


Simulating the Fate of Dimethyl Sulfide (DMS) in the Atmosphere: A Review of Emission and Chemical Parameterizations

 , , , ,
, , , ,
Abstract
1. Introduction
2. Estimation of the DMS Emission Inventories and Climatologies
3. Modeling of DMS Chemistry in the Atmosphere
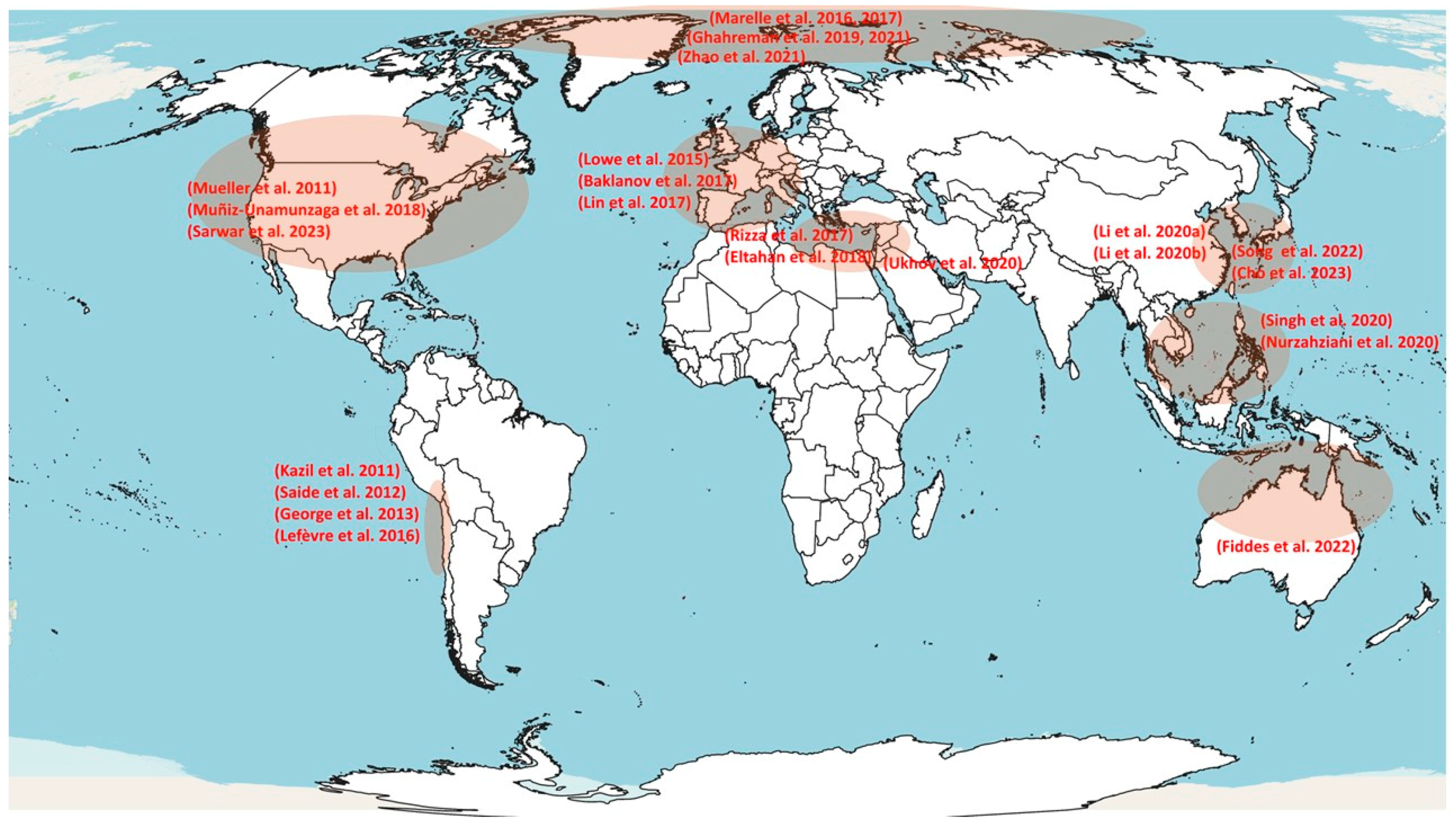
3.1. Regional Models
- Offline approach: This method estimates the DMS emission outside the air quality model, which is a crucial stage before modeling. The generated file must contain the geographic information of the analysis domain and temporal variation during the period of study. As a benefit, the obtained emission inventory can be applied to different models. One way to apply this method is to estimate DMS emissions from monitoring campaign studies, providing an extrapolation of those values. Considering this approach, the study of Kazil et al. (2011) [53] used the VOCALS-Rex (VAMOS Ocean-Cloud-Atmosphere-Land Study Regional Experiment) campaign, while Mueller et al. (2011) [54] applied constant values from Kloster et al. (2006) [55]. We highlight that the GOCART (Goddard Chemistry Aerosol Radiation and Transport) dataset was the preferred input in several studies [56,57,58,59,60].
- Inline approach: This method calculates DMS emissions within the atmospheric models during the simulation. This alternative requires the spatial distribution of the DMS seawater concentration, which is combined with meteorological variables and parameterizations to obtain the factor kw. In this approach, constant input values have been considered [61,62,63], giving variability to the inline flux emission only according to the selected parameterization as functions of the meteorological variables present in the zone. However, a method that is closer to reality is the application of time–space-varying DMS seawater concentration maps applied in several studies [9,10,16,64,65,66,67,68,69,70,71].
3.1.1. CMAQ
3.1.2. WRF-Chem
3.2. Global Models
GEOS-Chem
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3GP | Three main gas phase |
| AerChemMIP | Aerosols and 47 Chemistry Model Intercomparison Project |
| CAM-Chem | The Community Atmosphere Model with Chemistry |
| CAMS | Copernicus Atmosphere Monitoring Service |
| CanAM | The Canadian Atmospheric Model |
| CCN | Cloud condensation nuclei |
| CH3O2 | Methyl peroxy radical |
| Chl-a | Chlorophyll a |
| CMAQ | Community Multiscale Air Quality |
| CMIP6 | Coupled Model Intercomparison Project |
| Cg | Concentration of DMS in air |
| Cw | Concentration of DMS in water |
| DMS | Dimethyl sulfide |
| DMSO | Dimethyl sulfoxide |
| DMSP | Dimethysulfoniopropionate |
| ECCAD | Emissions of Atmospheric Compounds and Compilation |
| ECHAM | European Centre Hamburg general circulation model |
| EMD | DMS equivalent mixing depth |
| EMEP4UK | European Monitoring and Evaluation Programme for United Kingdom |
| Enviro-HIRLAM | Environment—High Resolution Limited Area Model |
| F | Sea–air flux |
| GEM-MACH | Global Environmental Multiscale—Modelling Air quality and Chemistry |
| GEOS-Chem | Goddard Earth Observing System—Chemistry |
| GOCART | Goddard Chemistry Aerosol Radiation and Transport |
| H | Henry’s law constant |
| HadGEM | Hadley Centre Global Environmental Model |
| H06 | Parameterization from Ho et al., (2006) [30] |
| HNO3 | Nitric acid |
| IOMB | International Ocean Model Benchmarking |
| IPCC | Intergovernmental Panel on Climate Change |
| IRR | Integrated Reaction Rate |
| ka | Total resistance to gas transfer on side of the air |
| kw | Total resistance to gas transfer on side of the sea |
| LM86 | Parameterization from Liss and Merlivat (1986) [26] |
| M09 | Parameterization from Marandino et al., (2009) [31] |
| MIROC | Model for Interdisciplinary Research on Climate |
| MSA | Methanesulfonic acid |
| N00 | Parameterization from Nightingale et al., (2000) [29] |
| NO3 | Nitrate radical |
| NorESM2 | The Norwegian Earth System Model version 2 |
| OH | Hydroxyl radical |
| PASE | Pacific Atmospheric Sulfur Experiment |
| R | Universal gas constant |
| REAM | The Regional chEmical trAnsport Model |
| Scn | Normalized Schmidt number |
| SO2 | Sulfur dioxide |
| VSL | Very short-lived |
| T | Seawater temperature |
| Ta | Temperature of the air |
| TOMAS | TwO-Moment Aerosol Sectional |
| U10 | Wind speed at 10 m |
| VOCALS-Rex | VAMOS Ocean-Cloud-Atmosphere-Land Study Regional Experiment |
| W14 | Parameterization from Wanninkhof et al., (2014) [32] |
| W92 | Parameterization from Wanninkhof (1992) [27] |
| WM99 | Parameterization from Wanninkhof et al., (1999) [28] |
| WRF-Chem | Weather Research and Forecasting coupled with Chemistry |
References
- Saltzman, E.S. Marine Aerosols. In Surface Ocean–Lower Atmosphere Processes; Geophysical Monograph Series; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 17–35. ISBN 9781118670330. [Google Scholar] [CrossRef]
- Hulswar, S.; Simó, R.; Galí, M.; Bell, T.G.; Lana, A.; Inamdar, S.; Halloran, P.R.; Manville, G.; Mahajan, A.S. Third Revision of the Global Surface Seawater Dimethyl Sulfide Climatology (DMS-Rev3). Earth Syst. Sci. Data 2022, 14, 2963–2987. [Google Scholar] [CrossRef]
- Mahowald, N.; Ward, D.S.; Kloster, S.; Flanner, M.G.; Heald, C.L.; Heavens, N.G.; Hess, P.G.; Lamarque, J.-F.; Chuang, P.Y. Aerosol Impacts on Climate and Biogeochemistry. Annu. Rev. Environ. Resour. 2011, 36, 45–74. [Google Scholar] [CrossRef]
- Simó, R. Production of Atmospheric Sulfur by Oceanic Plankton: Biogeochemical, Ecological and Evolutionary Links. Trends Ecol. Evol. 2001, 16, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.S.; Fadnavis, S.; Thomas, M.A.; Pozzoli, L.; Gupta, S.; Royer, S.-J.; Saiz-Lopez, A.; Simó, R. Quantifying the Impacts of an Updated Global Dimethyl Sulfide Climatology on Cloud Microphysics and Aerosol Radiative Forcing. J. Geophys. Res. Atmos. 2015, 120, 2524–2536. [Google Scholar] [CrossRef]
- Choi, Y.N.; Song, S.K.; Lee, S.H.; Moon, J.H. Estimation of Marine Dimethyl Sulfide Emissions from East Asian Seas and Their Impact on Natural Direct Radiative Forcing. Atmos. Environ. 2020, 222, 117165. [Google Scholar] [CrossRef]
- Woodhouse, M.T.; Mann, G.W.; Carslaw, K.S.; Boucher, O. Sensitivity of Cloud Condensation Nuclei to Regional Changes in Dimethyl-Sulphide Emissions. Atmos. Chem. Phys. 2013, 13, 2723–2733. [Google Scholar] [CrossRef]
- Thornhill, G.; Collins, W.; Olivié, D.; Skeie, R.B.; Archibald, A.; Bauer, S.; Checa-Garcia, R.; Fiedler, S.; Folberth, G.; Gjermundsen, A.; et al. Climate-Driven Chemistry and Aerosol Feedbacks in CMIP6 Earth System Models. Atmos. Chem. Phys. 2021, 21, 1105–1126. [Google Scholar] [CrossRef]
