Abstract
Atmospheric aqueous-phase reactions have been recognized as an important source of secondary organic aerosols (SOAs). However, the unclear reaction kinetics and mechanics hinder the in-depth understanding of the SOA sources and formation processes. This study selected ten different substituted phenolic compounds (termed as PhCs) emitted from biomass burning as precursors, to investigate the kinetics using OH oxidation reactions under simulated sunlight. The factors influencing reaction rates were examined, and the contribution of reactive oxygen species (ROS) was evaluated through quenching and kinetic analysis experiments. The results showed that the pseudo-first-order rate constants (kobs) for the OH oxidation of phenolic compounds ranged from 1.03 × 10−4 to 7.85 × 10−4 s−1 under simulated sunlight irradiation with an initial H2O2 concentration of 3 mM. Precursors with electron-donating groups (-OH, -OCH3, -CH3, etc.) exhibited higher electrophilic radical reactivity due to the enhanced electron density of the benzene ring, leading to higher reaction rates than those with electron-withdrawing groups (-NO2, -CHO, -COOH). At pH 2, the second-order reaction rate (kPhCs, OH) was lower than at pH 5. However, the kobs did not show dependence on pH. The presence of O2 facilitated substituted phenols’ photodecay. Inorganic salts and transition metal ions exhibited varying effects on reaction rates. Specifically, NO3− and Cu2+ promoted kPhCs, OH, Cl− significantly enhanced the reaction at pH 2, while SO42− inhibited the reaction. The kPhCs, OH were determined to be in the range of 109~1010 L mol−1 s−1 via the bimolecular rate method, and a modest relationship with their oxidation potential was found. Additionally, multiple substituents can suppress the reactivity of phenolic compounds toward •OH based on Hammett plots. Quenching experiments revealed that •OH played a dominant role in phenolic compound degradation (exceeding 65%). Electron paramagnetic resonance confirmed the generation of singlet oxygen (1O2) in the system, and probe-based quantification further explored the concentrations of •OH and 1O2 in the system. Based on reaction rates and concentrations, the atmospheric aqueous-phase lifetimes of phenolic compounds were estimated, providing valuable insights for expanding atmospheric kinetic databases and understanding the chemical transformation and persistence of phenolic substances in the atmosphere.
1. Introduction
Organic aerosol (OA) is a critical pollutant in the atmosphere. Besides primary emissions from biomass and fossil fuel combustion, gas- or aqueous-phase chemical reactions and aging processes are regarded as important sources of secondary organic aerosol (SOA) [1,2,3,4,5]. SOA accounts for 20–90% of the total OA and substantially impacts global climate, atmospheric visibility, and human health. Approximately 70% of the earth is covered or influenced by clouds. Aqueous-phase environments such as cloud droplets, fog droplets, and aerosol water serve as reaction media for soluble gases and aerosols, hence, aqueous-phase SOA (aqSOA) contributes significantly to atmospheric SOA. The formation of aqSOA predominantly results from aqueous-phase oxidation by O3, hydroxyl radicals (•OH), nitrate radicals (•NO3), and triplet states of organic matter (3C*) [6,7,8]. Among them, •OH is regarded as the dominant atmospheric oxidant owing to its strong oxidizing capacity, high electrophilicity, and low selectivity. Its reaction mechanism typically involves either hydrogen abstraction or electron transfer, depending on the precursor’s reduction potential and molecular structure.
Biomass burning (BB) emits significant amounts of both gaseous and particulate pollutants. Among them, phenolic compounds such as phenol, methoxyphenols, aromatic ketones, aromatic aldehydes, and other polysubstituted phenols are key products, with an estimated global annual emission of 4.7 Tg [9,10]. These compounds exhibit a wide range of solubilities, reflected in Henry’s law constants ranging from 40 to 3.1 × 108 mol·L−1 [11], resulting in varied partitioning behaviors into cloud and fog droplets. The aqueous-phase photochemical oxidation of phenolic compounds represents an important pathway for SOA formation. For phenols with large Henry’s law constants, the aqueous-phase oxidation in cloud droplets can be more significant than gas-phase oxidation.
In recent years, increasing attention has been given to the atmospheric aqueous-phase photochemical reactions of BB-derived phenols. Understanding the rates of both photochemical reactions and dark reactions in the atmospheric aqueous phase is crucial for accurately assessing SOA formation and its environmental impacts [10]. Nonetheless, modeling studies frequently overestimate •OH levels, resulting in overestimated SOA yield and underestimated atmospheric lifetimes when compared to field observations [12]. Arciva et al. [13] determined second-order rate constants for ·OH reactions with several highly substituted phenols and found that these rates were lower at pH 2 than at pH 5. Smith’s group [14] also investigated the aqueous-phase oxidation kinetics of highly substituted phenols under varying pH conditions. Kroflič et al. reported temperature-dependent (278.15–318.15 K) second-order rate constants for the reaction of •OH with four substituted phenols and conducted Hammett plots for mono- and di-substituted phenols [15]. Błaziak et al. [16] explored the •OH kinetics with carboxylic acids from α-pinene oxidation in aqueous solutions across various temperatures (278−318 K) and pH levels (2–8). Hoffmann et al. focused on the oxidation of substituted aromatic hydrocarbons in the tropospheric aqueous phase [17]. Hems et al. investigated the aqueous-phase photo-oxidation kinetics, reaction mechanism and optical properties of nitrophenols [18]. Tomaz et al. [19] observed that aqueous oxidation of BB smoke particles leads to the breakdown of high-molecular-weight compounds and the formation of low-molecular-weight organic acids, altering aerosol optical properties. Go et al. [10] demonstrated that mixtures of aromatic carbonyl compounds (e.g., vanillin, acetosyringone, syringaldehyde) produce more oxidized and less light-absorbing aqSOA than individual precursors. In recent years, our research group has also explored the aqSOA yields and optical properties of products formed from aqueous-phase oxidation of phenolic organics [20,21]. However, the role of reactive oxygen species (ROS) and the underlying reaction mechanisms remain unclear.
Previous studies have employed various methods to determine rate constants, including laser flash photolysis, bimolecular rate method, and theoretical calculations. However, these approaches often yield inconsistent results [22]. Additionally, most existing studies use quenching experiments to infer the relative contributions of ROS, typically providing only qualitative assessments (e.g., dominant vs. minor roles) rather than quantitative estimates of their absolute contributions to precursor degradation [23]. Moreover, when examining the degradation of phenolic precursors, the focus has primarily been on the influence of substituent structures on reaction rates, with limited attention to potential photosensitized hydrogen-abstraction self-reactions. Therefore, comprehensive investigations into reactive oxygen species (ROS) contributions and degradation pathways across structurally diverse phenols are urgently needed to deepen mechanistic understanding.
In this work, ten phenolic compounds with different substituents, commonly emitted from BB, were selected to explore the pseudo-first-order rate constants of their aqueous-phase oxidation by •OH. The effects of various influencing factors, including pH, saturated gases, oxidant concentration, and inorganic salts were systematically examined. In addition, a quantitative structure–activity relationship (QSAR) analysis was conducted to assess the correlation between reaction rate and precursor structure. By integrating ROS quantification, quenching experiments, and kinetic calculations, we quantified the absolute contributions of individual ROS species to phenolic degradation. These findings not only enhance our understanding of phenolic compounds’ stability and atmospheric lifetimes in the aqueous phase but also offer more accurate kinetic parameters for atmospheric models, thereby improving the reliability of SOA source apportionment.
2. Materials and Methods
2.1. Chemicals and Reagents
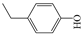
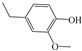
4-ethylphenol (4EP, ≥99%), 4-ethylguaiacol (4EG, ≥99%), 4-methyl-2,6-dimethoxyphenol (DMP, ≥99%), 2,6-dimethylphenol (SYR, ≥99%), catechol (CAT, ≥99%), 3-methylcatechol (3MC, ≥99%), 4-nitrocatechol (4NC, ≥99%), syringic acid (SA, ≥99%), 4-hydroxy-3-methoxypropiophenone (GA, ≥99%), acetosyringone (AS, ≥99%), phenol (PhOH, ≥99%), guaiacol (GUA, ≥99%), furfuryl alcohol (FFA, ≥99%), disodium hydrogen phosphate (99%), sodium dihydrogen phosphate (99%), benzoic acid (BA, ≥99%), sodium chloride (≥99%), sodium nitrate (≥99%), hydrogen peroxide (H2O2, 29–32%), and ammonium sulfate (GR grade) were from Alfa Aesar. Copper(II) sulfate pentahydrate (≥99%), potassium hydrogen phthalate (≥99%), sodium hydroxide (≥99%), and methanol (GR grade) were from Acros Organics. Sulfuric acid (GR grade) was from Sinopharm Group. Tert-Butyl alcohol (TBA, AR) was from Sigma-Aldrich. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO, 97%) and 2,2,6,6-tetramethylpiperidine (TEMP, 98%) were from Shanghai Anpel Laboratory Technologies Inc., Shanghai, China. Acetonitrile (HPLC grade) and methanol (MeOH, HPLC grade) were purchased from CNW Technologies GmbH, Düsseldorf, Germany. All solutions were prepared using ultrapure water from a Milli-Q purification system (Millipore, Burlington, MA, USA, resistivity ≥ 18.2 MΩ cm).
2.2. Photochemical Reaction Experiments
In this study, a photochemical reactor was used to investigate aqueous-phase photodegradation of ten phenolic compounds under simulated sunlight. The irradiation experiments were conducted using a Rayonet photoreactor (RPR-200) equipped with 16 lamps (2 bulbs at 300 nm, 7 bulbs at 350 nm, and 7 bulbs at 420 nm) to simulate solar irradiation. The spectral irradiation of the lamps was recorded using an Ocean Optics Maya2000 Pro spectrometer (see Figure S1 in the Supplementary Information), as previously described in detail [21]. The light intensity at the surface of the reaction solution was approximately 2400 μW/cm2 in the 290–320 nm (UVB) range, measured with a radiometer (Photoelectric Instrument Factory of Everfine Corporation, Hangzhou, China), and is comparable to natural sunlight levels.
