Occurrence of the d-Proline Chemotype in Enzyme Inhibitors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
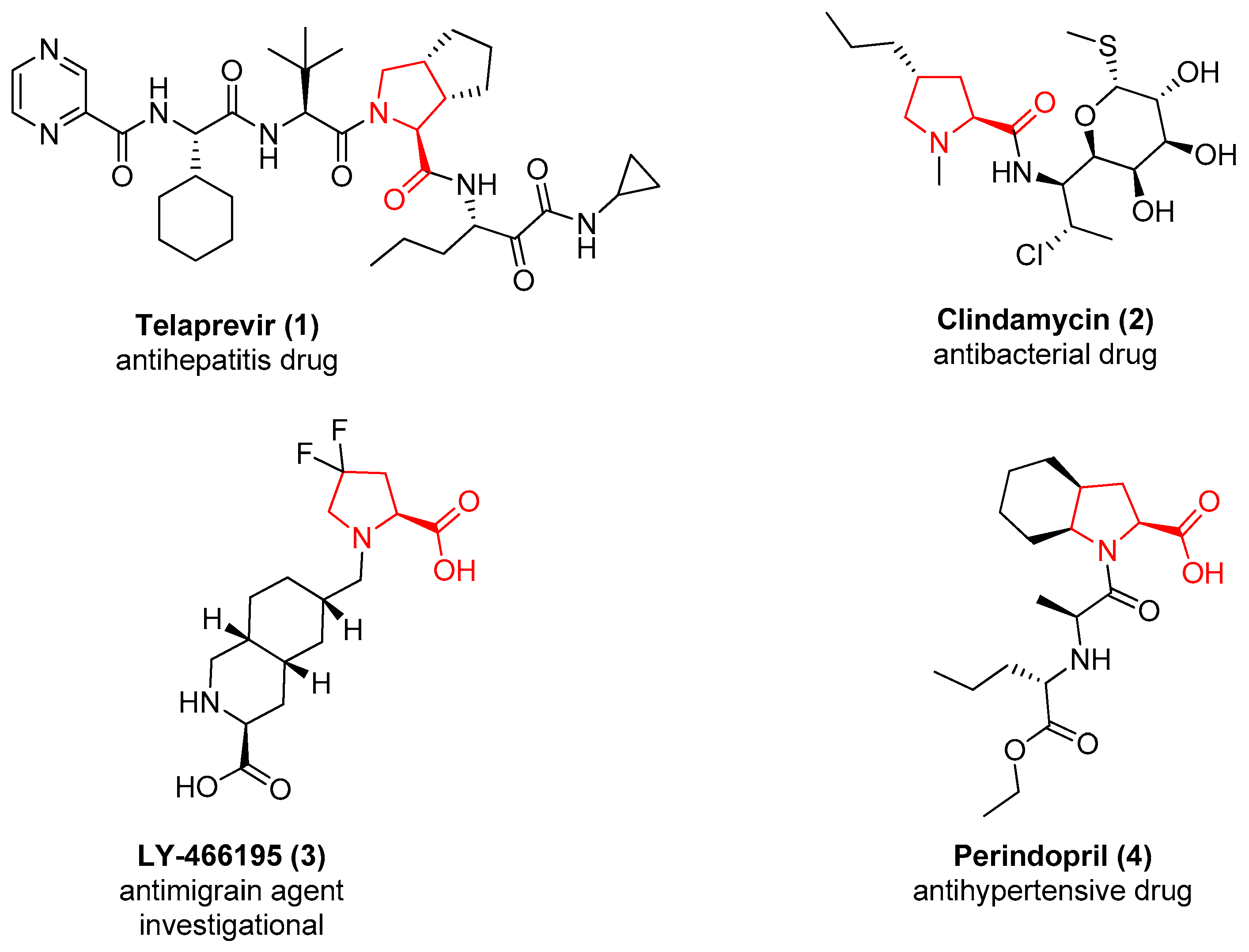
1. Introduction
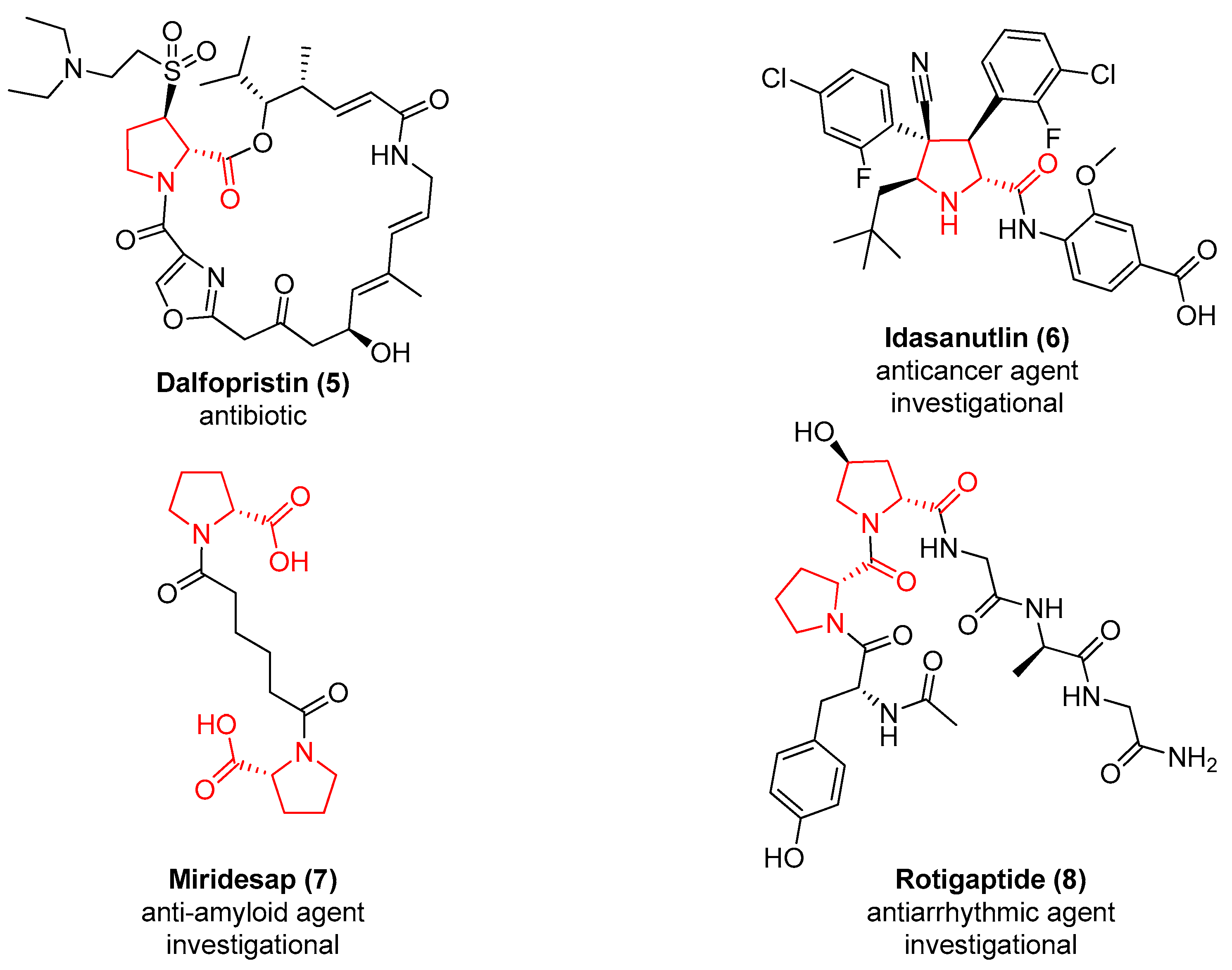
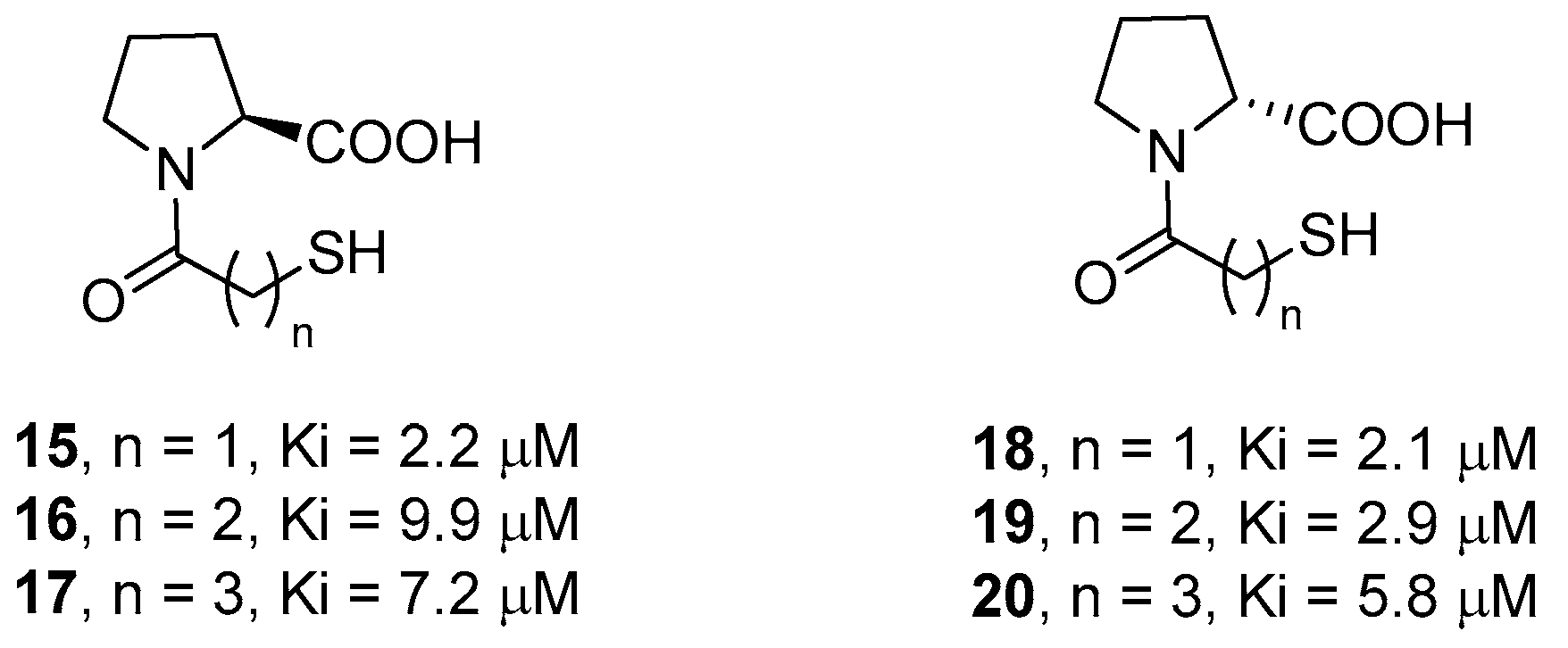
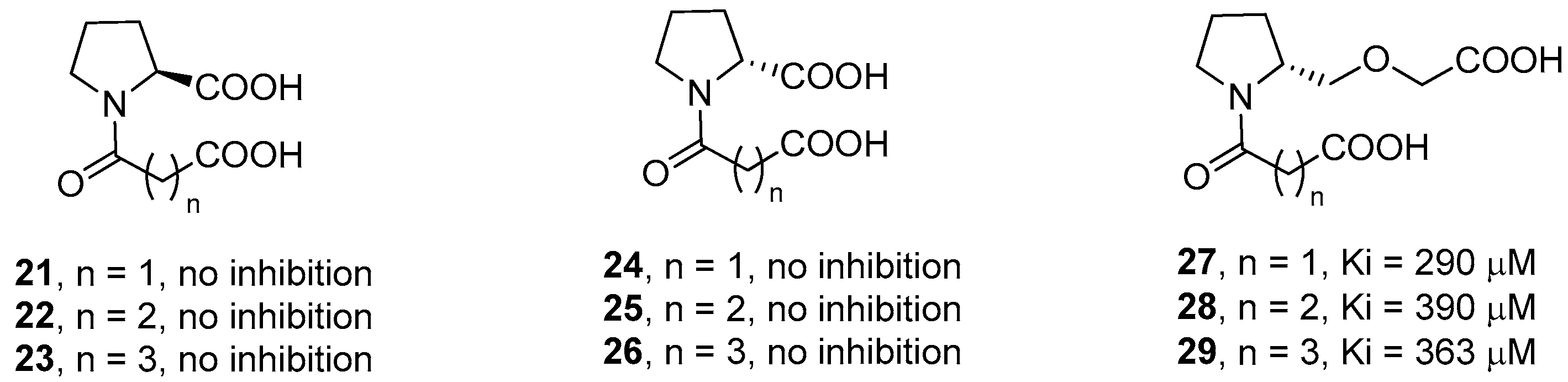
2. d-Proline Derivatives as Metallo-β-Lactamase (MBL) Inhibitors
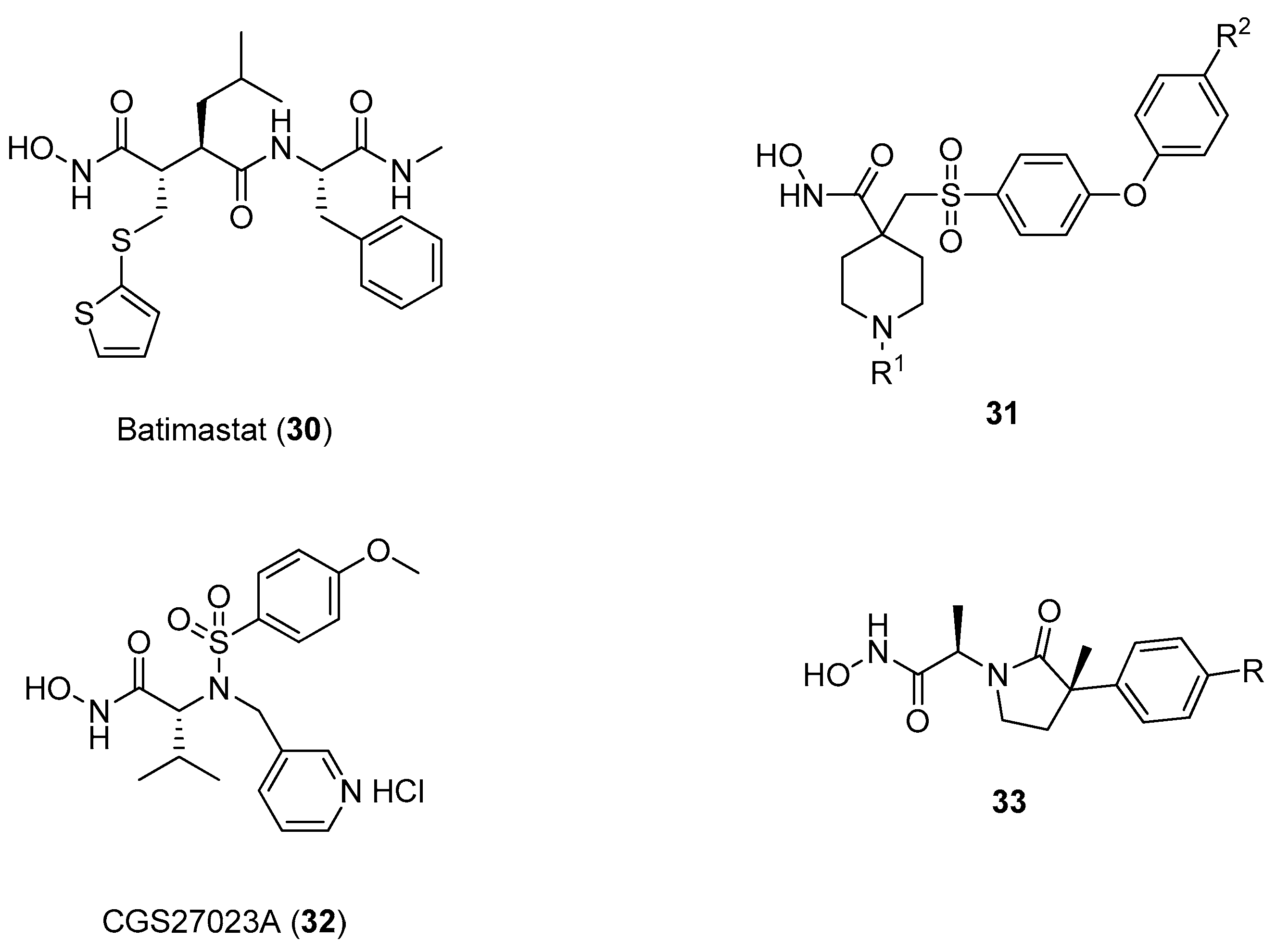
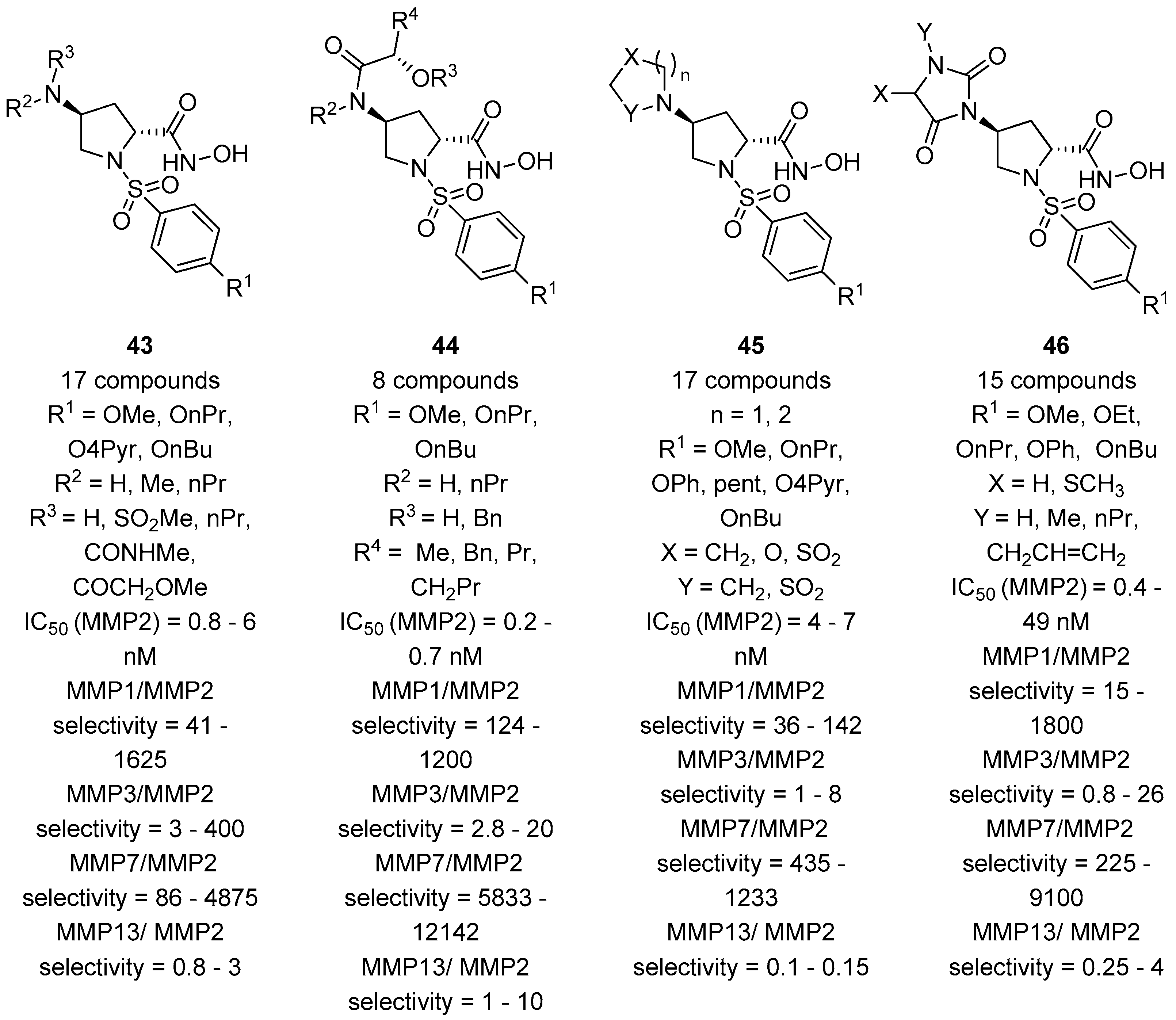
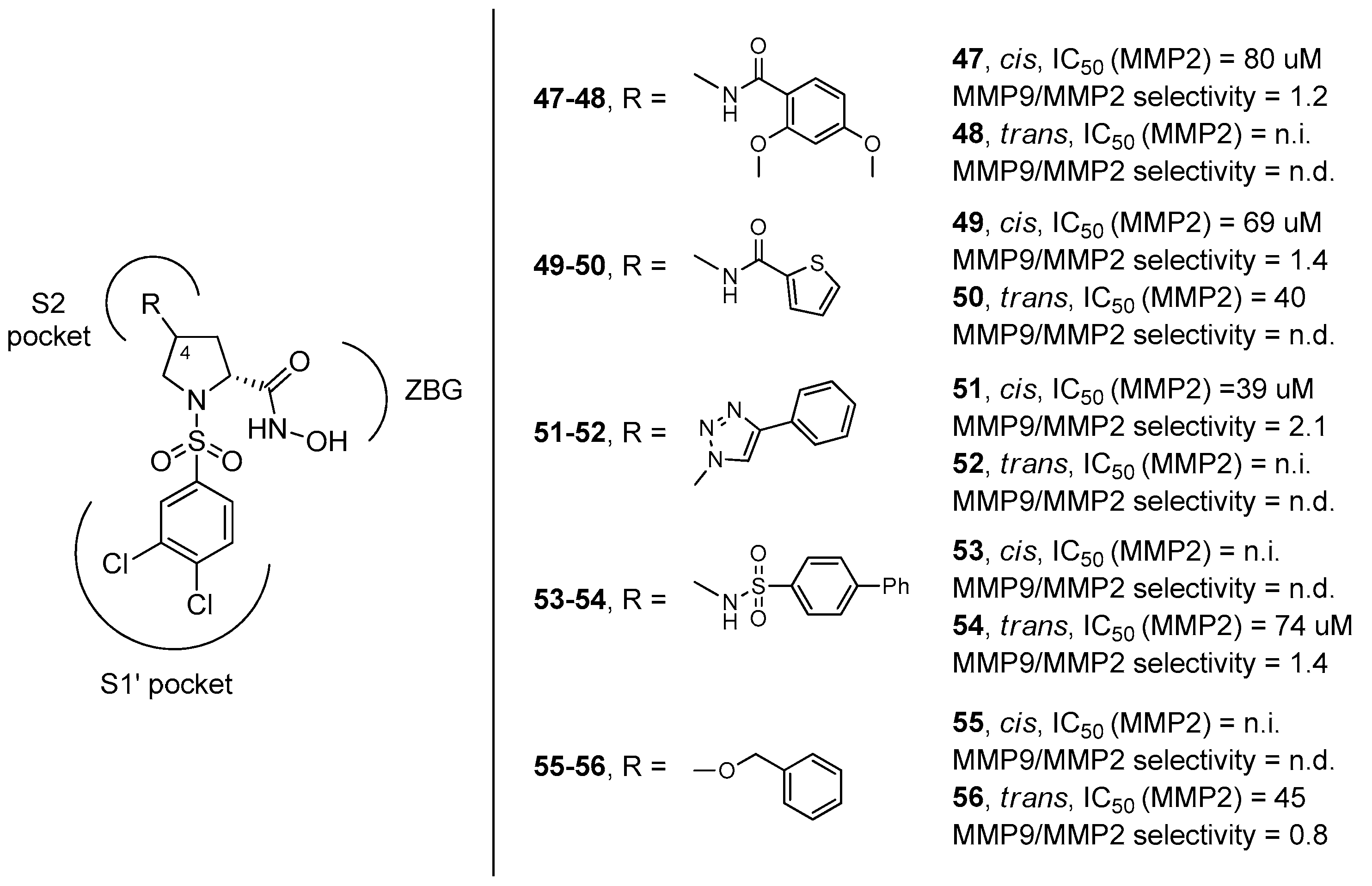
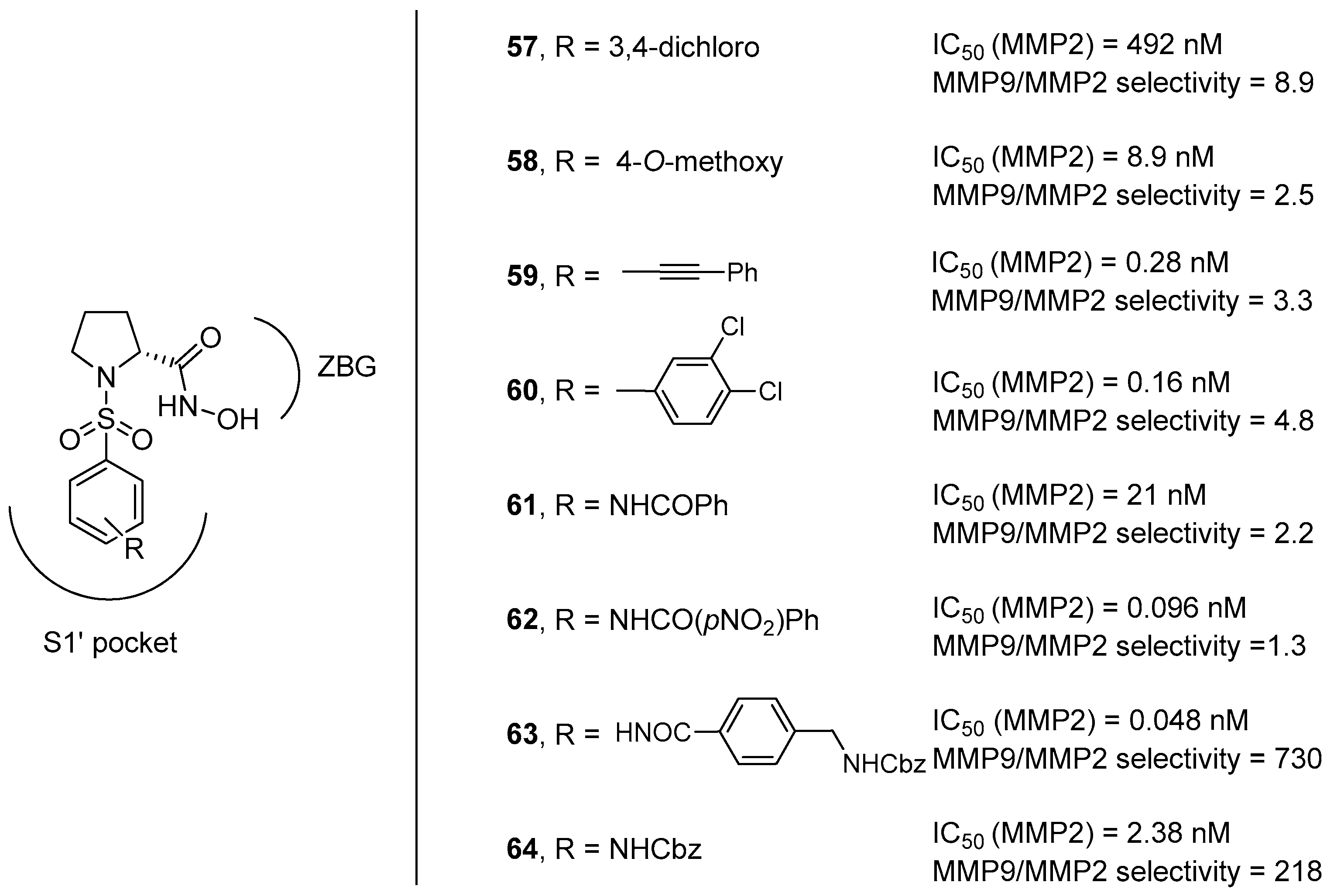
3. d-Proline Derivatives as Matrix Metalloproteases (MMP) Inhibitors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Genchi, G. An overview on D-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Veiga, P.; Piquet, S.; Maisons, A.; Furlan, S.; Courtin, P.; Chapot-Chartier, M.P.; Kulakauskas, S. Identification of an essential gene responsible for d-Asp incorporation in the Lactococcus lactis peptidoglycan crossbridge. Mol. Microbiol. 