|---|---|
| HID-1 n = 1 | Gas phase | −1.246 | 0.459 | −1.134 | −1.194 | −0.025 |
| PCM H2O | −1.252 | 0.480 | −1.192 | −1.208 | −0.043 | |
| Δ(PCM-gas) | −0.006 | 0.021 | −0.058 | −0.014 | ||
| HID-2 n = 2 | Gas phase | −1.266 | 0.478 | −1.133 | −1.193 | −0.014 |
| PCM H2O | −1.254 | 0.482 | −1.193 | −1.210 | −0.051 | |
| Δ(PCM-gas) | 0.012 | 0.004 | −0.060 | −0.017 | ||
| HID-3 n = 3 | Gas phase | −1.254 | 0.475 | −1.165 | −1.195 | 0.015 |
| PCM H2O | −1.250 | 0.488 | −1.210 | −1.207 | 0.017 | |
| Δ(PCM-gas) | 0.004 | 0.013 | −0.045 | −0.012 | ||
| HID-4 n = 4 | Gas phase | −1.260 | 0.495 | −1.166 | −1.193 | 0.032 |
| PCM H2O | −1.258 | 0.495 | −1.192 | −1.209 | 0.010 | |
| Δ(PCM-gas) | 0.002 | 0.001 | −0.026 | −0.016 | ||
| HID-5 n = 5 | Gas phase | −1.260 | 0.497 | −1.168 | −1.209 | 0.035 |
| PCM H2O | −1.251 | 0.488 | −1.194 | −1.212 | 0.023 | |
| Δ(PCM-gas) | 0.008 | −0.009 | −0.026 | −0.002 | ||
| HID-6 n = 6 | Gas phase | −1.262 | 0.507 | −1.174 | −1.206 | 0.022 |
| PCM H2O | −1.249 | 0.490 | −1.192 | −1.213 | 0.028 | |
| Δ(PCM-gas) | 0.013 | −0.017 | −0.018 | −0.008 |
| MP2/aug-cc-pVDZ | MP2/aug-cc-pVTZ | SAPT2/aug-cc-pVDZ | SAPT2/aug-cc-pVTZ | |||||
|---|---|---|---|---|---|---|---|---|
| Eint | per 1 H2O | Eint | per 1 H2O | Eint | per 1 H2O | Eint | per 1 H2O | |
| HIP-1 | −12.50 | −12.50 | −13.49 | −13.49 | −12.57 | −12.57 | −13.79 | −13.79 |
| HIP-2 | −21.03 | −10.52 | −22.47 | −11.23 | −21.02 | −10.51 | −22.76 | −11.38 |
| HIP-3 | −23.87 | −7.96 | −25.43 | −8.48 | −24.11 | −8.04 | −25.99 | −8.66 |
| HIP-4 | −37.99 | −9.50 | −40.23 | −10.06 | −37.88 | −9.47 | −40.54 | −10.13 |
| HIP-5 | −57.64 | −11.53 | −61.90 | −12.38 | −57.87 | −11.57 | ||
| HIP-6 | −58.74 | −9.79 | −62.72 | −10.45 | −58.78 | −9.80 | ||
| HIE-1 | −8.02 | −8.02 | −8.74 | −8.74 | −7.98 | −7.98 | −8.89 | −8.89 |
| HIE-2 | −12.84 | −6.42 | −14.16 | −7.08 | −12.78 | −6.39 | −14.44 | −7.22 |
| HIE-3 | −18.87 | −6.29 | −20.58 | −6.86 | −18.70 | −6.23 | −20.85 | −6.95 |
| HIE-4 | −23.38 | −5.85 | −25.63 | −6.41 | −23.43 | −5.86 | −26.26 | −6.57 |
| HIE-5 | −38.97 | −7.79 | −42.25 | −8.45 | −39.14 | −7.83 | ||
| HIE-6 | −40.74 | −6.79 | −44.39 | −7.40 | −41.33 | −6.89 | ||
| HID-1 | −10.09 | −10.09 | −11.19 | −11.19 | −10.31 | −10.31 | −11.70 | −11.70 |
| HID-2 | −14.93 | −7.46 | −16.56 | −8.28 | −15.16 | −7.58 | −17.15 | −8.58 |
| HID-3 | −21.62 | −7.21 | −23.64 | −7.88 | −21.87 | −7.29 | −24.41 | −8.14 |
| HID-4 | −29.12 | −7.28 | −31.80 | −7.95 | −29.05 | −7.26 | −32.29 | −8.07 |
| HID-5 | −36.72 | −7.34 | −40.10 | −8.02 | −36.53 | −7.31 | ||
| HID-6 | −43.71 | −7.28 | −47.70 | −7.95 | −43.87 | −7.31 | ||
| ELST | EXCH | IND | DISP | Total SAPT2 | |
|---|---|---|---|---|---|
| HIP-1 | −22.19 | 24.75 | −10.39 | −4.74 | −12.57 |
| HIP-2 | −34.12 | 33.95 | −12.95 | −7.89 | −21.02 |
| HIP-3 | −37.44 | 36.04 | −13.01 | −9.70 | −24.11 |
| HIP-4 | -55.27 | 50.20 | −20.16 | −12.66 | −37.88 |
| HIP−5 | −100.08 | 110.86 | −44.28 | −24.38 | −57.87 |
| HIP-6 | −96.58 | 102.61 | −40.69 | −24.12 | −58.78 |
| HIE-1 | −17.19 | 20.65 | −6.48 | −4.96 | −7.98 |
| HIE-2 | −26.68 | 32.45 | −9.27 | −9.28 | −12.78 |
| HIE-3 | −36.61 | 42.29 | −12.79 | −11.59 | −18.70 |
| HIE-4 | −46.72 | 55.55 | −16.98 | −15.28 | −23.43 |
| HIE-5 | −72.60 | 84.21 | −30.90 | −19.85 | −39.14 |
| HIE-6 | −78.58 | 94.95 | −34.93 | −22.78 | −41.33 |
| HID-1 | −23.95 | 29.14 | −9.87 | −5.63 | −10.31 |
| HID-2 | −31.15 | 37.99 | −11.90 | −10.10 | −15.16 |
| HID-3 | −45.01 | 52.18 | −17.34 | −11.69 | −21.87 |
| HID-4 | −52.01 | 61.66 | −21.06 | −17.65 | −29.05 |
| HID-5 | −65.31 | 76.76 | −26.04 | −21.94 | −36.53 |
| HID-6 | −74.77 | 88.46 | −30.94 | −26.63 | −43.87 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kizior, B.; Panek, J.J.; Jezierska, A. Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches. Symmetry 2020, 12, 1153. https://doi.org/10.3390/sym12071153
Kizior B, Panek JJ, Jezierska A. Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches. Symmetry. 2020; 12(7):1153. https://doi.org/10.3390/sym12071153
Chicago/Turabian StyleKizior, Beata, Jarosław J. Panek, and Aneta Jezierska. 2020. "Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches" Symmetry 12, no. 7: 1153. https://doi.org/10.3390/sym12071153
APA StyleKizior, B., Panek, J. J., & Jezierska, A. (2020). Microsolvation of Histidine—A Theoretical Study of Intermolecular Interactions Based on AIM and SAPT Approaches. Symmetry, 12(7), 1153. https://doi.org/10.3390/sym12071153