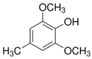
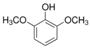
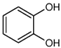
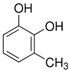
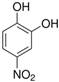
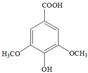
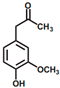
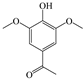
The ten substituted phenolic compounds were categorized into two main groups: (1) compounds containing electron-donating groups, such as 4-ethylphenol, 4-ethylguaiacol, 4-methyl-2,6-dimethoxyphenol, 2,6-dimethylphenol, catechol, 3-methylcatechol, and (2) compounds containing electron-withdrawing groups, such as 4-nitrocatechol, syringic acid, 4-hydroxy-3-methoxypropiophenone, acetosyringone. The molecular structures of these compounds are presented in Table 1. For the photodegradation experiments, approximately 20 mL of each reaction solution was transferred into a quartz reaction tube. A cooling fan positioned at the bottom of the photochemical reactor maintained the solution temperature at 298 K and ensured homogeneous mixing. Meanwhile, a dark control experiment was performed using an identical quartz tube wrapped in aluminum foil and filled with the same volume of reaction solution.

Table 1.
Chemical formula and second-order rate constants (kPhCs, OH) for the reaction of ten substituted phenolic compounds with •OH.
Unless otherwise specified, the initial concentrations of these phenolic compounds were 300 μmol·L−1, with 3 mmol·L−1 of H2O2 added as •OH source, and the pH was 5.0. For each set of experiments, dark reactions and direct photolysis were conducted as control groups. The results indicated that the loss of ten phenolic compounds under dark conditions was negligible. All phenolic compounds underwent direct photolysis under simulated sunlight (see Figure S2). To investigate the effects of different reaction parameters on the pseudo-first-order rate constant (kobs, s−1) of aqueous-phase oxidation, the oxidant concentration, solution pH, and the type of saturated gases (air, N2 and O2) bubbled into the solution were varied. All experiments were conducted in triplicate. Blank experiments consisting of direct photodegradation (PhCs with sunlight without H2O2) were run in parallel.
2.3. Analysis Method and Kinetics
2.3.1. Determination of Phenols Concentration
During irradiation, aliquots were periodically taken from both the illuminated and dark reaction tubes to measure the concentrations of substituted phenolic compounds with high-performance liquid chromatography (HPLC) equipped with a C18 column (150 mm × 4.6 mm, 5.0 μm). Detailed HPLC operating conditions are presented in the Supplementary Information. Photodegradation rates of substituted phenolic compounds were calculated based on their concentration at different irradiation times.
2.3.2. Kinetic Experiments
At designated time intervals, 2 mL of the reaction solution was withdrawn periodically using a pipette and filtered through a 0.45 μm polytetrafluoroethylene (PTFE) membrane filter into an HPLC sample vial. The concentration of phenolic compounds (PhCs) at each time point (t) was determined by HPLC analysis. The kobs was calculated according to Equation (1).
where [PhCs]0 is the initial concentration of PhCs, and [PhCs]t is the concentration at time t, expressed in μmol·L−1.
In this study, the bimolecular rate method suggested by Li et al. [24] was employed to determine the second-order rate constant of PhCs:
Here, PhCs refers to the substituted phenolic compounds, ref denotes the reference compound, and the square brackets indicate concentrations. kdir, PhCs represents the pseudo-first-order rate constants for direct photolysis of PhCs. Phenol was selected as the reference compound with known second-order rate constants with •OH of (1.8 ± 0.3) × 109 L mol−1 s−1 at pH 2 and (1.5 ± 0.5) × 1010 L mol−1 s−1 at pH 5 [15]. In our experiments, phenol was added at an initial concentration of 300 μM. After determining the concentrations of phenolic compounds and reference compounds at various reaction times, we plotted
The slope of the resulting fitted line was then multiplied by the known rate constant kref, OH to calculate the second-order rate constant kPhCs, OH for the reaction between •OH and each phenolic compound.
2.4. Determination of •OH and 1O2 Concentrations
In this work, two substituted phenolic compounds, 3MC and 4NC, were selected as representative compounds to evaluate the concentrations of ROS in the reaction system. DMPO and TEMP were chosen as the trapping agents for •OH and 1O2, respectively. A micro electron paramagnetic resonance (ESR) spectrometer was used for the qualitative detection of ROS.
To quantify •OH under simulated sunlight irradiation, BA was introduced into the system at concentrations of 5, 10, 15, 20, 30, and 50 μmol·L−1 as a probe molecule. A linear regression was performed between the reciprocal of the pseudo-first-order rate constant and the corresponding BA concentrations. The •OH concentration was then calculated by dividing the reciprocal of the y-intercept by the known second-order rate constants of BA with •OH (kBA, OH = 5.9 × 109 L mol−1 s−1, at 298 K) [25].
Under simulated sunlight irradiation, FFA (5.0 μmol·L−1) was used as a chemical probe in both H2O and D2O solution to determine the concentration of singlet oxygen (1O2) generated. The 1O2 concentration was calculated according to Equation (3) [26].
where kFFA, 1O2 is the second-order rate constant for the reaction between 1O2 with FFA (1.2 × 108 L mol−1 s−1, at 298 K); kEXP, D2O and kEXP, H2O are the first-order rate constants for FFA degradation under simulated sunlight irradiation in D2O and H2O, respectively (this work).χH2O and χD2O represent the mole fractions of H2O and D2O in the dilution experiments, respectively; D is the dilution factor applied in the experiments (typically 0.22); k’H2O and k’D2O are the first-order rate constants for 1O2* decay in 100% H2O and 100% D2O, respectively (2.2 × 105 s−1and 1.6 × 104 s−1, respectively).
To evaluate the contribution of different ROS to the degradation of phenolic compounds, quenching experiments were conducted using TBA and FFA as quenchers for •OH and 1O2, respectively. Considering that the quenchers and the target organic pollutant coexist in the reaction system, the quencher concentrations should be high enough to ensure complete scavenging of the generated reactive species.
2.5. Determination of Oxidation Potentials via Cyclic Voltammetry
Cyclic voltammetry (CV) was performed using a BASi EC Epsilon potentiostat with a three-electrode configuration: a 3 mm glassy carbon working electrode, an Ag/AgCl reference electrode (3 mol·L−1 KCl), and a 0.5 mm platinum wire counter electrode (BASi). Prior to each measurement, the working electrode was polished with 0.05 μm alumina slurry. Cyclic voltammograms were recorded between −0.8 V and 1.85 V with a scan speed of 0.05 V/s. Measurements were performed in deoxygenated pH 2 (0.2 mol·L−1 NaC1 + 0.01 mol·L−1 HCl) and pH 5 (0.1 mol·L−1 potassium hydrogen phthalate + 0.04 mol·L−1 NaOH) buffer solutions with 0.3 mmol·L−1 of phenolic compounds. Since all these compounds exhibited irreversible voltammetric behavior, the anodic peak potentials (E0*) were determined directly from the first scan of the cyclic voltammograms (scan rate of 0.05 V/s). The measured potentials were converted from the Ag/AgCl reference electrode scale to standard hydrogen electrode (SHE) by adding 0.209 V. The Gibbs free energy(ΔG°ox) under standard state (101.325 kPa, 298 K) can be calculated from the standard redox potential according to Equation (4) [3]:
where n is the number of electrons (1 here), F is Faraday’s constant (96,485.3365 C mol−1), and Eox is one-electron oxidation potential.
ΔG°ox = −nFEox
3. Result and Discussions
3.1. Rate Constants Between Phenolic Compounds with •OH
To determine the rate constants for the reaction of substituent phenolic compounds with •OH, we first investigated the direct photolysis efficiency of phenolic compounds in the absence of H2O2 (Figure S2). The results are consistent with previous findings [9], which reported 36–94% photolytic loss for six carbonyl-substituted phenols. As shown, the ten phenolic compounds exhibited varying degrees of direct photolysis, depending on their substituents. Among them, 4NC, CAT, SYR, and SA showed low photodegradation efficiencies (<10% loss within 40 min), which might be linked to the electron-withdrawing groups forming conjugated systems with the benzene ring, steric hindrance effect, and weak light absorption capabilities. In contrast, DMP and AS displayed higher direct photolysis efficiencies (>30% loss within 40 min). These compounds contain methyl (-CH3) and methoxy groups (-OCH3), which enhance light absorption and thus accelerate photolysis. Additionally, the relatively rapid photodegradation observed for AS and GA may be attributed to the possible formation of small amounts of H2O2 during the aqueous-phase photochemical reactions of aromatic carbonyl compounds. This accelerated photolysis may result from: (1) the formation of organic triplet excited states under simulated sunlight; (2) hydrogen abstraction from the parent compounds; or (3) subsequent reactions with O2 to generate HO2 radicals, which ultimately form H2O2 [27,28,29]. These processes may contribute to the faster direct photolysis observed in these compounds compared to other phenolic compounds.