- Muñiz-Unamunzaga, M.; Borge, R.; Sarwar, G.; Gantt, B.; de la Paz, D.; Cuevas, C.A.; Saiz-Lopez, A. The Influence of Ocean Halogen and Sulfur Emissions in the Air Quality of a Coastal Megacity: The Case of Los Angeles. Sci. Total Environ. 2018, 610–611, 1536–1545. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Zhao, J.; Sarwar, G.; Zhou, S.; Chen, Y.; Yang, G.; Saiz-Lopez, A. Regional and Urban-Scale Environmental Influences of Oceanic DMS Emissions over Coastal China Seas. Atmosphere 2020, 11, 849. [Google Scholar] [CrossRef]
- Saarikoski, S.; Reyes, F.; Vázquez, Y.; Tagle, M.; Timonen, H.; Aurela, M.; Carbone, S.; Worsnop, D.R.; Hillamo, R.; Oyola, P. Characterization of Submicron Aerosol Chemical Composition and Sources in the Coastal Area of Central Chile. Atmos. Environ. 2019, 199, 391–401. [Google Scholar] [CrossRef]
- Wohl, C.; Villamayor, J.; Galí, M.; Mahajan, A.S.; Fernández, R.P.; Cuevas, C.A.; Bossolasco, A.; Li, Q.; Kettle, A.J.; Williams, T.; et al. Marine Emissions of Methanethiol Increase Aerosol Cooling in the Southern Ocean. Sci. Adv. 2025, 10, eadq2465. [Google Scholar] [CrossRef]
- Barnes, I.; Hjorth, J.; Mihalopoulos, N. Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in the Atmosphere. Chem. Rev. 2006, 106, 940–975. [Google Scholar] [CrossRef]
- Breider, T.J.; Chipperfield, M.P.; Richards, N.A.D.; Carslaw, K.S.; Mann, G.W.; Spracklen, D.V. Impact of BrO on Dimethylsulfide in the Remote Marine Boundary Layer. Geophys. Res. Lett. 2010, 37, L02807. [Google Scholar] [CrossRef]
- Wilson, C.; Hirst, D.M. Kinetics of Gas Phase Oxidation of Reduced Sulfur Compounds. Prog. React. Kinet. 1996, 21, 69–132. [Google Scholar]
- Zhao, J.; Sarwar, G.; Gantt, B.; Foley, K.; Henderson, B.H.; Pye, H.O.T.; Fahey, K.M.; Kang, D.; Mathur, R.; Zhang, Y.; et al. Impact of Dimethylsulfide Chemistry on Air Quality over the Northern Hemisphere. Atmos. Environ. 2021, 244, 117961. [Google Scholar] [CrossRef]
- Chen, Q.; Sherwen, T.; Evans, M.; Alexander, B. DMS Oxidation and Sulfur Aerosol Formation in the Marine Troposphere: A Focus on Reactive Halogen and Multiphase Chemistry. Atmos. Chem. Phys. 2018, 18, 13617–13637. [Google Scholar] [CrossRef]
- Saiz-Lopez, A.; Fernandez, R.P.; Li, Q.; Cuevas, C.A.; Fu, X.; Kinnison, D.E.; Tilmes, S.; Mahajan, A.S.; Gómez Martín, J.C.; Iglesias-Suarez, F.; et al. Natural Short-Lived Halogens Exert an Indirect Cooling Effect on Climate. Nature 2023, 618, 967–973. [Google Scholar] [CrossRef]
- Lana, A.; Bell, T.G.; Simó, R.; Vallina, S.M.; Ballabrera-Poy, J.; Kettle, A.J.; Dachs, J.; Bopp, L.; Saltzman, E.S.; Stefels, J.; et al. An Updated Climatology of Surface Dimethlysulfide Concentrations and Emission Fluxes in the Global Ocean. Glob. Biogeochem. Cycles 2011, 25, GB1004. [Google Scholar] [CrossRef]
- Keller, M.D. Dimethyl Sulfide Production and Marine Phytoplankton: The Importance of Species Composition and Cell Size. Biol. Oceanogr. 1989, 6, 375–382. [Google Scholar]
- Curson, A.R.J.; Liu, J.; Bermejo Martínez, A.; Green, R.T.; Chan, Y.; Carrión, O.; Williams, B.T.; Zhang, S.-H.; Yang, G.-P.; Bulman Page, P.C.; et al. Dimethylsulfoniopropionate Biosynthesis in Marine Bacteria and Identification of the Key Gene in This Process. Nat. Microbiol. 2017, 2, 17009. [Google Scholar] [CrossRef] [PubMed]
- Gage, D.A.; Rhodes, D.; Nolte, K.D.; Hicks, W.A.; Leustek, T.; Cooper, A.J.L.; Hanson, A.D. A New Route for Synthesis of Dimethylsulphoniopropionate in Marine Algae. Nature 1997, 387, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Liss, P.S.; Slater, P.G. Flux of Gases across the Air-Sea Interface. Nature 1974, 247, 181–184. [Google Scholar] [CrossRef]
- Dacey, J.W.H.; Wakeham, S.G.; Howes, B.L. Henry’s Law Constants for Dimethylsulfide in Freshwater and Seawater. Geophys. Res. Lett. 1984, 11, 991–994. [Google Scholar] [CrossRef]
- McGillis, W.R.; Dacey, J.W.H.; Frew, N.M.; Bock, E.J.; Nelson, R.K. Water-Air Flux of Dimethylsulfide. J. Geophys. Res. Ocean. 2000, 105, 1187–1193. [Google Scholar] [CrossRef]
- Liss, P.S.; Merlivat, L. Air-Sea Gas Exchange Rates: Introduction and Synthesis BT—The Role of Air-Sea Exchange in Geochemical Cycling. In The Role of Air-Sea Exchange in Geochemical Cycling; Buat-Ménard, P., Ed.; Springer: Dordrecht, The Netherlands, 1986; pp. 113–127. ISBN 978-94-009-4738-2. [Google Scholar]
- Wanninkhof, R. Relationship between Wind Speed and Gas Exchange over the Ocean. J. Geophys. Res. Ocean. 1992, 97, 7373–7382. [Google Scholar] [CrossRef]
- Wanninkhof, R.; McGillis, W.R. A Cubic Relationship between Air-Sea CO2 Exchange and Wind Speed. Geophys. Res. Lett. 1999, 26, 1889–1892. [Google Scholar] [CrossRef]
- Nightingale, P.D.; Malin, G.; Law, C.S.; Watson, A.J.; Liss, P.S.; Liddicoat, M.I.; Boutin, J.; Upstill-Goddard, R.C. In Situ Evaluation of Air-Sea Gas Exchange Parameterizations Using Novel Conservative and Volatile Tracers. Glob. Biogeochem. Cycles 2000, 14, 373–387. [Google Scholar] [CrossRef]
- Ho, D.T.; Law, C.S.; Smith, M.J.; Schlosser, P.; Harvey, M.; Hill, P. Measurements of Air-Sea Gas Exchange at High Wind Speeds in the Southern Ocean: Implications for Global Parameterizations. Geophys. Res. Lett. 2006, 33, L16611. [Google Scholar] [CrossRef]
- Marandino, C.A.; de Bruyn, W.J.; Miller, S.D.; Saltzman, E.S. Open Ocean DMS Air/Sea Fluxes over the Eastern South Pacific Ocean. Atmos. Chem. Phys. 2009, 9, 345–356. [Google Scholar] [CrossRef]
- Wanninkhof, R. Relationship between Wind Speed and Gas Exchange over the Ocean Revisited. Limnol. Ocean. Methods 2014, 12, 351–362. [Google Scholar] [CrossRef]
- Joge, S.D.; Mahajan, A.S.; Hulswar, S.; Marandino, C.A.; Galí, M.; Bell, T.G.; Yang, M.; Simó, R. Dimethyl Sulfide (DMS) Climatologies, Fluxes, and Trends—Part 2: Sea–Air Fluxes. Biogeosciences 2024, 21, 4453–4467. [Google Scholar] [CrossRef]
- Saltzman, E.S.; King, D.B.; Holmen, K.; Leck, C. Experimental Determination of the Diffusion Coefficient of Dimethylsulfide in Water. J. Geophys. Res. Ocean. 1993, 98, 16481–16486. [Google Scholar] [CrossRef]
- Jones, A.; Roberts, D.L. An Interactive DMS Emissions Scheme for the Unified Model; Met Office: Exeter, UK, 2004. [Google Scholar]
- Bates, T.S.; Cline, J.D.; Gammon, R.H.; Kelly-Hansen, S.R. Regional and Seasonal Variations in the Flux of Oceanic Dimethylsulfide to the Atmosphere. J. Geophys. Res. Ocean. 1987, 92, 2930–2938. [Google Scholar] [CrossRef]