2006, 62, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Hamase, K.; Morikawa, A.; Zaitsu, K. D-amino acids in mammals and their diagnostic value. J. Chromatogr. B Anal. Technol. Biomed Life Sci. 2002, 781, 73–91. [Google Scholar] [CrossRef]
- Hamase, K. Sensitive two-dimensional determination of small amounts of D-amino acids in mammals and the study on their functions. Chem. Pharm. Bull. 2007, 55, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.A.; Berger, R.; Klomp, L.W.; de Koning, T.J. D-amino acids in the central nervous system in health and disease. Mol. Genet. Metab. 2005, 85, 168–180. [Google Scholar] [CrossRef]
- Bauer, D.; Hamacher, K.; Bröer, S.; Pauleit, D.; Palm, C.; Zilles, K.; Coenen, H.; Langen, K.J. Preferred stereoselective brain uptake of d-serine-a modulator of glutamatergic neurotransmission. Nucl. Med. Biol. 2005, 32, 793–797. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, A. D-aspartic acid: An endogenous amino acid with an important neuroendocrine role. Brain Res. Rev. 2007, 53, 215–234. [Google Scholar] [CrossRef]
- Friedman, M.; Levin, C.E. Nutritional and medicinal aspects of D-amino acids. Amino Acids 2012, 42, 1553–1582. [Google Scholar] [CrossRef]
- Sladojevich, F.; Guarna, A.; Trabocchi, A. Evaluation of stereochemically dense morpholine-based scaffolds as proline surrogates in β-turn peptides. Org. Biomol. Chem. 2010, 7, 916–924. [Google Scholar] [CrossRef]
- Sladojevich, F.; Trabocchi, A.; Guarna, A. Configurationally-driven folding of model tetrapeptides containing l- or d-morpholine-3-carboxylic acids as β-turn nucleators. Chirality 2009, 21, 584–594. [Google Scholar]
- Cini, N.; Trabocchi, A.; Menchi, G.; Bottoncetti, A.; Raspanti, S.; Pupi, A.; Guarna, A. Morpholine-based RGD-cyclopentapeptides as αvβ3/αvβ5 integrin ligands: Role of configuration towards receptor binding affinity. Biorg. Med. Chem. 2009, 17, 1542–1549. [Google Scholar] [CrossRef]
- Yoshimura, T.; Esak, N. Amino acid racemases: Functions and mechanisms. J. Biosci. Bioeng. 2003, 96, 103–109. [Google Scholar] [CrossRef]