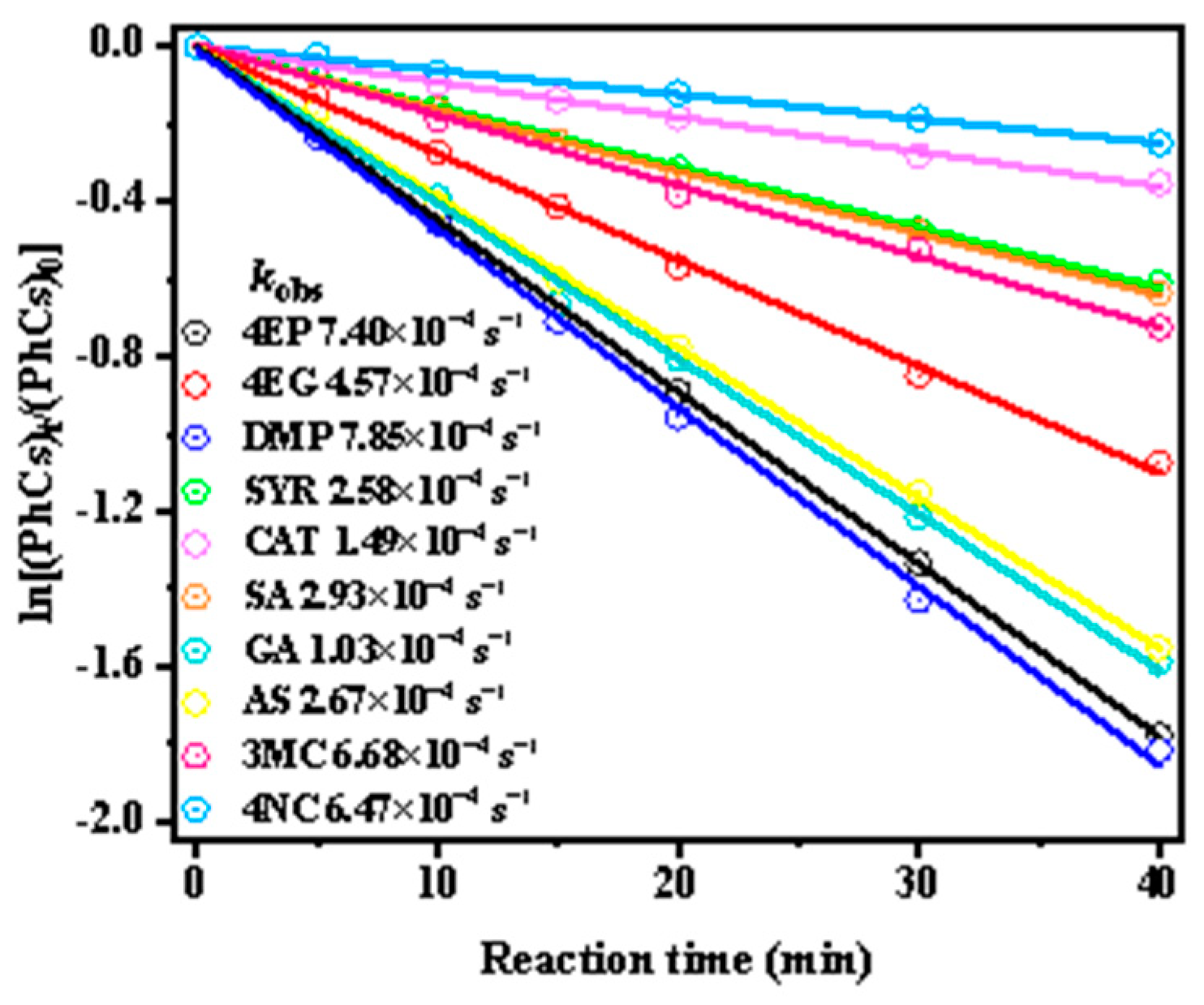
Next, we investigated the kinetics of phenolic compounds’ photooxidation by •OH. Figure 1 shows the decay kinetic curves and first-order rate constants for ten substituted phenolic compounds at pH 5, while the kobs were determined by fitting the data using Equation (1). The half-life (t1/2) time for 50% degradation of phenolic compounds during •OH oxidation is 150, 75, 48, 45, 40, 25, 20, 18, 16, 15, and 14 min, respectively, with photodegradation rates ordered as: 4NC < CAT < SYR < SA < 3MC < 4EG < AS < GA < 4EP < DMP. Among them, DMP degrades the fastest, while 4NC degrades the slowest. Theoretically, organic compounds undergo OH-induced photodegradation primarily via hydrogen abstraction and hydroxylation addition pathways. The likelihood of electron transfer is largely governed by the reduction potential and molecular structure of the precursor. Therefore, variations in the substituents on the aromatic ring significantly influence the reactivity of phenolic compounds with •OH, thereby affecting their degradation rates.
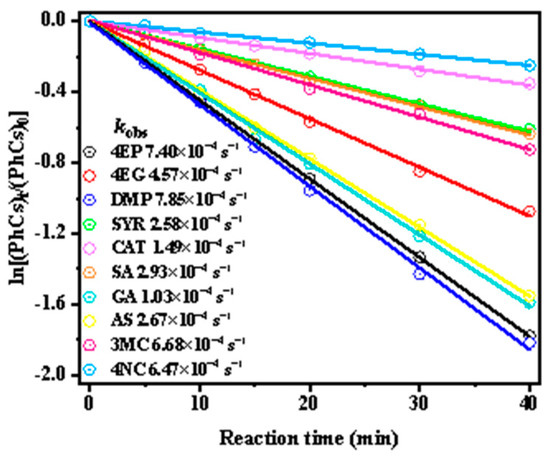
Figure 1.
Decay kinetic curves and first-order rate constants for ten substituted phenolic compounds at pH 5.
For instance, 3MC, which contains one additional -CH3 group compared to CAT, exhibited a higher rate constant (6.68 × 10−4 s−1 vs.1.49 × 10−4 s−1). Similarly, DMP, which has one more -CH3 group than SYR, showed enhanced reactivity. The susceptibility to electrophilic attack also varies with different substituents on the phenol ring, following the order: -OH > -OCH3 > -CH3. Electron-donating groups such as -CH3 and -OCH3, as found in 4EP, 4EG and DMP, increase the electron density on the aromatic ring and provide extra reactive sites, thereby favoring electrophilic substitution and resulting in higher kobs. In particular, the lone pair electrons on the O atom of the -OCH3 group enhance the conjugation of the aromatic ring and reactivity with •OH. Similar patterns were reported in the literature [11], where the electron-donating substituents (e.g., -OH, -CH3, -OCH3, -C2H5) facilitated electrophilic substitution and elevated the first-order rate constant. Conversely, electron-withdrawing groups such as -NO2, -COOH), and-CHO reduce the electron density of the aromatic ring, thus lowering its reactivity. Likewise, SA and 4NC, which possess -COOH and-NO2 groups, respectively, showed significantly decreased degradation rates due to their deactivating effects. Notably, kobs increased with the number of electron-donating substituents, as observed in the trend: kCAT (1.49 × 10−4 s−1) < kSYR (2.58 × 10−4 s−1) < kDMP (7.85 × 10−4 s−1).
3.2. Factors Affecting the First-Order Rate Constant
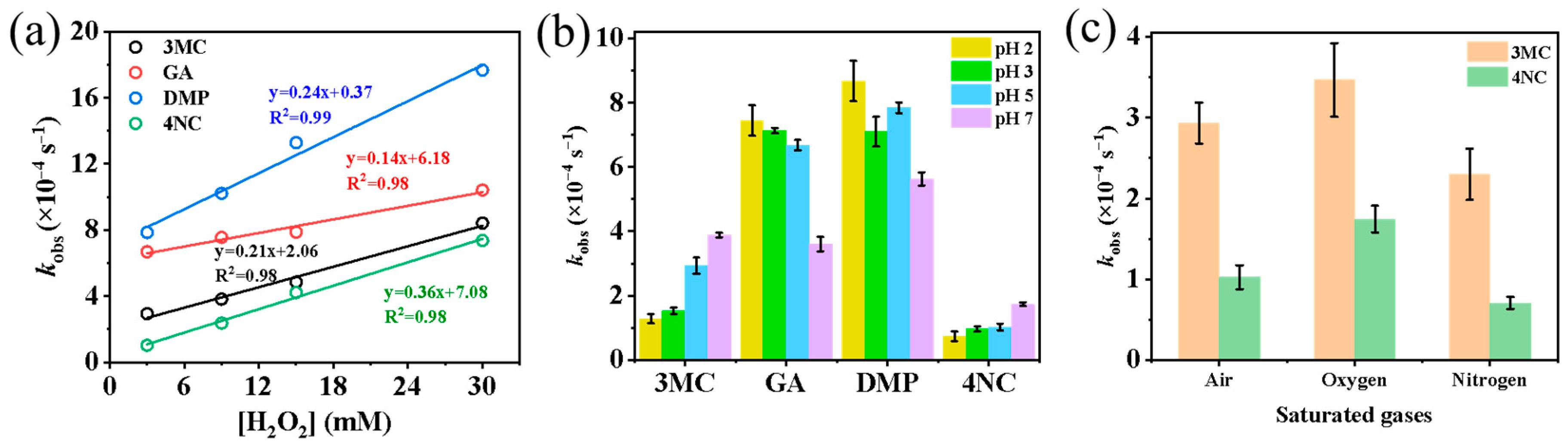
The influence of H2O2 concentration on the reaction rates was examined by adding 3, 9, 15, and 30 mmol·L−1 of H2O2 to the reaction system. As shown in Figure 2a and Figure S3, a strong linear relationship (R2 > 0.98) was observed between H2O2 concentration and the first-order rate constant of phenolic compounds. Previous studies [11] have also demonstrated that when H2O2 is present in large excess relative to phenols (molar ratio > 10:1), the reaction follows pseudo-first-order kinetics, with kobs being directly proportional to the concentration of H2O2 and, consequently, to •OH concentration. Therefore, the photodegradation rate of substituted phenols can be expressed as = −kobs[PhCs]0.
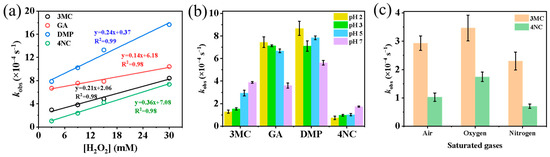
Figure 2.
Influencing factors of pseudo first-order rate constants: (a) H2O2 concentration; (b) solution pH value; (c) saturated gases (air, O2, N2).
The effect of pH value (adjusted to 2, 3, 5 and 7 using H2SO4 or NaOH) on reaction rates is presented in Figure 2b and Figure S4. The results show that for 3MC and 4NC, higher pH values are favorable for their degradation, indicating enhanced reaction rates under more alkaline conditions. In contrast, for the GA and DMP, increasing the pH leads to a decrease in the first-order rate constants.
Figure 2c and Figure S5 compare the first-order rate constants under three different saturated gases (air, O2 and N2). As shown in Figure 2c, the overall reaction rates for aqueous-phase oxidation follow the order of kO2 > kair > kN2, indicating that O2 is beneficial for the photodegradation of phenolic compounds. This enhancement is likely due to the role of O2 as an electron acceptor, facilitating the formation of highly ROS such as superoxide (O2•−) and hydroperoxyl radicals (•HO2). These reactive intermediates can further generate H2O2 and •OH radicals, thereby increasing the oxidative capacity of the system toward organic compounds.