- Erickson III, D.J.; Ghan, S.J.; Penner, J.E. Global Ocean-to-Atmosphere Dimethyl Sulfide Flux. J. Geophys. Res. Atmos. 1990, 95, 7543–7552. [Google Scholar] [CrossRef]
- Andreae, M.O. Ocean-Atmosphere Interactions in the Global Biogeochemical Sulfur Cycle. Mar. Chem. 1990, 30, 1–29. [Google Scholar] [CrossRef]
- Belviso, S.; Bopp, L.; Moulin, C.; Orr, J.C.; Anderson, T.R.; Aumont, O.; Chu, S.; Elliott, S.; Maltrud, M.E.; Simó, R. Comparison of Global Climatological Maps of Sea Surface Dimethyl Sulfide. Glob. Biogeochem. Cycles 2004, 18, GB3013. [Google Scholar] [CrossRef]
- Kettle, A.J.; Andreae, M.O.; Amouroux, D.; Andreae, T.W.; Bates, T.S.; Berresheim, H.; Bingemer, H.; Boniforti, R.; Curran, M.A.J.; DiTullio, G.R.; et al. A Global Database of Sea Surface Dimethylsulfide (DMS) Measurements and a Procedure to Predict Sea Surface DMS as a Function of Latitude, Longitude, and Month. Glob. Biogeochem. Cycles 1999, 13, 399–444. [Google Scholar] [CrossRef]
- Kettle, A.J.; Andreae, M.O. Flux of Dimethylsulfide from the Oceans: A Comparison of Updated Data Sets and Flux Models. J. Geophys. Res. Atmos. 2000, 105, 26793–26808. [Google Scholar] [CrossRef]
- Chen, G.; Davis, D.; Kasibhatla, P.; Bandy, A.; Thornton, D.; Blake, D. A Mass-Balance/Photochemical Assessment of DMS Sea-to-Air Flux as Inferred from NASA GTE PEM-West A and B Observations. J. Geophys. Res. Atmos. 1999, 104, 5471–5482. [Google Scholar] [CrossRef]
- Davis, D.; Chen, G.; Bandy, A.; Thornton, D.; Eisele, F.; Mauldin, L.; Tanner, D.; Lenschow, D.; Fuelberg, H.; Huebert, B.; et al. Dimethyl Sulfide Oxidation in the Equatorial Pacific: Comparison of Model Simulations with Field Observations for DMS, SO2, H2SO4(g), MSA(g), MS and NSS. J. Geophys. Res. Atmos. 1999, 104, 5765–5784. [Google Scholar] [CrossRef]
- Shon, Z.-H.; Kim, K.-H.; Swan, H.; Lee, G.; Kim, Y.-K. DMS Photochemistry during the Asian Dust-Storm Period in the Spring of 2001: Model Simulations vs. Field Observations. Chemosphere 2005, 58, 149–161. [Google Scholar] [CrossRef]
- Ogunro, O.O.; Elliott, S.M.; Wingenter, O.W.; Deal, C.; Fu, W.; Collier, N.; Hoffman, F.M. Evaluating Uncertainties in Marine Biogeochemical Models: Benchmarking Aerosol Precursors. Atmosphere 2018, 9, 184. [Google Scholar] [CrossRef]
- Wang, W.L.; Song, G.; Primeau, F.; Saltzman, E.S.; Bell, T.G.; Moore, K.K. Global Ocean Dimethyl Sulfide Climatology Estimated from Observations and an Artificial Neural Network. Biogeosciences 2020, 17, 5335–5354. [Google Scholar] [CrossRef]
- Galí, M.; Levasseur, M.; Devred, E.; Simó, R.; Babin, M. Sea-Surface Dimethylsulfide (DMS) Concentration from Satellite Data at Global and Regional Scales. Biogeosciences 2018, 15, 3497–3519. [Google Scholar] [CrossRef]
- Bock, J.; Michou, M.; Nabat, P.; Abe, M.; Mulcahy, J.P.; Olivié, D.J.L.; Schwinger, J.; Suntharalingam, P.; Tjiputra, J.; Van Hulten, M.; et al. Evaluation of Ocean Dimethylsulfide Concentration and Emission in CMIP6 Models. Biogeosciences 2021, 18, 3823–3860. [Google Scholar] [CrossRef]
- Grell, G.A.; Peckham, S.E.; Schmitz, R.; McKeen, S.A.; Frost, G.; Skamarock, W.C.; Eder, B. Fully Coupled “Online” Chemistry within the WRF Model. Atmos. Environ. 2005, 39, 6957–6975. [Google Scholar] [CrossRef]
- Byun, D.; Schere, K.L. Review of the Governing Equations, Computational Algorithms, and Other Components of the Models-3 Community Multiscale Air Quality (CMAQ) Modeling System. Appl. Mech. Rev. 2006, 59, 51–77. [Google Scholar] [CrossRef]
- Baklanov, A.; Korsholm, U.; Mahura, A.; Petersen, C.; Gross, A. ENVIRO-HIRLAM: On-Line Coupled Modelling of Urban Meteorology and Air Pollution. Adv. Sci. Res. 2008, 2, 41–46. [Google Scholar] [CrossRef]
- Gong, S.L.; Lavoué, D.; Zhao, T.L.; Huang, P.; Kaminski, J.W. GEM-AQ/EC, an on-Line Global Multi-Scale Chemical Weather Modelling System: Model Development and Evaluation of Global Aerosol Climatology. Atmos. Chem. Phys. 2012, 12, 8237–8256. [Google Scholar] [CrossRef]
- Kazil, J.; Wang, H.; Feingold, G.; Clarke, A.D.; Snider, J.R.; Bandy, A.R. Modeling Chemical and Aerosol Processes in the Transition from Closed to Open Cells during VOCALS-REx. Atmos. Chem. Phys. 2011, 11, 7491–7514. [Google Scholar] [CrossRef]
- Mueller, S.F.; Mao, Q.; Mallard, J.W. Modeling Natural Emissions in the Community Multiscale Air Quality (CMAQ) Model—Part 2: Modifications for Simulating Natural Emissions. Atmos. Chem. Phys. 2011, 11, 293–320. [Google Scholar] [CrossRef]
- Kloster, S.; Feichter, J.; Maier-Reimer, E.; Six, K.D.; Stier, P.; Wetzel, P. DMS Cycle in the Marine Ocean-Atmosphere System—A Global Model Study. Biogeosciences 2006, 3, 29–51. [Google Scholar] [CrossRef]
- Rizza, U.; Barnaba, F.; Miglietta, M.M.; Mangia, C.; Di Liberto, L.; Dionisi, D.; Costabile, F.; Grasso, F.; Gobbi, G.P. WRF-Chem Model Simulations of a Dust Outbreak over the Central Mediterranean and Comparison with Multi-Sensor Desert Dust Observations. Atmos. Chem. Phys. 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Eltahan, M.; Shokr, M.; Sherif, A.O. Simulation of Severe Dust Events over Egypt Using Tuned Dust Schemes in Weather Research Forecast (WRF-Chem). Atmosphere 2018, 9, 246. [Google Scholar] [CrossRef]
- Singh, P.; Sarawade, P.; Adhikary, B. Transport of Black Carbon from Planetary Boundary Layer to Free Troposphere during the Summer Monsoon over South Asia. Atmos. Res. 2020, 235, 104761. [Google Scholar] [CrossRef]
- Ukhov, A.; Mostamandi, S.; Krotkov, N.; Flemming, J.; da Silva, A.; Li, C.; Fioletov, V.; McLinden, C.; Anisimov, A.; Alshehri, Y.M.; et al. Study of SO2 Pollution in the Middle East Using MERRA-2, CAMS Data Assimilation Products, and High-Resolution WRF-Chem Simulations. J. Geophys. Res. Atmos. 2020, 125, e2019JD031993. [Google Scholar] [CrossRef]
- Nurzahziani; Surussavadee, C.; Noosook, T. High-Resolution Biomass Burning Aerosol Transport Simulations in the Tropics. Atmosphere 2020, 11, 91. [Google Scholar] [CrossRef]
- Saide, P.E.; Spak, S.N.; Carmichael, G.R.; Mena-Carrasco, M.A.; Yang, Q.; Howell, S.; Leon, D.C.; Snider, J.R.; Bandy, A.R.; Collett, J.L.; et al. Evaluating WRF-Chem Aerosol Indirect Effects in Southeast Pacific Marine Stratocumulus during VOCALS-REx. Atmos. Chem. Phys. 2012, 12, 3045–3064. [Google Scholar] [CrossRef]
- George, R.C.; Wood, R.; Bretherton, C.S.; Painter, G. Development and Impact of Hooks of High Droplet Concentration on Remote Southeast Pacific Stratocumulus. Atmos. Chem. Phys. 2013, 13, 6305–6328. [Google Scholar] [CrossRef]