- Ollivaux, C.; Soyez, D.; Toullec, J.Y. Biogenesis of d-amino acid containing peptides/proteins: Where, when and how? J. Pept. Sci. 2014, 20, 595–612. [Google Scholar] [CrossRef]
- Mauger, A.B. Naturally Occurring Proline Analogues. J. Nat. Prod. 1996, 59, 1205–1211. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. The n → π * interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef]
- Sanchez-Navarro, M.; Teixido, M.; Giralt, E. Jumping hurdles: Peptides able to overcome biological barriers. Acc. Chem. Res. 2017, 50, 1847–1854. [Google Scholar] [CrossRef]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef]
- Kraus, A. Proline and lysine residues provide modulatory switches in amyloid formation: Insights from prion protein. Prion 2016, 10, 57–62. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Khare, P.; Nagar, H.K.; Raghuwanshi, N.; Srivastava, R. Hydroxyproline: A potential biochemical marker and its role in the pathogenesis of different diseases. Curr. Protein. Pept. Sci. 2016, 17, 596–602. [Google Scholar] [CrossRef]
- Dienstag, J.L. Antiviral Drugs against Hepatitis Viruses. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Philadelphia, PA, USA, 2015; Volume 1, pp. 563–575. [Google Scholar]
- Smieja, M. Current indications for the use of clindamycin: A critical review. Can. J. Infect. Dis. 1998, 9, 22–28. [Google Scholar] [CrossRef]
- Weiss, B.; Alt, A.; Ogden, A.M.; Gates, M.; Dieckman, D.K.; Clemens-Smith, A.; Ho, K.H.; Jarvie, K.; Rizkalla, G.; Wright, R.A.; et al. Pharmacological characterization of the competitive GLUK5 receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J. Pharm. Exp. 2006, 318, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Butlin, M.; Avolio, A.P. Persistent effect of early, brief angiotensin-converting enzyme inhibition on segmental pressure dependency of aortic stiffness in spontaneously hypertensive rats. J. Hypertens. 2012, 30, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Drug Bank. Available online: https://www.drugbank.ca (accessed on 20 February 2019).