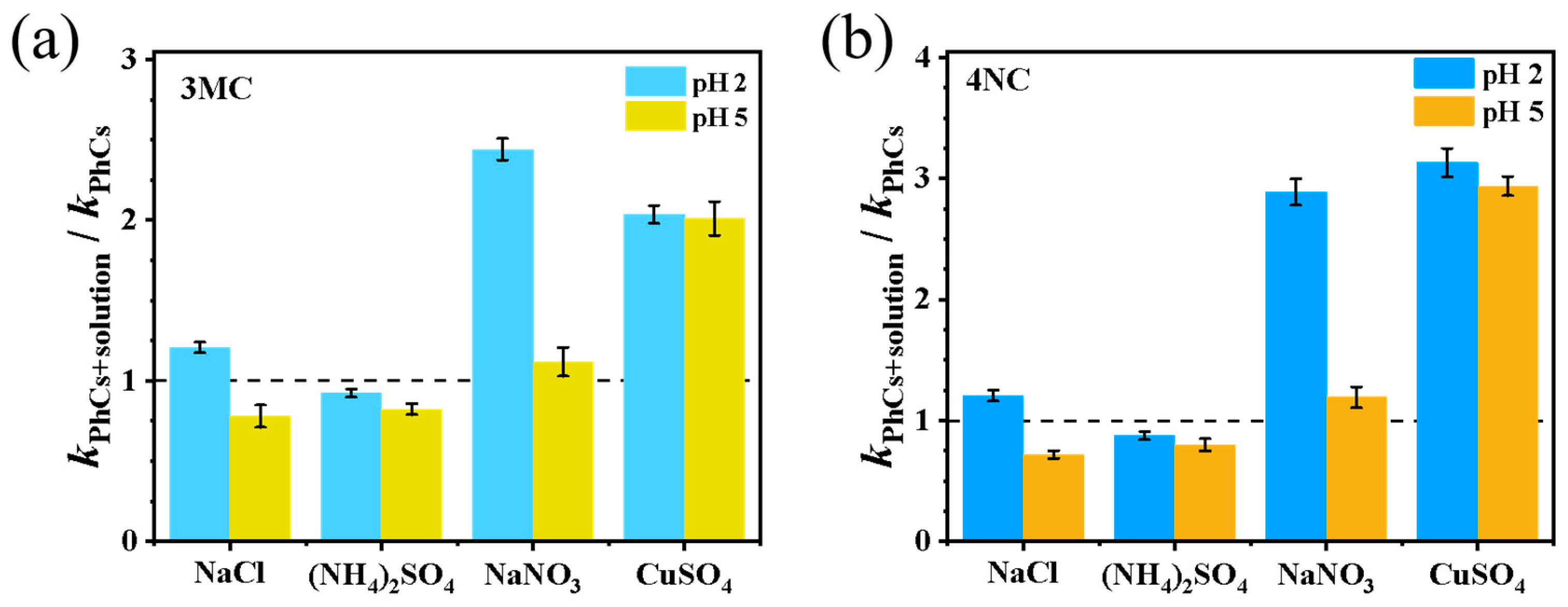
Transition metals such as Fe and Cu, as well as inorganic ions (e.g., NO3−) are abundant in atmospheric aerosols, with Fe concentrations in cloud droplets typically ranging from 10−6 to 10−3 mol·L−1 [30]. Figure 3 shows the normalized rate constants in the presence of various inorganic ions and metals. As shown, the addition of NO3− promotes the reaction, particularly under strong acidity conditions. This enhancement is attributed to the photolysis of NO3− in the presence of H+, leading to the generation of •OH [3]. However, the degree of enhancement observed here is lower than that reported in reference [13], where 0.5 mol·L−1 NH4NO3 increased the degradation efficiency of guaiacyl acetone by 11–20 times, suggesting precursor-dependent effects. SO42− inhibits the reaction by approximately 20%, which is primarily attributed to the increased ionic strength. The elevated ionic strength may reduce the reactivity of radicals through diffusion limitations. However, a quenching to OH effect from SO42− cannot be entirely ruled out.
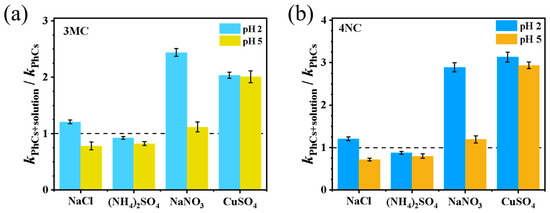
Figure 3.
Influence of inorganic ions on the pseudo-first-order rate constants: (a) 3MC and (b) 4NC.
The effect of NaCl on the photodegradation is strongly pH-dependent. At pH 2, Cl− can be converted into •Cl via a series of reactions with hydroxyl radicals, as shown in reactions (5) and (6):
•OH + Cl−→ClOH•−
ClOH•− + H+→H2O2 + •Cl
The rate constant for reaction (5) has been reported to range from 1.0 to 4.3 × 109 L mol−1 s−1 [31]. The generation of reactive chlorine species significantly enhanced photodegradation efficiency, leading to a twofold increase in the observed first-order rate constant under acidic conditions. In contrast, at pH 5, the ionic strength effect dominates, and NaCl exhibits a slight inhibitory effect on photodegradation [12,29]. Fe3+ significantly enhances the reaction rate by generating Fe2+ and •OH through the Fenton reaction with H2O2. In this study, the reaction proceeded too rapidly after Fe addition to quantify the pseudo-first-order rate constant, and thus the data were not included in Figure 3. Cu2+ also promotes the reaction by participating in a Fenton-like mechanism, where O2•− reduces Cu2+ to Cu+, leading to •OH generation. Notably, the effect of Cu2+ is not significantly related to the pH, differing from the findings in reference [32], which reported that at pH 2, O2•− mainly exists in the form of •HO2 with lower oxidation ability, thereby slowing the Fenton-like reaction.
3.3. Second-Order Rate and Structure–Activity Relationship
3.3.1. Second-Order Rate Constants of Substituted Phenols with OH
According to Equation (2), the second-order rate constants for the reactions of ten substituted phenols with •OH are summarized in Table 1 and depicted in Figure S6. As shown in Figure S6, the intercepts of the linear fits were close to zero for all phenolic compounds, indicating negligible contributions from secondary reactions. The second-order rate constants (kPhCs, OH) range from 109 to 1010 L mol −1 s−1. At pH 2, the kPhCs, OH range from 2.45 × 109 to 1.39 × 1010 L mol−1 s−1, which are lower than the values observed at pH 5 (1.21 × 1010 to 6.03 × 1010 L mol−1 s−1), in agreement with findings reported by Arciva et al. [13]. Previous studies [3] have also shown the effects of acidity on •OH-mediated oxidation. The observed pH dependence is unlikely to result from deprotonation-induced changes in the electronic structure, as the pKa values of most phenolic compounds studied are significantly higher than 5. The mechanisms for this change in reactivity with acidity are unclear yet. We inferred that the enhanced reaction rates at pH 5 may be attributed to improved reactivity or extended lifetimes of •OH under mildly acidic conditions. Additionally, lower concentrations of H+ at pH 5 may reduce the scavenging of •OH, increasing its availability for reaction with phenols. Notably, GA and AS, acting as photosensitizers, exhibited exceptionally high second-order rate constants, further emphasizing the role of molecular structure and functionality in determining reactivity.
The electronic effects of substituents play a crucial role in determining reactivity, with compounds bearing electron-donating groups generally having higher activity than those with electron-withdrawing substituents. Theoretically, among electron-donating groups, the -OH group exhibits a stronger electron-donating effect than the -OCH3. Based on hydrogen bond dissociation energies, the order of bond strength is CH < CH2 < CH3 < OH, implying that compounds with -OH substitution should exhibit higher reactivity and larger second-order rate constants than those with -OCH3 groups. Nevertheless, a previous study [18] reported that nitrocatechol, which contains one more OH group, has a slightly lower second-order rate constant than nitroguaiacol, which contains an extra OCH3 group. This suggests that factors beyond simple electronic effects—such as intramolecular hydrogen bonding (e.g., in GUA, where adjacent -OCH3 and -OH groups may be stabilized through hydrogen bonding [21]) or steric hindrance from ortho/para substituents [33]—can also influence the overall reactivity.
3.3.2. Relationship Between Second-Order Rate Constant and Selected Parameters
A quantitative structure–activity relationship (QSAR) was developed between the second-order rate constant for phenolic compounds reacting with OH (kPhCs, OH) and their oxidation potential (Eox), as measured by cyclic voltammetry, for ten phenols listed in Table 2. Figure S7 illustrates the relationship between Eox and log kPhCs, OH: log kPhCs, OH = −1.43 Eox + 11.15 (r2 = 0.42) at pH 2 and log kPhCs, OH = −0.65 Eox + 10.81 (r2 = 0.62) at pH 5. A stronger correlation at pH 5 suggests that oxidation potential is a more reliable predictor of reactivity under near-neutral conditions. Theoretically, an increase in Eox implies greater resistance to oxidation, which corresponds to a lower reactivity with •OH. Previous studies have also reported only a modest correlation between Eox and second-order rate constants [3,13]. The absence of a strong correlation implies that additional molecular characteristics—such as electronic effects, steric hindrance, and bond dissociation enthalpy—may significantly influence the reactivity of phenolic compounds with •OH.
Table 2.
Redox potentials of ten phenols.
This study reveals that phenols exhibit spontaneous oxidation (ΔG°ox < 0) at both pH 2.0 and 5.0, with a stronger thermodynamic drive at pH 2.0 due to enhanced proton-coupled electron transfer efficiency under elevated H+ concentrations, consistent with Nernstian redox principles. However, extreme acidity paradoxically diminishes the predictive capacity of thermodynamic parameters (e.g., oxidation potential) for reactivity, as excessive H+ induces kinetic bottlenecks through solvation alterations, intermediate stabilization, or steric effects, decoupling electron transfer from redox potentials [33]. Consequently, pH 5.0 demonstrates superior second-order kinetics and stronger correlations between oxidation potential and reactivity compared to pH 2.0, highlighting the critical interplay between thermodynamic favorability and kinetic accessibility in governing phenolic oxidation processes.