- Lowe, D.; Archer-Nicholls, S.; Morgan, W.; Allan, J.; Utembe, S.; Ouyang, B.; Aruffo, E.; Le Breton, M.; Zaveri, R.A.; Di Carlo, P.; et al. WRF-Chem Model Predictions of the Regional Impacts of N2O5 Heterogeneous Processes on Night-Time Chemistry over North-Western Europe. Atmos. Chem. Phys. 2015, 15, 1385–1409. [Google Scholar] [CrossRef]
- Li, S.; Sarwar, G.; Zhao, J.; Zhang, Y.; Zhou, S.; Chen, Y.; Yang, G.; Saiz-Lopez, A. Modeling the Impact of Marine DMS Emissions on Summertime Air Quality over the Coastal East China Seas. Earth Space Sci. 2020, 7, e2020EA001220. [Google Scholar] [CrossRef]
- Song, S.K.; Shon, Z.H.; Bae, M.S.; Cho, S.B.; Moon, S.H.; Kim, H.S.; Son, Y.B.; Lee, C. Effects of Natural and Anthropogenic Emissions on the Composition and Toxicity of Aerosols in the Marine Atmosphere. Sci. Total Environ. 2022, 806, 150928. [Google Scholar] [CrossRef]
- Cho, S.-B.; Song, S.-K.; Shon, Z.-H.; Moon, S.-H. Evaluation of Air Quality Simulation with a Coupled Atmosphere-Ocean Model: A Case Study on Natural Marine and Biogenic Emissions. Sci. Total Environ. 2023, 882, 163021. [Google Scholar] [CrossRef]
- Sarwar, G.; Kang, D.; Henderson, B.H.; Hogrefe, C.; Appel, W.; Mathur, R. Examining the Impact of Dimethyl Sulfide Emissions on Atmospheric Sulfate over the Continental U.S. Atmosphere 2023, 14, 660. [Google Scholar] [CrossRef]
- Fiddes, S.L.; Woodhouse, M.T.; Utembe, S.; Schofield, R.; Alexander, S.P.; Alroe, J.; Chambers, S.D.; Chen, Z.; Cravigan, L.; Dunne, E.; et al. The Contribution of Coral-Reef-Derived Dimethyl Sulfide to Aerosol Burden over the Great Barrier Reef: A Modelling Study. Atmos. Chem. Phys. 2022, 22, 2419–2445. [Google Scholar] [CrossRef]
- Marelle, L.; Raut, J.-C.; Law, K.S.; Berg, L.K.; Fast, J.D.; Easter, R.C.; Shrivastava, M.; Thomas, J.L. Improvements to the WRF-Chem 3.5.1 Model for Quasi-Hemispheric Simulations of Aerosols and Ozone in the Arctic. Geosci. Model. Dev. 2017, 10, 3661–3677. [Google Scholar] [CrossRef]
- Marelle, L.; Thomas, J.L.; Raut, J.-C.; Law, K.S.; Jalkanen, J.-P.; Johansson, L.; Roiger, A.; Schlager, H.; Kim, J.; Reiter, A.; et al. Air Quality and Radiative Impacts of Arctic Shipping Emissions in the Summertime in Northern Norway: From the Local to the regional Scale. Atmos. Chem. Phys. 2016, 16, 2359–2379. [Google Scholar] [CrossRef]
- Lefèvre, J.; Menkes, C.; Bani, P.; Marchesiello, P.; Curci, G.; Grell, G.A.; Frouin, R. Distribution of Sulfur Aerosol Precursors in the SPCZ Released by Continuous Volcanic Degassing at Ambrym, Vanuatu. J. Volcanol. Geotherm. Res. 2016, 322, 76–104. [Google Scholar] [CrossRef]
- Zaveri, R.A. Development and Evaluation of a Comprehensive Tropospheric Chemistry Model for Regional and Global Applications Development and Evaluation of a Comprehensive Tropospheric Chemistry Model for Regional and Global Applications. Ph.D. Thesis, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA, 1997. [Google Scholar]
- Zaveri, R.A.; Easter, R.C.; Fast, J.D.; Peters, L.K. Model for Simulating Aerosol Interactions and Chemistry (MOSAIC). J. Geophys. Res. Atmos. 2008, 113, D13204. [Google Scholar] [CrossRef]
- Baklanov, A.; Smith Korsholm, U.; Nuterman, R.; Mahura, A.; Nielsen, K.P.; Sass, B.H.; Rasmussen, A.; Zakey, A.; Kaas, E.; Kurganskiy, A.; et al. Enviro-HIRLAM Online Integrated Meteorology—Chemistry Modelling System: Strategy, Methodology, Developments and Applications (v7.2). Geosci. Model. Dev. 2017, 10, 2971–2999. [Google Scholar] [CrossRef]
- Tarrasón, L.; Turner, S.; Fløisand, I. Estimation of Seasonal Dimethyl Sulphide Fluxes over the North Atlantic Ocean and Their Contribution to European Pollution Levels. J. Geophys. Res. Atmos. 1995, 100, 11623–11639. [Google Scholar] [CrossRef]
- Lin, C.; Heal, M.R.; Vieno, M.; MacKenzie, I.A.; Armstrong, B.G.; Butland, B.K.; Milojevic, A.; Chalabi, Z.; Atkinson, R.W.; Stevenson, D.S.; et al. Spatiotemporal Evaluation of EMEP4UK-WRF v4.3 Atmospheric Chemistry Transport Simulations of Health-Related Metrics for NO2, O3, PM10, and PM2.5 for 2001–2010. Geosci. Model. Dev. 2017, 10, 1767–1787. [Google Scholar] [CrossRef]
- Ghahreman, R.; Gong, W.; Galí, M.; Norman, A.L.; Beagley, S.R.; Akingunola, A.; Zheng, Q.; Lupu, A.; Lizotte, M.; Levasseur, M.; et al. Dimethyl Sulfide and Its Role in Aerosol Formation and Growth in the Arctic Summer—A Modelling Study. Atmos. Chem. Phys. 2019, 19, 14455–14476. [Google Scholar] [CrossRef]
- Ghahreman, R.; Gong, W.; Beagley, S.R.; Akingunola, A.; Makar, P.A.; Leaitch, W.R. Modeling Aerosol Effects on Liquid Clouds in the Summertime Arctic. J. Geophys. Res. Atmos. 2021, 126, e2021JD034962. [Google Scholar] [CrossRef]
- Appel, K.W.; Bash, J.O.; Fahey, K.M.; Foley, K.M.; Gilliam, R.C.; Hogrefe, C.; Hutzell, W.T.; Kang, D.; Mathur, R.; Murphy, B.N.; et al. The Community Multiscale Air Quality (CMAQ) Model Versions 5.3 and 5.3.1: System Updates and Evaluation. Geosci. Model. Dev. 2021, 14, 2867–2897. [Google Scholar] [CrossRef]
- Hynes, A.J.; Wine, P.H.; Semmes, D.H. Kinetics and Mechanism of Hydroxyl Reactions with Organic Sulfides. J. Phys. Chem. 1986, 90, 4148–4156. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, R.F.; Kerr, J.A.; Troe, J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Supplement IV. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data 1992, 21, 1125–1568. [Google Scholar] [CrossRef]
- Simó, R.; Dachs, J. Global Ocean Emission of Dimethylsulfide Predicted from Biogeophysical Data. Glob. Biogeochem. Cycles 2002, 16, 26-1–26-10. [Google Scholar] [CrossRef]
- Hezel, P.J.; Alexander, B.; Bitz, C.M.; Steig, E.J.; Holmes, C.D.; Yang, X.; Sciare, J. Modeled Methanesulfonic Acid (MSA) Deposition in Antarctica and Its Relationship to Sea Ice. J. Geophys. Res. Atmos. 2011, 116, D23214. [Google Scholar] [CrossRef]
- Gray, B.A.; Wang, Y.; Gu, D.; Bandy, A.; Mauldin, L.; Clarke, A.; Alexander, B.; Davis, D.D. Sources, Transport, and Sinks of SO2 over the Equatorial Pacific during the Pacific Atmospheric Sulfur Experiment. J. Atmos. Chem. 2011, 68, 27–53. [Google Scholar] [CrossRef]
- Leaitch, W.R.; Sharma, S.; Huang, L.; Toom-Sauntry, D.; Chivulescu, A.; Macdonald, A.M.; Von Salzen, K.; Pierce, J.R.; Bertram, A.K.; Schroder, J.C.; et al. Dimethyl Sulfide Control of the Clean Summertime Arctic Aerosol and Cloud. Elementa 2013, 1, 000017. [Google Scholar] [CrossRef]
- Mungall, E.L.; Croft, B.; Lizotte, M.; Thomas, J.L.; Murphy, J.G.; Levasseur, M.; Martin, R.V.; Wentzell, J.J.B.; Liggio, J.; Abbatt, J.P.D. Dimethyl Sulfide in the Summertime Arctic Atmosphere: Measurements and Source Sensitivity Simulations. Atmos. Chem. Phys. 2016, 16, 6665–6680. [Google Scholar] [CrossRef]