- Allington, D.R.; Rivey, M.P. Quinupristin/dalfopristin: A therapeutic review. Clin. Ther. 2001, 23, 24–44. [Google Scholar] [CrossRef]
- Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers 2018, 10, 170. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Tennent, G.A.; Hutchinson, W.L.; Gallimore, J.R.; Lachmann, H.J.; Goodman, H.J.B.; Offer, M.; Millar, D.J.; Petrie, A.; Hawkins, P.N.; et al. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br. J. Haematol. 2010, 148, 760–767. [Google Scholar] [CrossRef]
- Pedersen, C.M.; Venkatasubramanian, S.; Vase, H.; Hyldebrandt, J.A.; Contractor, H.; Schmidt, M.R.; Bøtker, H.E.; Cruden, N.L.; Newby, D.E.; Kharbanda, R.K.; et al. Rotigaptide protects the myocardium and arterial vasculature from ischaemia reperfusion injury. Br. J. Clin. Pharmacol. 2016, 81, 1037–1045. [Google Scholar] [CrossRef]
- König, S.; Marco, H.; Gäde, G. The hypertrehalosemic neuropeptides of cicadas are structural isomers—evidence by ion mobility mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 6415–6420. [Google Scholar] [CrossRef] [PubMed]
- Reina-San-Martin, B.; Degrave, W.; Rougeot, C.; Cosson, A.; Chamond, N.; Cordeiro-Da-Silva, A.; Arala-Chaves, M.; Coutinho, A.; Minoprio, P. A B-cell mitogen from a pathogenic trypanosome is a eukaryotic proline racemase. Nat. Med. 2000, 6, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Coatnoan, N.; Berneman, A.; Chamond, N.; Minoprio, P. Proline racemases: Insights into Trypanosoma cruzi peptides containing D-proline. Mem. Inst. Oswaldo Cruz 2009, 104, 295–300. [Google Scholar] [CrossRef]
- Chamond, N.; Goytia, M.; Coatnoan, N.; Barale, J.C.; Cosson, A.; Degrave, W.M.; Minoprio, P. Trypanosoma cruzi proline racemases are involved in parasite differentiation and infectivity. Mol. Microbiol. 2005, 58, 46–60. [Google Scholar] [CrossRef]
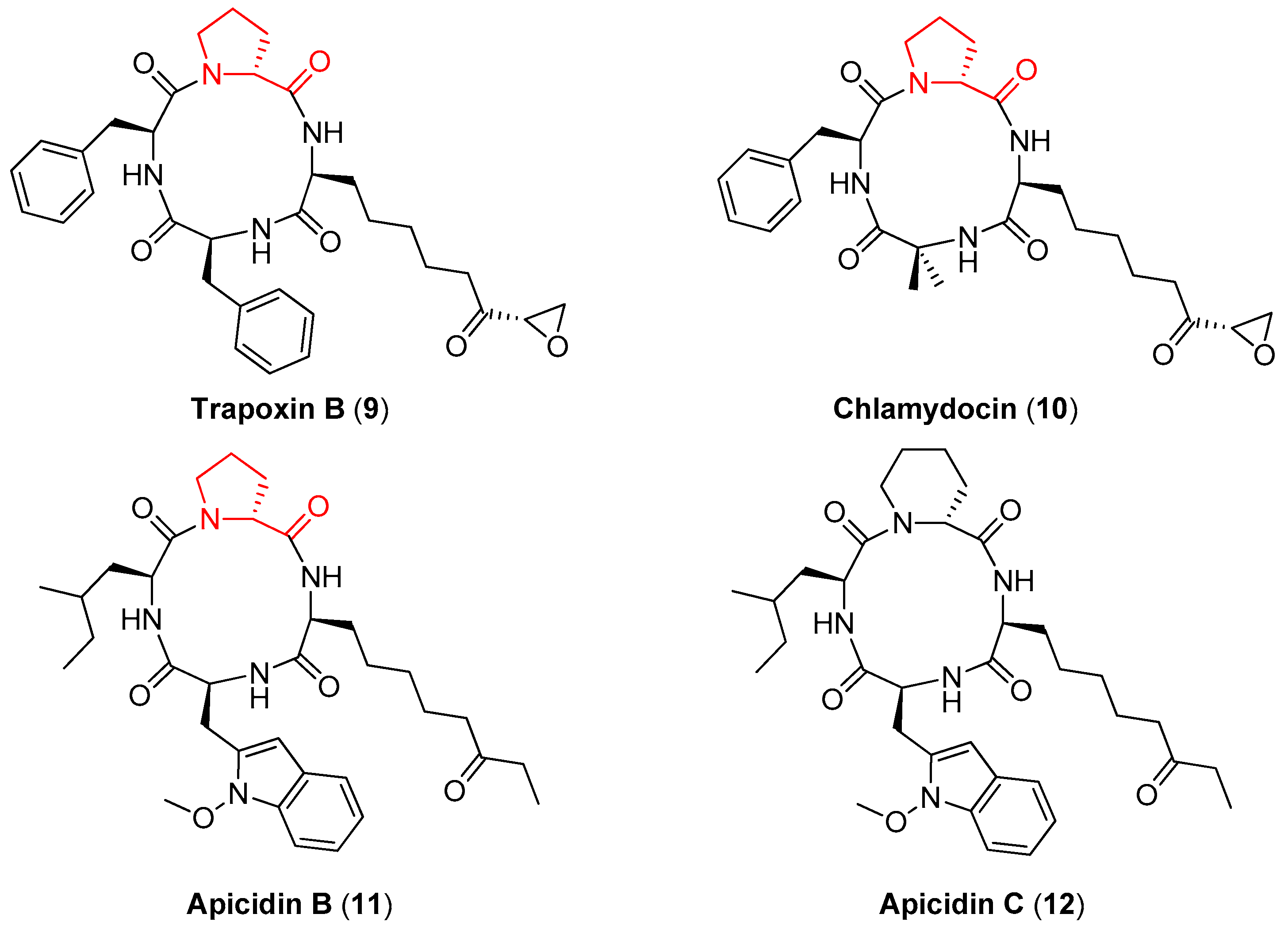
- Islam, M.S.; Bhuiyan, M.P.I.; Islam, M.N.; Nsiama, T.K.; Oishi, N.; Kato, T.; Nishino, N.; Ito, A.; Yoshida, M. Evaluation of functional groups on amino acids in cyclic tetrapeptides in histone deacetylase inhibition. Amino Acids 2012, 42, 2103–2110. [Google Scholar] [CrossRef]
- Yoshida, M.; Matsuayama, A.; Komatsu, Y.; Nishino, N. From discovery to the coming generation of histone deacetylase inhibitors. Curr. Med. Chem. 2003, 10, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Zink, D.L.; Liesch, J.M.; Dombrowski, A.W.; Darkin-Rattray, S.J.; Schmatz, D.M.; Goet, M.A. Structure, Histone Deacetylase, and Antiprotozoal Activities of Apicidins B and C, Congeners of Apicidin with Proline and Valine Substitutions. Org. Lett. 2001, 3, 2815–2818. [Google Scholar] [CrossRef] [PubMed]
- Trabocchi, A.; Guarna, A. Cyclic α-Amino Acids as Proline Mimetics. In Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs, 1st ed.; Trabocchi, A., Guarna, A., Eds.; John Wiley and Sons, Ltd: Chichester, UK, 2014; pp. 165–190. [Google Scholar]
- Luppi, G.; Lanci, D.; Trigari, V.; Garavelli, M.; Garelli, A.; Tomasini, A. Development and conformational analysis of a pseudoproline-containing turn mimic. J. Org. Chem. 2003, 68, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Schutkowski, M.; Drakenberg, T. cis–trans-Amide isomerism of the 3,4-dehydroproline residue, the ‘unpuckered’ proline. J. Am. Chem. Soc. 1997, 119, 8403–8408. [Google Scholar] [CrossRef]