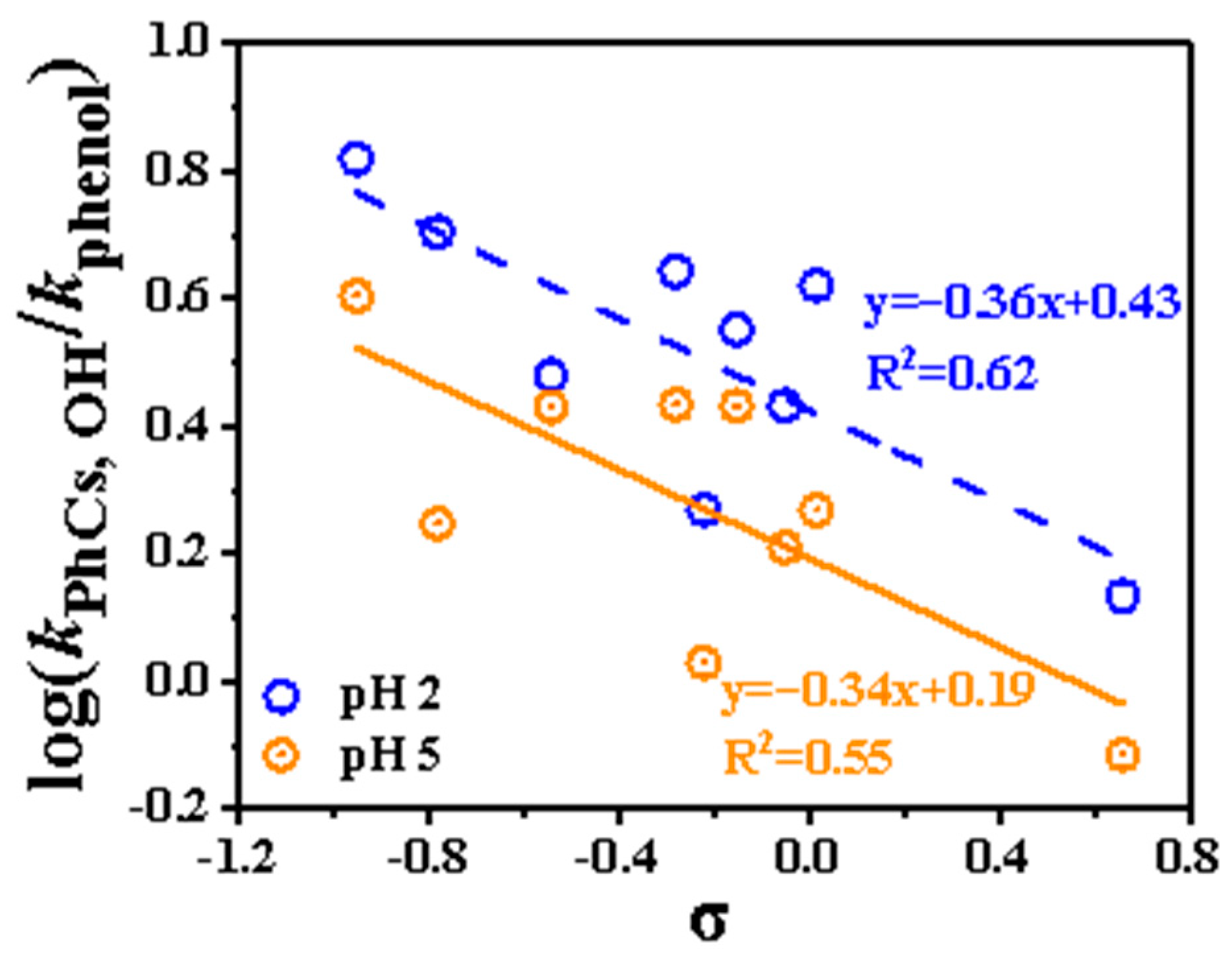
In the 1930s, Hammett observed a relationship between the electronic effect of substituents on a benzene ring and the reaction rate and described it using a simple empirical formula: log k = log k0 + ρσ. Here, k and k0 represent the rate constants of substituted and unsubstituted aromatic compounds (with phenol used as a reference in this study), σ is the substituent constant and ρ is a reaction-specific constant defined by Hammett. This relationship forms the basis for discussing the influence of substituents on reaction rates. In this study, the σ values were obtained from the previous literature [15,34,35] and listed in Table S1. For the phenolic compounds with multiple substituents, the overall σ value was calculated as the sum of the contributions of all substituents.
The Hammett plot (Figure 4) for substituted phenols was constructed and applied to predict the reaction rate constants of other substituted phenols at 298 K. Linear regression analysis, expressed as log kPhCs, OH/kphenol = A+ ρσ, yielded ρ values of −0.36 and −0.34 at pH 2 and pH 5, respectively. The ρ value reflects the sensitivity of the reaction center’s charge distribution to substituent effects. A more negative ρ indicates a greater sensitivity to substituent variations. As shown in Figure 4, the obtained ρ values were comparable to those reported in previous results [15]. The correlation at pH 5 (R2 = 0.55) is slightly worse than that at pH 2 (R2 = 0.62). It is worth noting that compounds such as SYR, GA, and CAT deviate significantly from the predicted trend. This deviation may be attributed to the complex interplay of multiple functional groups, which can influence the reactivity of substituted phenols toward •OH in ways not captured by the Hammett model. Therefore, the applicability of this relationship is limited for certain multifunctional phenolic compounds.
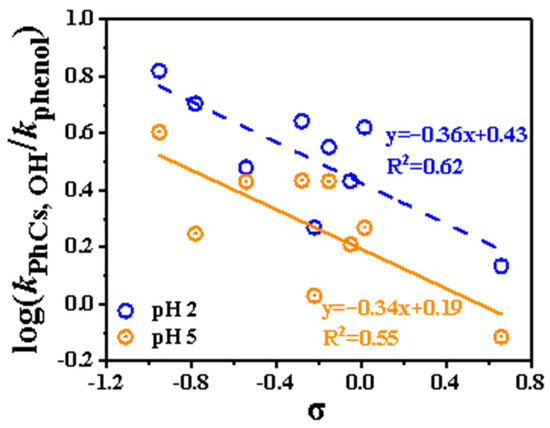
Figure 4.
Hammett plot (298 K) for correlation between log kPhCs, OH/kphenol with Hammett constants (σ). The dashed line represents the linear relationship at pH 2 (blue symbols), and the solid line represents the relationship at pH 5 (orange symbols).
Overall, the presence of multiple substituents can reduce the reactivity of phenolic compounds toward •OH, as indicated by the Hammett plots.
Lastly, the choice of reference compounds also has a significant impact on the rate constants. In this study, phenol, catechol and guaiacol were selected as reference substances. Their second-order rate constants with •OH at 298 K at pH 2 and pH 5 are summarized in Table S2. Table 3 presents second-order rate constants of 3MC and 4NC using these three reference substances. As shown, the calculated kPhCs, OH values vary depending on the chosen reference compounds. For instance, at pH 2, the kPhCs, OH for 3MC ranges from 4.87 to 9.78 × 109 L mol−1 s−1, and for 4NC from 2.45 to 5.32 × 109 L mol−1 s−1. These results are consistent with previously reported values, such as (1.4 ± 0.1) × 1010 L mol−1 s−1 for 3MC at pH 3 and (5 ± 1) × 109 L mol−1 s−1 for 4NC at pH 7 [23]. All three reference compounds exhibited minimal direct photodegradation under our light source (<10% within 60 min, Figure S8), with phenol showing the lowest extent (~3%). Due to the high direct photolysis rates of catechol and guaiacol, as well as their excessively high reactivity with •OH, they are not particularly suitable as reference compounds for the two phenolic compounds (3MC and 4NC) investigated in this study [36]. Therefore, phenol was primarily used as the reference compound in the subsequent experiments to ensure the reliability of the reported second-order rate constants. In future studies, if reference compounds with higher susceptibility to direct photodegradation are selected, corrections for direct photolysis will be explicitly incorporated.
Table 3.
Second-order rate constants of 3MC and 4NC using different reference compounds (unit: L mol−1 s−1).
3.4. Qualitative and Quantitative Evaluations of ROS Role
3.4.1. Determination of [•OH]ss and [1O2]ss
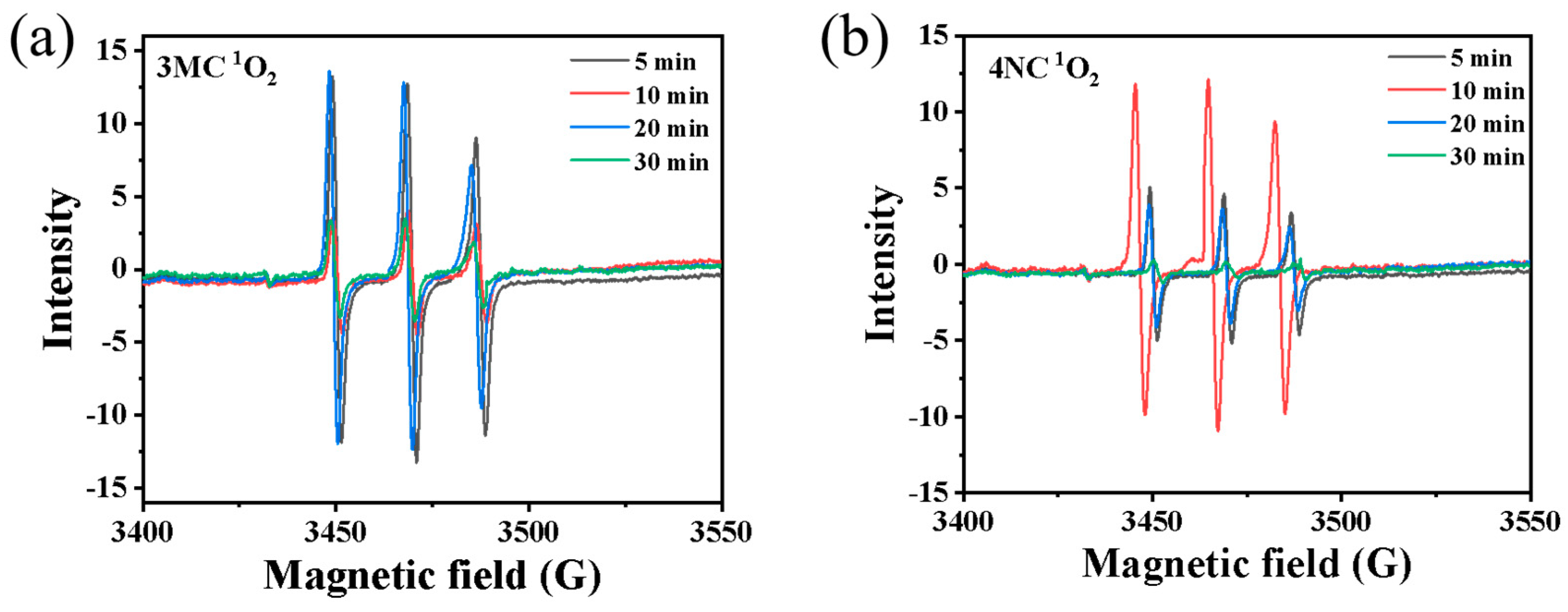
The spin trap DMPO was employed to detect OH radicals. However, due to the detection limit of the Micro ESR spectrometer, no •OH signal was observed. In contrast, spin trap TEMP was used to capture 1O2, forming the TEMP-1O2 spin adduct (TEMPO). The characteristic ESR signal with 1:1:1 triplet pattern, indicative of 1O2, was successfully detected by the Micro ESR spectrometer, as shown in Figure 5. This result confirms the possible presence of 1O2 in the reaction system.