- Ghahremaninezhad, R.; Norman, A.-L.; Croft, B.; Martin, R.V.; Pierce, J.R.; Burkart, J.; Rempillo, O.; Bozem, H.; Kunkel, D.; Thomas, J.L.; et al. Boundary Layer and Free-Tropospheric Dimethyl Sulfide in the Arctic Spring and Summer. Atmos. Chem. Phys. 2017, 17, 8757–8770. [Google Scholar] [CrossRef]
- Hodshire, A.L.; Campuzano-Jost, P.; Kodros, J.K.; Croft, B.; Nault, B.A.; Schroder, J.C.; Jimenez, J.L.; Pierce, J.R. The Potential Role of Methanesulfonic Acid (MSA) in Aerosol Formation and Growth and the Associated Radiative Forcings. Atmos. Chem. Phys. 2019, 19, 3137–3160. [Google Scholar] [CrossRef]
- Meng, J.; Martin, R.V.; Li, C.; van Donkelaar, A.; Tzompa-Sosa, Z.A.; Yue, X.; Xu, J.-W.; Weagle, C.L.; Burnett, R.T. Source Contributions to Ambient Fine Particulate Matter for Canada. Environ. Sci. Technol. 2019, 53, 10269–10278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ma, W.; Bilsback, K.R.; Pierce, J.R.; Zhou, S.; Chen, Y.; Yang, G.; Zhang, Y. Simulating the Radiative Forcing of Oceanic Dimethylsulfide (DMS) in Asia Based on Machine Learning Estimates. Atmos. Chem. Phys. 2022, 22, 9583–9600. [Google Scholar] [CrossRef]
- Kim, A.-H.; Yum, S.S.; Lee, H.; Chang, D.Y.; Shim, S. Polar Cooling Effect Due to Increase of Phytoplankton and Dimethyl-Sulfide Emission. Atmosphere 2018, 9, 384. [Google Scholar] [CrossRef]
- Bodas-Salcedo, A.; Mulcahy, J.P.; Andrews, T.; Williams, K.D.; Ringer, M.A.; Field, P.R.; Elsaesser, G.S. Strong Dependence of Atmospheric Feedbacks on Mixed-Phase Microphysics and Aerosol-Cloud Interactions in HadGEM3. J. Adv. Model. Earth Syst. 2019, 11, 1735–1758. [Google Scholar] [CrossRef]
- Revell, L.E.; Kremser, S.; Hartery, S.; Harvey, M.; Mulcahy, J.P.; Williams, J.; Morgenstern, O.; McDonald, A.J.; Varma, V.; Bird, L.; et al. The Sensitivity of Southern Ocean Aerosols and Cloud Microphysics to Sea Spray and Sulfate Aerosol Production in the HadGEM3-GA7.1 Chemistry—Climate Model. Atmos. Chem. Phys. 2019, 19, 15447–15466. [Google Scholar] [CrossRef]
- Bhatti, Y.A.; Revell, L.E.; McDonald, A.J. Influences of Antarctic Ozone Depletion on Southern Ocean Aerosols. J. Geophys. Res. Atmos. 2022, 127, e2022JD037199. [Google Scholar] [CrossRef]
- Takemura, T. Return to Different Climate States by Reducing Sulphate Aerosols under Future CO2 Concentrations. Sci. Rep. 2020, 10, 21748. [Google Scholar] [CrossRef]
- Hajima, T.; Watanabe, M.; Yamamoto, A.; Tatebe, H.; Noguchi, M.A.; Abe, M.; Ohgaito, R.; Ito, A.; Yamazaki, D.; Okajima, H.; et al. Development of the MIROC-ES2L Earth System Model and the Evaluation of Biogeochemical Processes and Feedbacks. Geosci. Model. Dev. 2020, 13, 2197–2244. [Google Scholar] [CrossRef]
- Phoenix, D.; Khodayari, A.; Wuebbles, D.; Stewart, K. Aviation Impact on Air Quality Present Day and Mid-Century Simulated in the Community Atmosphere Model (CAM). Atmos. Environ. 2019, 196, 125–132. [Google Scholar] [CrossRef]
- Tjiputra, J.F.; Schwinger, J.; Bentsen, M.; Morée, A.L.; Gao, S.; Bethke, I.; Heinze, C.; Goris, N.; Gupta, A.; He, Y.C.; et al. Ocean Biogeochemistry in the Norwegian Earth System Model Version 2 (NorESM2). Geosci. Model. Dev. 2020, 13, 2393–2431. [Google Scholar] [CrossRef]
- Elliott, S. Dependence of DMS Global Sea-Air Flux Distribution on Transfer Velocity and Concentration Field Type. J. Geophys. Res. Biogeosci 2009, 114, G02001. [Google Scholar] [CrossRef]
- Xu, L.; Cameron-Smith, P.; Russell, L.M.; Ghan, S.J.; Liu, Y.; Elliott, S.; Yang, Y.; Lou, S.; Lamjiri, M.A.; Manizza, M. DMS Role in ENSO Cycle in the Tropics. J. Geophys. Res. Atmos. 2016, 121, 13513–537558. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Smith, S.J.; Easter, R.; Ma, P.-L.; Qian, Y.; Yu, H.; Li, C.; Rasch, P.J. Global Source Attribution of Sulfate Concentration and Direct and Indirect Radiative Forcing. Atmos. Chem. Phys. 2017, 17, 8903–8922. [Google Scholar] [CrossRef]
- Tesdal, J.-E.; Christian, J.R.; Monahan, A.H.; von Salzen, K. Sensitivity of Modelled Sulfate Aerosol and Its Radiative Effect on Climate to Ocean DMS Concentration and Air—Sea Flux. Atmos. Chem. Phys. 2016, 16, 10847–10864. [Google Scholar] [CrossRef]
- Mahmood, R.; von Salzen, K.; Norman, A.-L.; Galí, M.; Levasseur, M. Sensitivity of Arctic Sulfate Aerosol and Clouds to Changes in Future Surface Seawater Dimethylsulfide Concentrations. Atmos. Chem. Phys. 2019, 19, 6419–6435. [Google Scholar] [CrossRef]
- Xausa, F.; Paasonen, P.; Makkonen, R.; Arshinov, M.; Ding, A.; Denier Van Der Gon, H.; Kerminen, V.-M.; Kulmala, M. Advancing Global Aerosol Simulations with Size-Segregated Anthropogenic Particle Number Emissions. Atmos. Chem. Phys. 2018, 18, 10039–10054. [Google Scholar] [CrossRef]
- Hoffmann, E.H.; Heinold, B.; Kubin, A.; Tegen, I.; Herrmann, H. The Importance of the Representation of DMS Oxidation in Global Chemistry-Climate Simulations. Geophys. Res. Lett. 2021, 48, e2021GL094068. [Google Scholar] [CrossRef]
- Khan, M.A.H.; Gillespie, S.M.P.; Razis, B.; Xiao, P.; Davies-Coleman, M.T.; Percival, C.J.; Derwent, R.G.; Dyke, J.M.; Ghosh, M.V.; Lee, E.P.F.; et al. A Modelling Study of the Atmospheric Chemistry of DMS Using the Global Model, STOCHEM-CRI. Atmos. Environ. 2016, 127, 69–79. [Google Scholar] [CrossRef]
- Chin, M.; Ginoux, P.; Kinne, S.; Torres, O.; Holben, B.N.; Duncan, B.N.; Martin, R.V.; Logan, J.A.; Higurashi, A.; Nakajima, T. Tropospheric Aerosol Optical Thickness from the GOCART Model and Comparisons with Satellite and Sun Photometer Measurements. J. Atmos. Sci. 2002, 59, 461–483. [Google Scholar] [CrossRef]
- Burkholder, J.B.; Sander, S.P.; Abbatt, J.P.D.; Barker, J.R.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; Orkin, V.L.; Wilmouth, D.M.; Wine, P.H. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies. Evaluation Number 18; NASA Jet Propulsion Laboratory (JPL) Publication: La Cañada Flintridge, CA, USA, 2015. [Google Scholar]

{kind=link}
| Parameterization and Reference | Type | Equation |
|---|---|---|
| LM86 [26] | Linear | |
| W92 [27] | Quadratic | |
| WM99 [28] | Cubic | |
| N00 [29] | Quadratic | |
| H06 [30] | Quadratic | |
| M09 [31] | Linear | |
| W14 [32] | Quadratic |
| Model | Source of the DMS Emission Inventory | DMS Chemistry | Main Findings | Reference |
|---|---|---|---|---|