- Beausoleil, E.; L’Archevêque, B.; Bélec, L.; Atfani, M.; Lubell, W.D. 5-tert-Butylproline. J. Org. Chem. 1996, 61, 9447–9454. [Google Scholar] [CrossRef]
- Cornaglia, G.; Giamarellou, H.; Rossolini, G.M. Metallo-β-lactamases: A last frontier for -lactams? Lancet Infect. Dis. 2011, 11, 381–393. [Google Scholar] [CrossRef]
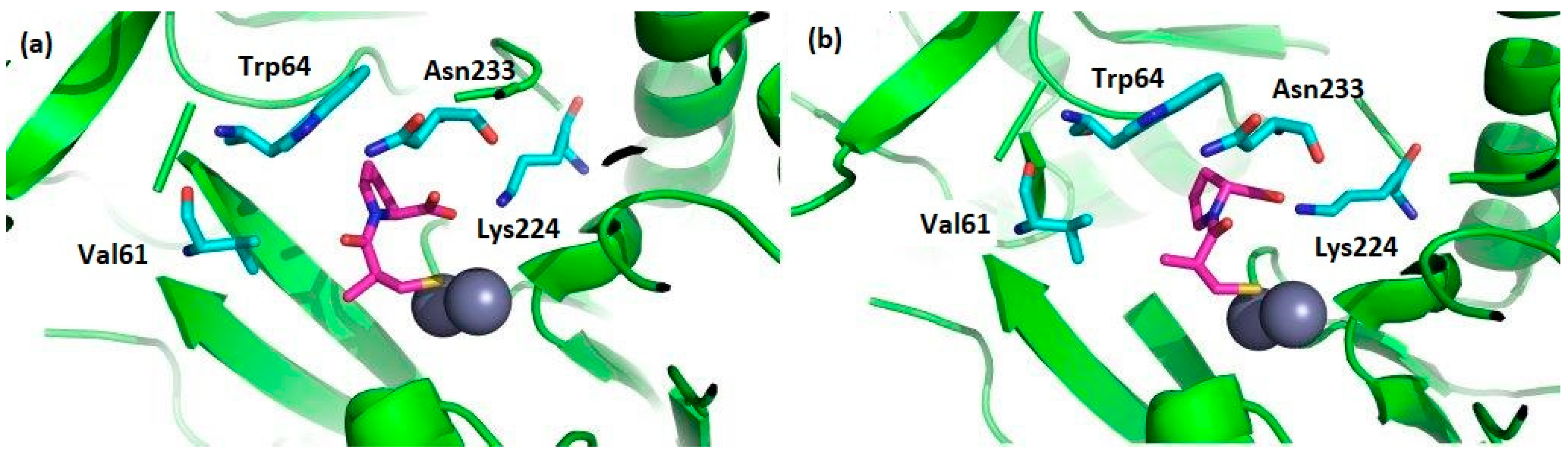
- Heinz, U.; Bauer, R.; Wommer, S.; Meyer-Klaucke, W.; Papamichaels, C.; Bateson, J.; Adolph, H.W. Coordination Geometries of Metal Ions in d- or l-Captopril-inhibited Metallo-β-lactamases. J. Biol. Chem. 2003, 278, 20659–20666. [Google Scholar] [CrossRef]
- Karsisiotis, A.I.; Damblon, C.F.; Roberts, G.C.K. A Variety of Roles for versatile zinc in Metallo-β-lactamases. Metallomics 2014, 6, 1181–1197. [Google Scholar] [CrossRef]
- Brem, J.; van Berkel, S.S.; Zollman, D.; Lee, S.Y.; Gileadi, O.; McHugh, P.J.; Walsh, T.R.; McDonough, M.A.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase Inhibition by Captopril Stereoisomers. Antimicrob. Agents Chemother. 2015, 60, 142–150. [Google Scholar] [CrossRef]
- Faridoon; Hussein, W.M.; Vella, P.; Nazar, U.I.; Ollis, D.L.; Schenk, G.; McGeary, R.P. 3-Mercapto-1,2,4-triazoles and N-acylated Thiosemicarbazides as Metallo-β-lactamase Inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 380–386. [Google Scholar] [CrossRef]
- Page, M.I.; Badarau, A. The Mechanism of Catalysis by Metallo-β-Lactamases. Bioinorg. Chem. Appl. 2008, 1–14. [Google Scholar] [CrossRef]
- Bebrone, C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharm. 2007, 74, 1686–1701. [Google Scholar] [CrossRef]
- Goto, M.; Takahashi, T.; Yamashita, F.; Koreeda, A.; Mori, H.; Ohta, M.; Arakawa, Y. Inhibition of the Metallo-β-lactamase Produced from Serratia marcescens by Thiol Compounds. Biol. Pharm. Bull. 1997, 20, 1136–1140. [Google Scholar] [CrossRef]
- Yusof, Y.; Tan, D.T.C.; Arjomandi, O.K.; Schenk, G.; McGeary, R.P. Captopril Analogues as Metallo-β-lactamase Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1589–1593. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Vihinen, P.; Ala-aho, R.; Kahari, V.M. Matrix metalloproteinases as therapeutic targets in cancer. Curr. Cancer Drug Targets 2005, 5, 203–220. [Google Scholar] [CrossRef]
- Fingleton, B. Targeting monocyte chemoattractant protein-1 signalling in disease. Expert Opin. Tar. 2003, 7, 385–390. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix Metalloproteinase Inhibitors and Cancer—Trials and Tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Li, N.G.; Shi, Z.H.; Tang, Y.P.; Duan, J.A. Selective matrix metalloproteinase inhibitors for cancer. Curr. Med. Chem. 2009, 16, 3805–3827. [Google Scholar] [CrossRef]
- Beattie, G.J.; Young, H.A.; Smyth, J.F. Phase I study of intra-peritoneal metalloproteinase inhibitor BB-94 in patients with malignant ascites. Ann. Oncol. 1994, 5, 72a. [Google Scholar]
- Rothenberg, M.L.; Nelson, A.R.; Hande, K.R. New Drugs on the Horizon: Matrix Metalloproteinase Inhibitors. Oncologist 1998, 3, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Levitt, N.C.; Eskens, F.A.; O’Byrne, K.J.; Propper, D.J.; Denis, L.J.; Owen, S.J.; Choi, L.; Foekens, J.A.; Wilner, S.; Wood, J.M.; et al. A phase one pharmacokinetic study of CGS27023A, a matrix metalloproteinase inhibitor. Proc. Am. Soc. Clin. Oncol. 1998, 17, 213a. [Google Scholar]