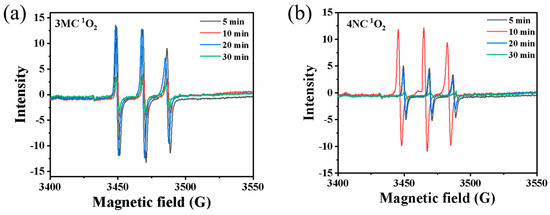
Figure 5.
EPR spectra obtained upon photodegradation of (a) 3 MC and (b) 4NC (0.3 mmol·L−1) in presence of H2O2 (3 mmol·L−1) under different reaction times.
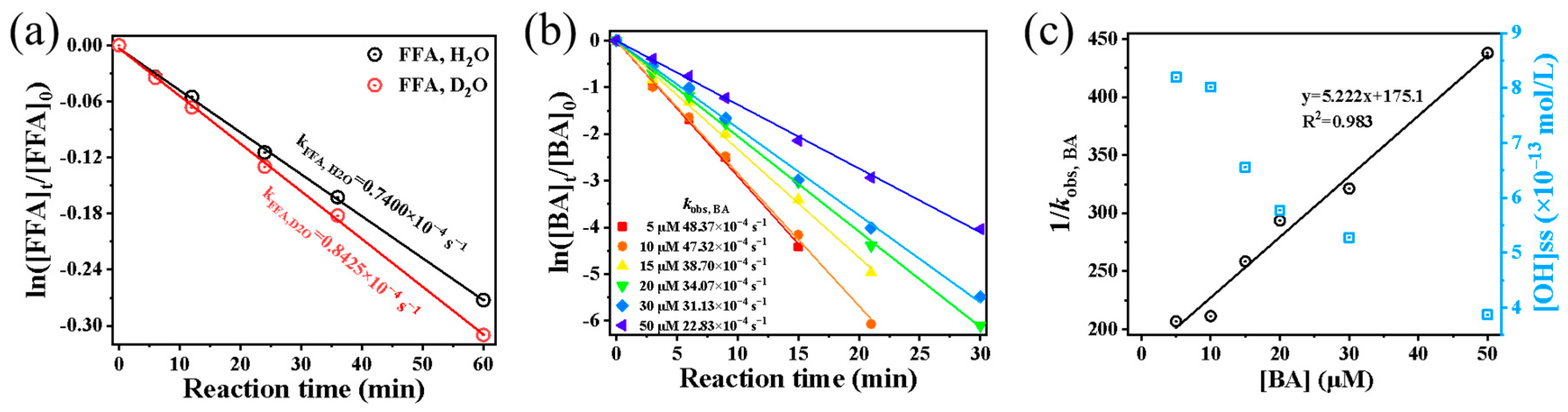
To further quantify [1O2]ss, FFA was employed as a chemical probe to capture singlet oxygen [37]. The corresponding results are presented in Figure 6a. By comparing FFA decay rates in systems containing H2O2 + FFA + D2O and H2O2 + FFA + H2O, the steady-state concentration of 1O2 was calculated. The resulting [1O2]ss was 6.336 × 10−11 mol·L−1, which is approximately twice the concentration typically observed in fog water ((0.11–3) × 10−11 mol·L−1) and about three orders of magnitude higher than that in aerosol water ((1.1–4.5) × 10−14 mol·L−1) [38].
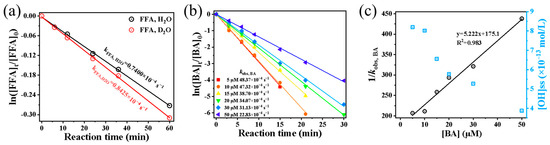
Figure 6.
(a) Comparative study of first-order degradation kinetics for FFA in H2O and D2O systems; (b) determination of first-order rate constants for benzoic acid (BA) as a function of concentration; (c) [OH]ss measured at varying BA concentrations, with linear correlation between BA concentration and the reciprocal of measured rate constants (1/kobs, BA).
Typically, the steady-state concentration of ROS in a system can be estimated by dividing the pseudo-first-order rate constant by its second-order rate constant [39,40]. However, this approach assumes the presence of only a single ROS. In systems where multiple ROS coexist due to chain reactions, each species contributes to the overall kobs through its respective second-order reaction rate with the precursor. Consequently, the simple division method becomes invalid in such cases. In this study, both the ESR qualitative method and the FFA chemical probe method confirmed the presence of 1O2 in the system, along with likely superoxide anion radicals (O2•−). Given these findings, we further employed the probe method to quantify the steady-state concentration of •OH ([OH]ss) via benzoic acid (BA) as a probe (kBA, OH = 5.9 × 109 L mol−1 s−1) [18]. BA concentrations (5, 10, 20, 30, 40, and 50 μmol·L−1) were tested to measure decay rates and [OH]ss (Figure 6b). Although low probe concentrations were used to minimize interference with the reaction, some influence on [•OH]ss measurements remained. To address this, we applied a modified method [25], wherein the reciprocal of the pseudo-first-order decay rate (1/kobs, BA), was linearly regressed against BA concentration (Figure 6c). The steady-state concentration was then calculated by taking the reciprocal of the intercept and dividing it by kobs, BA, yielding [•OH]ss of 9.681 × 10−13 mol·L−1 in the system. This value aligns with estimates obtained at low BA concentrations and falls within the typical •OH range reported for cloud/fog droplets (10−15–10−12 mol·L−1) [29], confirming the environmental relevance of the applied H2O2 dosage.
3.4.2. Contribution of ROS to Phenolic Compounds Photodegradation
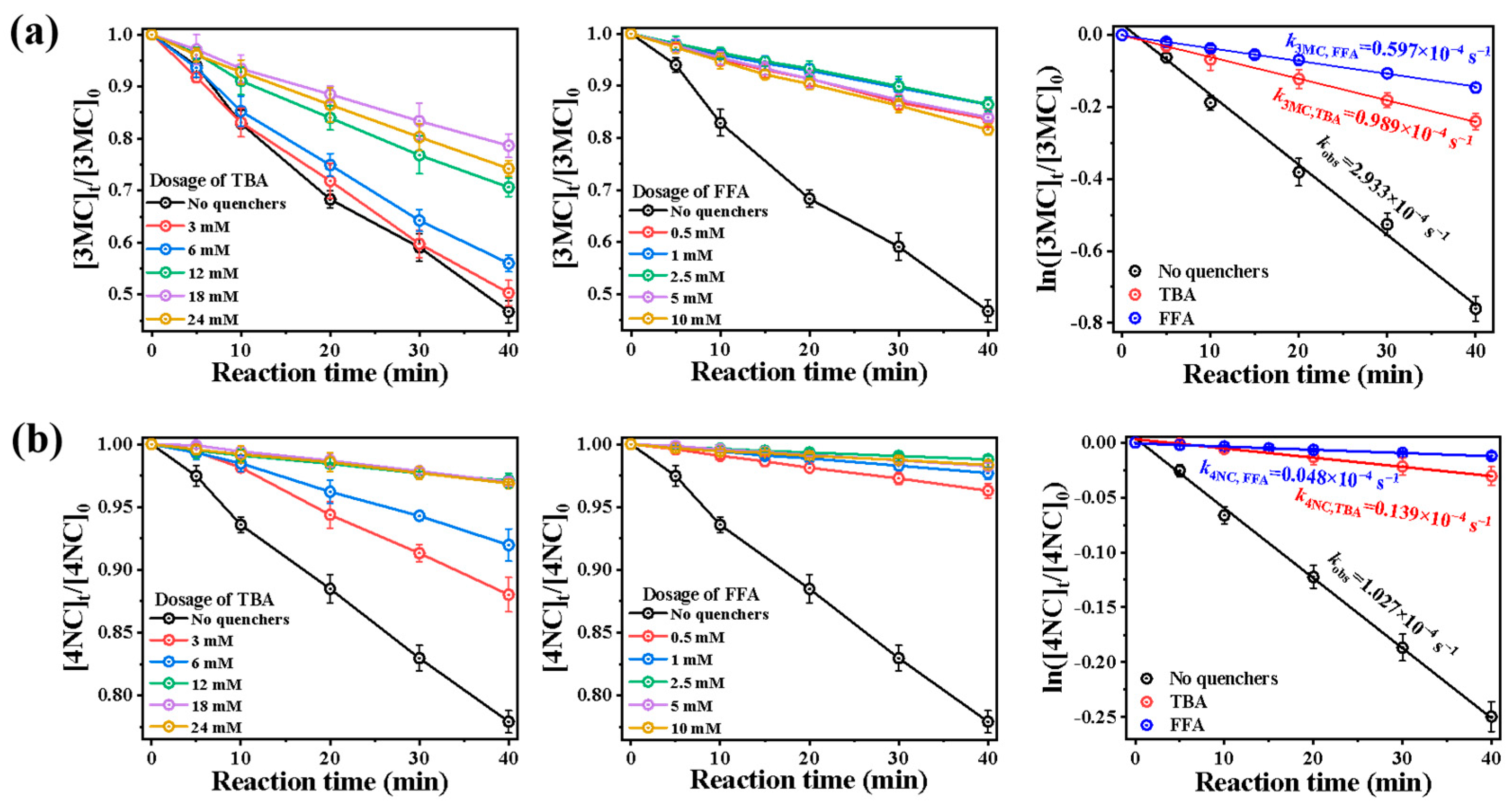
Figure 7 presents the photolysis of 4NC and 3MC with the addition of different quencher concentrations. Quenching experiments using TBA and FFA were conducted to assess ROS contributions (Figure 7). Among them, Figures (a,b) and (d,e) depict the time-dependent photodegradation profiles with varying amounts of quenchers. The addition of quenchers reduced the pseudo-first-order degradation rates for both compounds. The quenching effect reached saturation at 18 mmol·L−1 for TBA and 2.5 mmol·L−1 for FFA, with higher concentrations showing negligible additional impact. These concentrations were, therefore, established as optimal quencher dosages. At these optimal dosages, by using Equation (1), we plot ln(Ct/C0) versus time t. Before the addition of quenchers, the pseudo-first-order rate of 3MC was 2.933 × 10−4 s−1. After adding TBA and FFA, these rates decreased to 0.989 × 10−4 and 0.597 × 10−4 s−1, respectively. Similarly, before the addition of quenchers, the pseudo-first-order rate of 4NC was 1.027 × 10−4 s−1. After adding TBA and FFA, they quickly decreased to 0.139 × 10−4 and 0.048 × 10−4 s−1.