| CMAQ (v4.6) | Constant value from [55]. | Reaction with radicals OH and NO3 and halogen Cl. | The aerosol sulfate increased by nearly 2 µg·m3 in ocean and 0.1–0.2 µg·m3 in inland areas, attributable to DMS emissions and its chemistry. | [56] |
| CMAQ (v5.1) | DMS seawater concentration from [19] using the parameterization of LM86 [26]. | Reaction with halogen compounds BrO and IO. | An increase of 10% in secondary organic aerosol mean concentration due to aerosol acidity and sulfate aerosol formation. Otherwise, the ozone and nitrogen dioxide (NO2) concentrations diminished up to 5 ppbv and 2.5 ppbv. | [9] |
| CMAQ (v5.2) | DMS seawater concentration from [19] and local Chinese database using the parameterization of LM86, N00, W92 [26,27,29]. | Reaction with radicals OH and NO3 and halogen Cl. Also, the reactions with halogen compounds IO, BrO, and ClO were considered. | The inclusion of marine emissions of DMS and halogens increased by 4% the formation of sulfate aerosols on the coast and by 9% at sea. | [64] |
| CMAQ (v5.2) | DMS seawater concentration from [19] and local Chinese database using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3 and halogen Cl. Also, the reactions with halogen compounds IO, BrO, and ClO were considered. | The DMS emissions increased SO2 concentrations with the highest contribution in summer (0.63 μg/m3) and lowest in winter (0.12 μg/m3). The contribution of DMS to the SO2 levels in Shanghai was mostly around 0.08 μg/m3. The inclusion of DMS reduced O3 relatively larger in the open sea than in the urban area of Shanghai. | [10] |
| CMAQ (v5.3) | DMS seawater concentration from [19] using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3 and halogen Cl. Also, the reactions with halogen compounds IO, BrO, and ClO were considered. | Total of 63.5% of DMS was oxidized by OH (33.0% via the abstraction channel and 30.5% via the addition channel). Also, the oxidation of DMS by NO3 accounted for 11.8%. Therefore, BrO, Cl, IO, and ClO oxidation pathways contributed 16.0%, 8.2%, 0.4%, and 0.1%, respectively, to the total DMS oxidation. | [16] |
| CMAQ (v5.3.2) | DMS concentration was estimated using a DMS empirical algorithm constructed using chlorophyll a (Chl-a) concentrations, mixing layer depth, and using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3 and halogen Cl. Also, the reactions with halogen compounds IO, BrO, and ClO were considered. | DMS contribution to the total PM2.5 mass was approximately 1%, showing more influence in the coastal area than the open ocean. | [65] |
| CMAQ (v5.3.2) | DMS concentration was estimated using a DMS empirical algorithm constructed using chlorophyll a (Chl-a) concentrations, mixing layer depth, and using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3 and halogen Cl. Also, the reactions with halogen compounds IO, BrO, and ClO were considered. | Significant contributions of DMS emissions to PM2.5 concentrations in coastal areas. In addition, the mean contributions of DMS oxidation to SO2 concentrations over marine and coastal areas were approximately 14.5% and 13.1%, respectively | [66] |
| CMAQ (v5.4) | DMS seawater concentration from [19] using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3 and halogen Cl. The reactions of DMS with BrO and IO were not included in this study. | DMS emissions increased annual mean sulfate by 0.055 µg/m3 over the land area of the modeling domain. The inclusion of DMS emissions increased sulfate concentrations by 36% over seawater and 9% over land. | [67] |
| WRF-Chem | Not reported. | Chemical reactions reported in [72]. | Added the coral-reef-derived DMS contribution, showing insignificant change (around 1%) to the total sulfate aerosol mass. | [73] |
| WRF-Chem | Constant value of DMS concentration in the seawater from VOCALS-Rex campaign. | Reaction with radicals OH and NO3, without considering the formation of MSA. | The results were consistent with the surface measurement at the shipboard and showed good agreement with aircraft data. | [53] |
| WRF-Chem | Constant value of DMS concentration in the seawater using the parameterization of LM86 [26]. | Chemical reactions reported in [72]. | The study overestimated the DMS emissions due to overestimating the modeled DMS in the ocean due to the atmosphere transfer velocity, generating high biases in the modeled marine boundary layer and increasing the cloud-driven SO2-to-sulfate conversion. | [61] |
| WRF-Chem | Constant value of DMS concentration in the seawater using the parameterization of N00 [29]. | Reaction with radicals OH and NO3, without considering the formation of MSA. | An overestimation of DMS flux was obtained but showed its impact in the albedo. Also, the simulation tests suggested that DMS influenced the aerosol number and size distribution. | [62] |
| WRF-Chem | Constant value without details about the parameterization applied. | Reaction with radicals OH and NO3 and halogen Cl. Also, the reaction with halogen compound BrO was considered. | A nighttime NO3 oxidation of DMS showed its relevance compared to daytime OH reaction. This particular finding probed the biases found before regarding DMS chemistry. | [63] |
| WRF-Chem | DMS seawater concentration from [19,40] using the parameterization of LM86, N00 [26,29]. | Reaction with radicals OH and NO3. | This study updated the climatology and parameterization in the configuration of the WRF-Chem model. The parameterization of N00 showed higher (double) MSA surface concentrations. Meanwhile, large uncertainties were present due to the avoided emissions of BrO from volcanic plumes. | [71] |
| WRF-Chem | Constant value of DMS concentration in the sea using the parameterization of N00 [29]. | Not reported. | Did not analyze the effect of marine emissions. | [70] |
| WRF-Chem | Constant value of DMS concentration in the sea using the parameterization of N00 [29]. | Reaction with radicals OH and NO3. | A high source contribution (90–100%) of DMS to surface SO2 in the Artic was reported. Meanwhile, the inclusion of DMS emissions increased 2–4% the concentrations of PM10. Adding DMS slightly reduced (−2 ppbv) the surface ozone over the ocean, even when several improvements in the performance of the model for O3 and surface sulfate were applied. The simple gas phase chemistry had an impact on overestimation in some cases. | [69] |
| WRF-Chem | GOCART database | Reaction with radicals OH and NO3. | Dust and particulate matter were analyzed, revealing an overestimation of PM2.5 when the dust module was considered, but neither discussion was done about the effect of marine emissions. | [56] |