- Jenkins, C.L.; McCloskey, A.I.; Guzei, I.A.; Eberhardt, E.S.; Raines, R.T. O-acylation of hydroxyproline residues: Effect on peptide-bond isomerization and collagen stability. Biopolymers 2005, 80, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vitagliano, L.; Berisio, R.; Mazzarella, L.; Zagari, A. Structural bases of collagen stabilization induced by proline hydroxylation. Biopolymers 2001, 58, 459–464. [Google Scholar] [CrossRef]
- Hanessian, S.; MacKay, D.B.; Moitessier, N. Design and Synthesis of Matrix Metalloproteinase Inhibitors Guided by Molecular Modeling. Picking the S1 Pocket Using Conformationally Constrained Inhibitors. J. Med. Chem. 2001, 44, 3074–3082. [Google Scholar] [CrossRef]
- Natchus, M.G.; Bookland, R.G.; De, B.; Almstead, N.G.; Pikul, S.; Janusz, M.J.; Heitmeyer, S.A.; Hookfin, E.B.; Hsieh, L.C.; Dowty, M.E.; et al. Development of new hydroxamate matrix metalloproteinase inhibitors derived from functionalized 4-aminoprolines. J. Med. Chem. 2000, 43, 4948–4963. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; Calugi, C.; Ruzzolini, J.; Menchi, G.; Calorini, L.; Guarna, A.; Trabocchi, A. A study of a D-proline peptidomimetic inhibitor of melanoma and endothelial cell invasion through activity towards MMP-2 and MMP-9. Med. Chem. Comm. 2015, 6, 277–282. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Jiang, Y.; Feng, J.; Li, X.; Zhang, Y.; Xu, W. Design, synthesis and preliminary evaluation of α-sulfonyl γ-(glycinyl-amino)proline peptidomimetics as matrix metalloproteinase inhibitors. Bioorg. Med. Chem. 2014, 22, 3055–3064. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Innocenti, R.; Di Francescantonio, T.; Menchi, G.; Bianchini, F.; Contini, A.; Trabocchi, A. Identification of highly potent and selective MMP2 inhibitors addressing the S1’ subsite with d-proline-based compounds. Bioorg. Med. Chem. 2019, 27, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, D.V.; Wagner, S.; Riemann, B.; Hermann, S.; Schmidt, F.; Becker-Pauly, C.; Rose-John, S.; Schäfers, M.; Holl, R. Novel Potent Proline-Based Metalloproteinase Inhibitors: Design, (Radio)Synthesis, and First in Vivo Evaluation as Radiotracers for Positron Emission Tomography. J. Med. Chem. 2016, 59, 9541–9559. [Google Scholar] [CrossRef]
- Paumier, J.M.; Thinakaran, G. Matrix metalloproteinase 13, a new target for therapy in Alzheimer’s disease. Genes Dis. 2019, 16, 1–2. [Google Scholar] [CrossRef] [PubMed]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenci, E.; Trabocchi, A. Occurrence of the d-Proline Chemotype in Enzyme Inhibitors. Symmetry 2019, 11, 558. https://doi.org/10.3390/sym11040558
Lenci E, Trabocchi A. Occurrence of the d-Proline Chemotype in Enzyme Inhibitors. Symmetry. 2019; 11(4):558. https://doi.org/10.3390/sym11040558
Chicago/Turabian StyleLenci, Elena, and Andrea Trabocchi. 2019. "Occurrence of the d-Proline Chemotype in Enzyme Inhibitors" Symmetry 11, no. 4: 558. https://doi.org/10.3390/sym11040558
APA StyleLenci, E., & Trabocchi, A. (2019). Occurrence of the d-Proline Chemotype in Enzyme Inhibitors. Symmetry, 11(4), 558. https://doi.org/10.3390/sym11040558