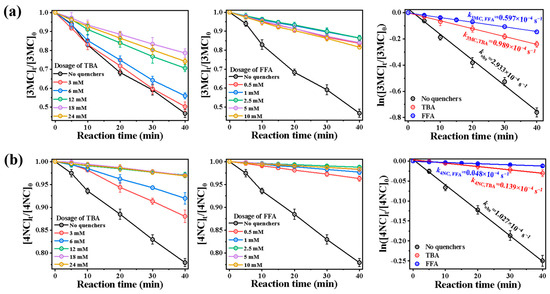
Figure 7.
Influence of different quenchers on the photodegradation of (a) 3MC and (b) 4NC.
Theoretically, TBA usually primarily quenches •OH with kTBA, OH = (3.8–7.6) × 108 L mol−1 s−1, and FFA quenches both •OH (kFFA, OH = 1.5 × 1010 L mol−1 s−1) and 1O2 (kFFA, 1O2 = 1.2 × 108 L mol−1 s−1) [25]. Suppose the first-order rate constant without quenchers is kPhCs, and the rate constants after adding TBA and FFA are labeled as kPhCs+TBA and kPhCs+FFA, respectively. Then, the contributions of •OH and 1O2 can be calculated by (kPhCs − kPhCs+TBA)/kPhCs and (kPhCs+TBA − kPhCs+FFA)/kPhCs, respectively. kPhCs+FFA/kPhCs can be attributed to the contribution of other factors, including direct photolysis or other free radicals. Therefore, for 3MC, the contribution of •OH, 1O2 and other free radicals was 66.3%, 13.4%, and 20.4%, respectively. For 4NC, the contribution of •OH, 1O2 and other free radicals was 86.5%, 8.9% and 4.7%. These results demonstrate that •OH serves as the primary oxidant (>65% contribution) in both systems, with significantly higher involvement in 4NC degradation.
3.5. Estimation of the Lifespan of Phenols in the Atmosphere
The aqueous-phase reactions of phenols in clouds and fog generate SOA, including light-absorbing BrC, which affects ambient air quality. The atmospheric lifetime of these phenols is critical in determining their environmental impact. Based on the measured second-order rate constants, combined with [•OH]ss in cloud droplets and wet aerosols [32], we estimated the atmospheric lifetimes of phenolic compounds (). As shown in Table 4, these lifetimes fall within the typical range for phenolic compounds in cloud and fog water, with slower-reacting phenols exhibiting longer lifetimes. Additionally, in urban environments, they showed longer lifetimes (5.7 h–32.4 h), while shorter lifetimes (<1.1 h) in marine regions due to different •OH concentrations. Since the rate constants are a function of temperature, lifetime also varied with temperature, which would be considered in future work.
Table 4.
Atmospheric aqueous-phase lifetimes of ten phenols in urban, remote and marine areas.
In conclusion, aqueous-phase oxidation under wet aerosols or cloud droplet conditions significantly governs phenolic compound lifetimes and SOA formation, highlighting its critical role in atmospheric chemistry and environmental impacts.
4. Conclusions
This study systematically investigated the aqueous-phase oxidation kinetics of ten phenolic compounds, focusing on the key influencing factors and the roles of ROS. Using comprehensive qualitative and quantitative analysis of •OH and 1O2, combined with quenching experiments, we elucidated the contribution of different ROS during the oxidation process. The main findings are summarized as follows:
Substituents on the benzene ring significantly influence phenolic reactivity. Electron-donating groups (e.g., -OH, -CH3, -OCH3) enhance the electron density of the benzene ring, facilitating electrophilic substitution reactions and leading to higher kobs as evidenced by the reactivity trend: DMP > 4EP > 4EG. Conversely, electron-withdrawing groups (e.g., -COOH) decrease the electron density, thereby inhibiting the reaction rate (e.g., SA exhibited lower reactivity). Additionally, multiple environmental factors, including H2O2 concentration, pH, saturating gases, and inorganic salts, were found to modulate the photodegradation kinetics of phenols. Among inorganic ions, Cu2+ and NO3− enhanced reaction rates, whereas Cl− exhibited a pH-dependent promotional effect under acidic conditions. In contrast, SO42− inhibited the reaction, likely due to increased ionic strength.
The second-order rate constants (kPhCs, OH) for the reaction between the ten phenolic compounds and •OH were determined to range from 109 to 1010 M−1 s−1. QSAR models were developed, demonstrating a strong correlation at pH 2 but only a moderate correlation at pH 5. Using chemical probes, [OH]ss and [1O2]ss were determined as 9.681 × 10−13 mol·L−1 and 6.336 × 10−11 mol·L−1, respectively. Quenching experiments confirmed that •OH was the dominant oxidant in phenolic degradation, contributing 66.3% for 3MC and 86.5% for 4NC. Based on the obtained kPhCs, OH values and [OH]ss, the atmospheric lifetimes of the ten phenolic compounds were estimated, providing insights into their environmental persistence.
This study elucidated the synergistic mechanism between the substituent electronic effects and environmental factors in governing phenolic oxidation kinetic, providing a theoretical foundation for predicting atmospheric transformation pathways and aqSOA formation. Future research should focus on the reactivity and toxicity of phenolic compounds with varying substituents. Additionally, the interference effects of the coexistence of multiple pollutants and real atmospheric components on the degradation pathways should be investigated. These investigations will enable more accurate prediction of atmospheric behavior and ecological impacts of phenolic compounds in complex environmental systems.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/atmos16050567/s1, Figure S1. Irradiation intensity of simulated sunlight in this study; Figure S2. Decay kinetic curves with first-order rate constants of ten phenolic compounds at: (a) pH 2; and (b) pH 5; Figure S3. Representative plot of the photodecay of four phenolic compounds with reaction time under different dosages of H2O2; Figure S4. Representative plot of decay of four phenols with reaction time under different pH; Figure S5. Influence of saturated gases on the decay kinetics of: (a) 3MC; and (b) 4NC; Figure S6. Second-order kinetic curves of the reaction between phenols and ·OH at: (a) pH 2; and (b) pH 5; Figure S7. Relationship between one-electron oxidation potential and measured second-order rate constant; Figure S8. Direct photolysis rates of three reference compounds (300 μM); Table S1. Hammett substituent constants (σ) for ten phenols taken from other worksa, b; Table S2. Second-order rate constants of different references at 298 K.
Author Contributions
D.H.: conceptualization, methodology, software, writing—original draft preparation, funding acquisition; Z.W., X.D.: data curation, software, investigation, formal analyses; E.A., Z.Z.: review and editing, project administration; Z.Y.: review and editing, supervision, project administration, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge support from the Natural Science Foundation of Jiangsu Province (BK20221405). We are also thankful for the Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX23_1646, SJCX24_1808).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon request.