| WRF-Chem | GOCART database | Reaction with radicals OH and NO3. | Did not analyze the effect of marine emissions. | [57] |
| WRF-Chem | GOCART database | Reaction with radicals OH and NO3. | Did not analyze the effect of marine emissions. | [58] |
| WRF-Chem | GOCART database | Reaction with radicals OH and NO3. | Did not analyze the effect of marine emissions. | [59] |
| WRF-Chem | GOCART database | Reaction with radicals OH and NO3. | Did not analyze the effect of marine emissions. | [60] |
| WRF-Chem | DMS seawater concentration from [19] using the parameterization of LM86 [26]. | Chemical reactions reported in [72]. | First research studying the role of coral-reef-derived DMS at sub-daily timescales. The authors used the version designed with 30 DMS oxidation pathway reactions. The inclusion of coral-reef-derived DMS resulted in no significant change in sulfate aerosol mass or total aerosol number. DMS removal by BrO or Cl2 was not considered. | [68] |
| Enviro-HIRLAM | Used the parameterization of N00 [29] without details about the climatology used. | Reaction with radicals OH and NO3. | An underestimation for PM2.5 was obtained, especially in coastal cities like Bilbao in Spain. Meanwhile, this study did not analyze the effect of DMS. | [74] |
| EMEP4UK-WRF | Monthly emission fields of DMS-derived SO2 were taken from the work of [75]. | Not reported. | Did not analyze the effect of DMS. | [76] |
| GEM-MACH | DMS seawater concentration from [19] and a satellite DMS concentration dataset from [47] using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3. | The addition of DMS compartment to the GEM-MACH model resulted in a significant increase in atmospheric SO2 for some regions of the Canadian Arctic (up to 100%). This study did not consider the impacts of halogens in the DMS chemistry. The climatology data of Lana et al. (2011) [19] do not well reflect the marine source in the Arctic due to the very limited observations available. | [77] |
| GEM-MACH | DMS seawater concentration from [19] and a satellite DMS concentration dataset from [47] using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3. | Dimethyl sulfide oxidation led to increased aerosols between 60 and 200 nm and a 50% increase in droplet number in some regions of the Arctic. | [78] |
| Model | Source of the DMS Emission Inventory | DMS Chemistry | Main Findings | Reference |
|---|---|---|---|---|
| GEOS-Chem | DMS seawater concentration from [19,40,82] using the parameterization of N00 [29]. | Reaction with radicals OH and NO3 and the halogen compound BrO. | A small contribution (11–30%) of sea ice extent to DMS emissions, but this was responsible for 26–62% of MSA deposition at the Antarctic coast and 36–95% inland. Also, the lifetime of DMS and MSA was varied if its reaction with BrO was considered or not. | [83] |
| GEOS-Chem-REAM | Not reported. | Reaction with radical OH and the halogen compound BrO. | By adding 1 pptv BrO, a reduction of 13% to DMS to SO2 conversion efficiency was obtained. | [84] |
| GEOS-Chem-TOMAS | DMS seawater concentration from [40] using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3. | The results showed overprediction of MSA concentrations compared to observed registries at Alert station argued by overestimation of DMS emissions. Other explanations could be associated with missing reactions in the chemical mechanism that could sink DMS in that region. The results showed the main contribution to nuclei cloud droplets (CCNs) from DMS during July–August in the region. | [85] |
| GEOS-Chem | DMS seawater concentration from [19] improved with the 21-day ship track measurements, using the parameterization of LM86 [26]. | The study applied DMS oxidation only by reaction with OH and NO3 according to Equations (1)–(3) following [85]. | The non-marine sources could contribute additional episodes of DMS in the Arctic region, although local marine sources of DMS dominated. Authors observed higher emissions during the monitoring campaign compared to the data reported in [19]. | [86] |
| GEOS-Chem | DMS seawater concentration from [19] improved with the 21-day ship track measurements using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3. | First attempt at comparison between aircraft measurements and simulated DMS at different levels of altitude. The model overpredicted the measurements at lower altitudes (below 1500 m). | [87] |
| GEOS-Chem | DMS seawater concentration from [19,40] without details about the parameterization applied. | Reaction with radicals OH and NO3, with the halogen compound BrO, with the halogen Cl, and with O3 (in multiphase). | The main results suggested a global oxidation of DMS in the gas phase by OH (66%), NO3 (16%), and BrO (12%). This paper denoted the presence (around 86%) of DMS in tropospheric burden below 2 km. The reaction with BrO was considerable in the Southern Ocean and Antarctica during winter. | [17] |
| GEOS-Chem-TOMAS | DMS seawater concentration from [19] without details about the parameterization applied. | Reaction with radicals OH and NO3. | The authors focused on the effect of MSA on the direct and indirect radiative force by the aerosols formed in 2014, obtaining negligible results (<−0.1 W m−2). However, uncertainties in DMS emission inventory and MSA transformation highlight the need for future studies to improve understanding in this area. | [88] |
| GEOS-Chem | The authors did not expose details about the DMS emission inventory. | The authors did not expose the DMS chemistry included in the model. | The results indicated low apportionment of DMS to particulate matter. | [89] |
| GEOS-Chem-TOMAS | DMS seawater concentration from [19] and DMS concentrations predicted by XGBoost using the parameterization of N00 [29]. | The authors did not expose the DMS chemistry included in the model. | DMS contributed around 88% to sulfate in remote oceanic areas. Authors used machine learning techniques to improve the DMS concentrations in seawater for the emission inventory. Meanwhile, the authors suggested more marine and atmospheric observational data for further model evaluation. | [90] |
| HadGEM2-AO | DMS seawater concentration from [41] without details about the parameterization applied. | The authors did not expose the DMS chemistry included in the model. | DMS flux confirmed effects regarding temperature, cloud fraction, and radiation, especially in polar regions. Authors emphasized the urgent need for more studies about the synergy of the phytoplankton activity and the climate responses due to DMS flux. | [91] |
| HadGEM3-GA7.1 | DMS seawater concentration from [19] using the parameterization of LM86 [26]. | The authors did not expose the DMS chemistry included in the model. | The model responses were analyzed with combinations of seawater concentrations of DMS and parameterizations for sea–air gas transfer. Also, the authors used a scenario where the emission of DMS was multiplicated by the factor 1.7. The analysis was restricted to the region between 50° S and 70° S, considering the strong seasonal cycle of DMS emissions. | [92] |
| HadGEM3-GA7.1 | DMS seawater concentration from [19] using the parameterization of LM86 [26]. | Reaction with radicals OH and NO3, with the halogen compound BrO, with the halogen Cl, and with O3 (in multiphase). | The main finding of the study was the inclusion of halogen in the DMS chemistry. As a result, the updated pathway led to a 20% increase in the number concentration of cloud condensation nuclei and cloud droplets, showing better performance with observations, especially in the Southern Ocean between 1989 and 2008. | [93] |
| UKESM1, BCC-ESM1, GFDL-CM4, HadGEM3-GC3.1-LL, MRI-ESM2-0, NorESM2, GFDL-ESM4, MPI-ESM-1-2-HAM, MIROC-ES2L | No details about the climatology data applied, but the authors informed the use of the parameterization of LM86, W92, W14, N00 [26,27,29,32]. | Not reported. | This paper focused on the wind speed changes impact on the DMS flux. Increased wind speeds over the summertime Southern Ocean resulted in increases of 0.82% in atmospheric DMS on average. | [94] |
| MIROC | Not reported. | Not reported. | Did not analyze the effect of marine emissions. | [95] |
| MIROC-ESM2 | Not reported. | Not reported. | Did not analyze the effect of marine emissions. | [96] |
| CAM-Chem (version5) | AeroCom dataset | Not reported. | Did not analyze the effect of marine emissions. | [97] |
| NorESM2 | DMS seawater concentration from [19] with the parameterization of W14 [32]. The flux of DMS was considered only unidirectional from the sea to the air. | Not reported. | Did not analyze the effect of marine emissions. | [98] |
| CESM | Used the method of [99] without details about the seawater concentration applied. | Only the DMS oxidation pathway with OH was used. | Showed the influence of El Niño–Southern Oscillation (ENSO) cycle on the DMS removal process in the air. Also, the authors found that DMS lifetime was about 3 days, and the SO2 formation was 52% from the DMS oxidation, mainly increasing the cloud condensation nuclei formation over the eastern Pacific Ocean. | [100] |
| CESM-CAM5 | Not reported. | Not reported. | This work found the source receptor apportionment of sulfate from 16 regions, being DMS from Southern Hemisphere responsible for 17–84% of the seasonal sulfate. DMS found to account for 16% of global mean sulfate concentrations. | [101] |
| CESM-CAM4 | Not reported. | Reaction with radicals OH and NO3, with the halogen compounds BrO, with the halogen Cl. | The addition of MeSH emissions showed competition with DMS for oxidants. | [12] |
| CanAM4.1 | DMS seawater concentration from [19,40,41] using the parameterizations of LM86 and N00 [26,29]. | Reaction with radicals OH and NO3. | The global mean radiative effect of sulfate found to be approximately linearly proportional to the global mean surface flux of DMS. Included DMS emissions from the terrestrial biosphere, but they were not analyzed for their source effect. Meanwhile, more than 40% of sulfur originating from DMS emissions found to be responsible of the sulfate concentrations in the open ocean. | [102] |
| CanAM4.3 | DMS seawater concentration from [19] using the parameterization of N00 [29]. | Reaction with radicals OH and NO3. | The model reproduced the seasonal variations, but an underestimation of DMS emissions was distinguished due to the omission of the sea ice melting source. Also, the authors found large uncertainties for nucleation parameterizations. The simulation overestimated the DMS source contribution to sulfate aerosol concentration at the Alert station. | [103] |
| ECHAM-HAM | DMS seawater concentration from [55] without details about the parameterization applied. | Not reported. | The results showed no effect of marine emissions during the simulation for one year and focused on anthropogenic emissions. | [104] |
| ECHAM-HAMMOZ | Not reported. | Reaction with radicals OH and NO3, with the halogen Cl. | The sulfate offset found to be higher when the MSA formation potential due to the reactive uptake was lower, that is, higher DMS-to-SO2 oxidation. | [105] |
| STOCHEM-CRI | DMS seawater concentration from [19] without details about the parameterization applied. | Reaction with radicals OH and NO3, with the halogen compound BrO, with the halogen Cl. | One of the main findings was 7.9% of the DMS reacted with BrO. BrO oxidation contributed significantly in the high latitudes of the Southern Hemisphere. A large amount of DMS was removed via reaction with Cl, specifically in the remote Southern Hemisphere ocean. | [106] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pino-Cortés, E.; Martínez, M.; Gómez, K.; Taboada, F.G.; Fu, J.S.; Sarwar, G.; Fernandez, R.P.; Joge, S.D.; Mahajan, A.S.; Höfer, J. Simulating the Fate of Dimethyl Sulfide (DMS) in the Atmosphere: A Review of Emission and Chemical Parameterizations. Atmosphere 2025, 16, 350. https://doi.org/10.3390/atmos16030350
Pino-Cortés E, Martínez M, Gómez K, Taboada FG, Fu JS, Sarwar G, Fernandez RP, Joge SD, Mahajan AS, Höfer J. Simulating the Fate of Dimethyl Sulfide (DMS) in the Atmosphere: A Review of Emission and Chemical Parameterizations. Atmosphere. 2025; 16(3):350. https://doi.org/10.3390/atmos16030350
Chicago/Turabian StylePino-Cortés, Ernesto, Mariela Martínez, Katherine Gómez, Fernando González Taboada, Joshua S. Fu, Golam Sarwar, Rafael P. Fernandez, Sankirna D. Joge, Anoop S. Mahajan, and Juan Höfer. 2025. "Simulating the Fate of Dimethyl Sulfide (DMS) in the Atmosphere: A Review of Emission and Chemical Parameterizations" Atmosphere 16, no. 3: 350. https://doi.org/10.3390/atmos16030350
APA StylePino-Cortés, E., Martínez, M., Gómez, K., Taboada, F. G., Fu, J. S., Sarwar, G., Fernandez, R. P., Joge, S. D., Mahajan, A. S., & Höfer, J. (2025). Simulating the Fate of Dimethyl Sulfide (DMS) in the Atmosphere: A Review of Emission and Chemical Parameterizations. Atmosphere, 16(3), 350. https://doi.org/10.3390/atmos16030350