Conflicts of Interest
Author Xuanli Dai was employed by the company Jiangsu Xinrui Environmental Monitoring Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Li, Z.; Wang, B.; Wang, T.T.; Zhang, D.; Song, X.S.; Yuan, M.H.; Lu, X.; Wang, W.J.; Yin, S.S.; Zhang, R.Q. Pollution characteristics, sources, and environmental impacts of VOCs in Zhengzhou city during summer based on photochemical loss. Environ. Sci. 2024, 45, 5157–5167. [Google Scholar]
- Zhang, X.; Zhang, N.; Wei, R.; Su, W.K.; He, J.L.; Song, K.L.; Zhang, M.M.; Zhao, J.W.; Meng, K.; Zhang, Y.P. Pollution characteristics, chemical reactivity, and source apportionment of VOCs in the high-tech zone of Shijiazhuang in autumn. Environ. Sci. 2024, 45, 7021–7030. [Google Scholar]
- Ma, L.; Guzman, C.; Niedek, C.; Tran, T.; Zhang, Q.; Anastasio, C. Kinetics and mass yields of aqueous secondary organic aerosol from highly substituted phenols reacting with a triplet excited state. Environ. Sci. Technol. 2021, 55, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Worland, R.; Jiang, W.; Niedek, C.; Guzman, C.; Bein, K.J.; Zhang, Q.; Anastasio, C. Predicting photooxidant concentrations in aerosol liquid water based on laboratory extracts of ambient particles. Atmos. Chem. Phys. 2023, 23, 8805–8821. [Google Scholar] [CrossRef]
- McNeill, V.F. Aqueous organic chemistry in the atmosphere: Sources and chemical processing of organic aerosols. Environ. Sci. Technol. 2015, 49, 1237–1244. [Google Scholar] [CrossRef]
- Kaur, R.; Anastasio, C. Light absorption and the photoformation of hydroxyl radical and singlet oxygen in fog waters. Atmos. Environ. 2017, 164, 387–397. [Google Scholar] [CrossRef]
- Tao, Y.; Chen, Y.T.; Li, W.L.; Zhang, X.Y.; Ye, Z.L.; Gai, X.L. Liquid—Phase oxidation of dissolved organic matter (DOM) in aerosols. Environ. Sci. 2021, 42, 2659–2667. [Google Scholar]
- Ye, Z.L.; Hu, D.D.; Wang, Z.X.; Wang, H.; Ge, X. Aqueous photochemical aging of water-soluble smoke particles from crop straws burning. Atmos. Environ. 2025, 340, 120897. [Google Scholar] [CrossRef]
- Smith, J.D.; Kinney, H.; Anastasio, C. Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products. Atmos. Environ. 2016, 126, 36–44. [Google Scholar] [CrossRef]
- Go, B.R.; Li, Y.J.; Huang, D.D.; Chan, C.K. Aqueous—Phase photoreactions of mixed aromatic carbonyl photosensitizers yield more oxygenated, oxidized, and less light-absorbing secondary organic aerosol (SOA) than single systems. Environ. Sci. Technol. 2024, 58, 7924–7936. [Google Scholar] [CrossRef]
- Cai, B.; Wang, Y.; Yang, X.; Li, Y.; Zhai, J.; Zeng, Y.; Ye, J.; Zhu, L.; Fu, T.-M.; Zhang, Q. Rapid aqueous-phase dark reaction of phenols with nitrosonium ions: Novel mechanism for atmospheric nitrosation and nitration at low pH. Proc. Natl. Acad. Sci. USA Nexus 2024, 3, 385. [Google Scholar] [CrossRef]
- Arakaki, T.; Anastasio, C.; Kuroki, Y.; Nakajima, H.; Okada, K.; Kotani, Y.; Handa, D.; Azechi, S.; Kimura, T.; Tsuhako, A.; et al. A General scavenging rate constant for reaction of hydroxyl radical with organic carbon in atmospheric waters. Environ. Sci. Technol. 2013, 47, 8196–8203. [Google Scholar] [CrossRef] [PubMed]
- Arciva, S.; Niedek, C.; Mavis, C.; Yoon, M.; Sanchez, M.E.; Zhang, Q.; Anastasio, C. Aqueous ·OH oxidation of highly substituted phenols as a source of secondary organic aerosol. Environ. Sci. Technol. 2022, 56, 9959–9967. [Google Scholar] [CrossRef]
- Yu, L.; Smith, J.; Laskin, A.; George, K.M.; Anastasio, C.; Laskin, J.; Dillner, A.M.; Zhang, Q. Molecular transformations of phenolic SOA during photochemical aging in the aqueous phase: Competition among oligomerization, functionalization, and fragmentation. Atmos. Chem. Phys. 2016, 16, 4511–4527. [Google Scholar] [CrossRef]
- Kroflič, A.; Schaefer, T.; Huš, M.; Le, H.P.; Otto, T.; Herrmann, H. OH radicals reactivity towards phenol-related pollutants in water: Temperature dependence of the rate constants and novel insights into the [OH–phenol] adduct formation. Phys. Chem. Chem. Phys. 2019, 22, 1324–1332. [Google Scholar] [CrossRef]
- Błaziak, A.; Schaefer, T.; Rudziński, K.; Herrmann, H. Photo-oxidation of α-pinene oxidation products in atmospheric waters—pH- and temperature-dependent kinetic studies. J. Phys. Chem. A 2024, 128, 4507–4516. [Google Scholar] [CrossRef]
- Hoffmann, E.H.; Tilgner, A.; Wolke, R.; Böge, O.; Walter, A.; Herrmann, H. Oxidation of substituted aromatic hydrocarbons in the tropospheric aqueous phase: Kinetic mechanism development and modelling. Phys. Chem. Chem. Phys. 2018, 20, 10960–10977. [Google Scholar] [CrossRef] [PubMed]
- Hems, R.F.; Abbatt, J.P.D. Aqueous phase photo—Oxidation of brown carbon nitrophenols: Reaction kinetics, mechanism, and evolution of light absorption. ACS Earth Space Chem. 2018, 2, 225–234. [Google Scholar] [CrossRef]
- Tomaz, S.; Cui, T.; Chen, Y.; Sexton, K.G.; Roberts, J.M.; Warneke, C.; Yokelson, R.J.; Surratt, J.D.; Turpin, B.J. Photochemical cloud processing of primary wildfire emissions as a potential source of secondary organic aerosol. Environ. Sci. Technol. 2018, 52, 11027–11037. [Google Scholar] [CrossRef]
- Chen, Y.; Li, N.; Li, X.; Tao, Y.; Luo, S.; Zhao, Z.; Ma, S.; Huang, H.; Chen, Y.; Ye, Z.; et al. Secondary organic aerosol formation from 3C⁎-initiated oxidation of 4-ethylguaiacol in atmospheric aqueous-phase. Sci. Total. Environ. 2020, 723, 137953. [Google Scholar] [CrossRef]
- Li, X.D.; Tao, Y.; Zhu, L.; Ma, S.; Luo, S.; Zhao, Z.; Sun, N.; Ge, X.; Ye, Z. Optical and chemical properties and oxidative potential of aqueous-phase products from OH and 3C∗-initiated photooxidation of eugenol. Atmos. Chem. Phys. 2022, 22, 7793–7814. [Google Scholar] [CrossRef]
- He, L.H.; Schaefer, T.; Otto, T.; Kroflič, A.; Herrmann, H. Kinetic and theoretical study of the atmospheric aqueous-phase reactions of OH radicals with methoxyphenolic compounds. J. Phys. Chem. A 2019, 123, 7828–7838. [Google Scholar] [CrossRef]
- Li, F.H.; Zhou, S.Z.; Du, L.; Zhao, J.; Hang, J.; Wang, X. Aqueous—Phase chemistry of atmospheric phenolic compounds: A critical review of laboratory studies. Sci. Total Environ. 2023, 856, 158895. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tsona, N.T.; Li, J.; Du, L. Aqueous-phase oxidation of syringic acid emitted from biomass burning: Formation of light-absorbing compounds. Sci. Total. Environ. 2021, 765, 144239. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yu, Y.F.; Lei, X.; Liang, X.; Cheng, S.; Ouyang, G.; Yang, X. Assessing the Use of probes and quenchers for understanding the reactive species in advanced oxidation processes. Environ. Sci. Technol. 2023, 57, 5433–5444. [Google Scholar] [CrossRef] [PubMed]
- Tratnyek, P.G.; Hoigne, J. Oxidation of substituted phenols in the environment: A QSAR analysis of rate constants for reaction with singlet oxygen. Environ. Sci. Technol. 1991, 25, 1596–1604. [Google Scholar] [CrossRef]
- Gligorovski, S.; Strekowski, R.; Barbati, S.; Vione, D. The environmental implications of hydroxyl radical (OH). Chem. Rev. 2015, 115, 13051–13092. [Google Scholar] [CrossRef]
- Anastasio, C.; Faust, B.C.; Rao, C.J. Aromatic Carbonyl Compounds as Aqueous-phase photochemical sources of hydrogen peroxide in acidic sulfate aerosols, fogs, and clouds. 1. non-Phenolic Methoxybenzaldehydes and Methoxyacetophenones with Reductants (Phenols). Environ. Sci. Technol. 1997, 31, 218–232. [Google Scholar] [CrossRef]
- Xie, Y.X.; Deng, Q.X.; He, B.W.; Liu, S.; He, J.; Wang, Y.; Li, X.; Yu, Z.; Pang, H.; Gligorovski, S. Acidity of atmospheric waters induces enhanced H2O2 production through photosensitized chemistry of phenolic substances. ACS Earth Space Chem. 2025, 9, 169–177. [Google Scholar] [CrossRef]
- Deguillaume, L.; Leriche, M.; Desboeufs, K.; Mailhot, G.; George, C.; Chaumerliac, N. Transition metals in atmospheric liquid phases: Sources, reactivity, and sensitive parameters. Chem. Rev. 2005, 105, 3388–3431. [Google Scholar] [CrossRef]
- Wang, J.L.; Wang, S.Z. Reactive species in advanced oxidation processes: Formation, identification and reaction mechanism. Chem. Eng. J. 2020, 401, 126158. [Google Scholar] [CrossRef]
- Herrmann, H.; Hoffmann, D.; Schaefer, T.; Bräuer, P.; Tilgner, A. Tropospheric Aqueous-phase free-radical chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools. ChemPhysChem 2010, 11, 3796–3822. [Google Scholar] [CrossRef]
- Sandhiya, L.; Kolandaivel, P.; Senthilkumar, K. Mechanism and kinetics of the atmospheric oxidative degradation of dimethylphenol isomers initiated by OH radical. J. Phys. Chem. A 2013, 117, 4611–4626. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Takahata, Y.; Chong, D. Estimation of Hammett sigma constants of substituted benzenes through accurate density-functional calculation of core-electron binding energy shifts. Int. J. Quantum Chem. 2005, 103, 509–515. [Google Scholar] [CrossRef]
- Bernhardt, P.C.; Dabbs, J.M., Jr.; Fielden, J.A.; Lutter, C.D. Testosterone changes during vicarious experiences of winning and losing among fans at sporting events. Physiol. Behav. 1998, 65, 59–62. [Google Scholar] [CrossRef]
- Anastasio, C.; McGregor, K.G. Chemistry of fog waters in California’s Central Valley: 1. In situ photoformation of hydroxyl radical and singlet molecular oxygen. Atmos. Environ. 2001, 35, 1079–1089. [Google Scholar] [CrossRef]
- Leresche, F.; Salazar, J.R.; Pfotenhauer, D.J.; Hannigan, M.P.; Majestic, B.J.; Rosario-Ortiz, F.L. Photochemical aging of atmospheric particulate matter in the aqueous phase. Environ. Sci. Technol. 2021, 55, 13152–13163. [Google Scholar] [CrossRef]
- Ma, L.; Worland, R.; Tran, T.; Anastasio, C. Evaluation of probes to measure oxidizing organic triplet excited states in aerosol liquid water. Environ. Sci. Technol. 2023, 57, 6052–6062. [Google Scholar] [CrossRef]
- Kaur, R.; Anastasio, C. First measurements of organic triplet excited states in atmospheric waters. Environ. Sci. Technol. 2018, 52, 5218–5226. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).