
Determination of PM1 Sources at a Prague Background Site during the 2012–2013 Period Using PMF Analysis of Combined Aerosol Mass Spectra

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Measurement Site
2.2. Instrumentation and Sampling
2.2.1. Data Preparation
2.3. Data Analysis
3. Results and Discussion
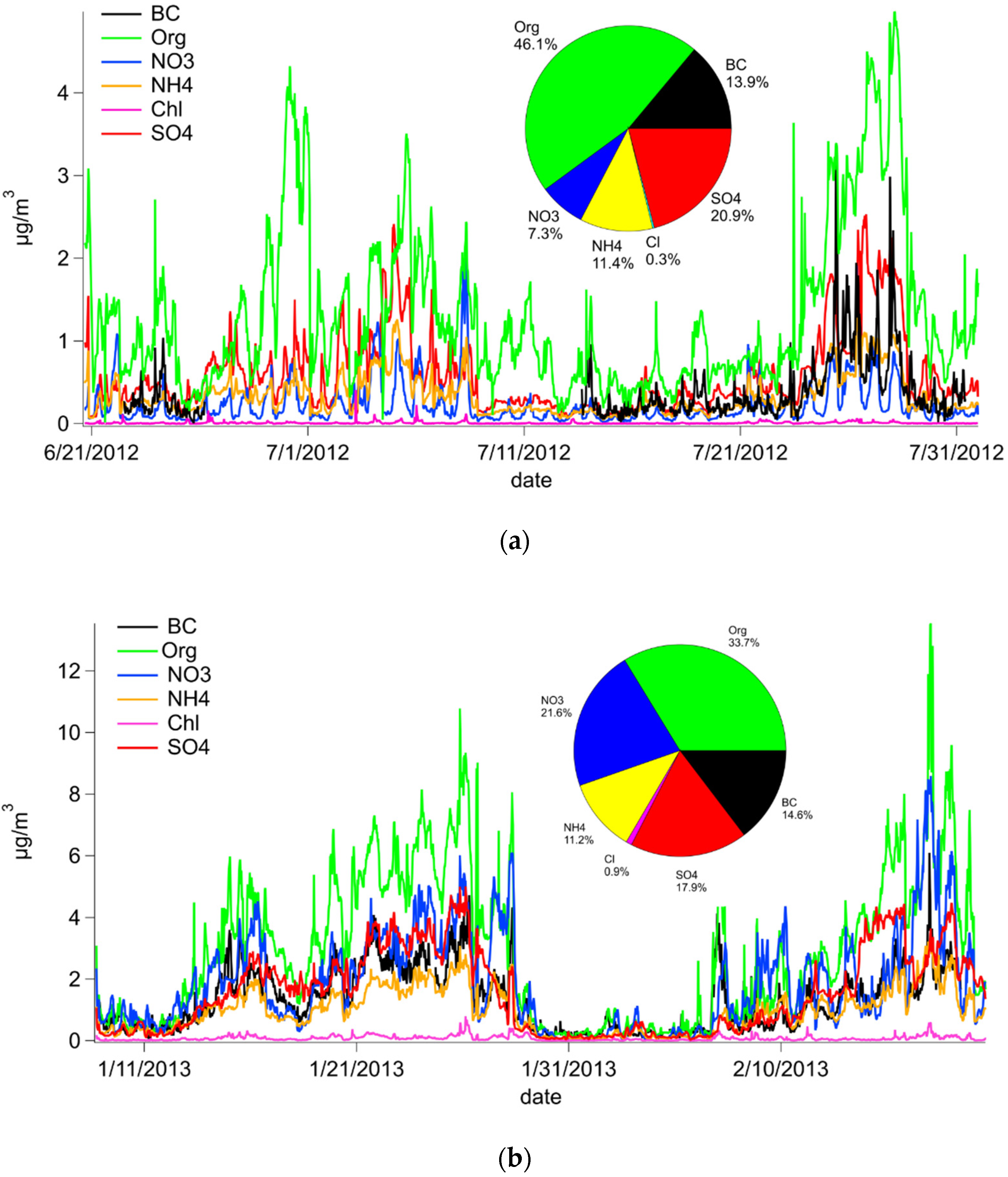
3.1. PM1 Chemical Composition
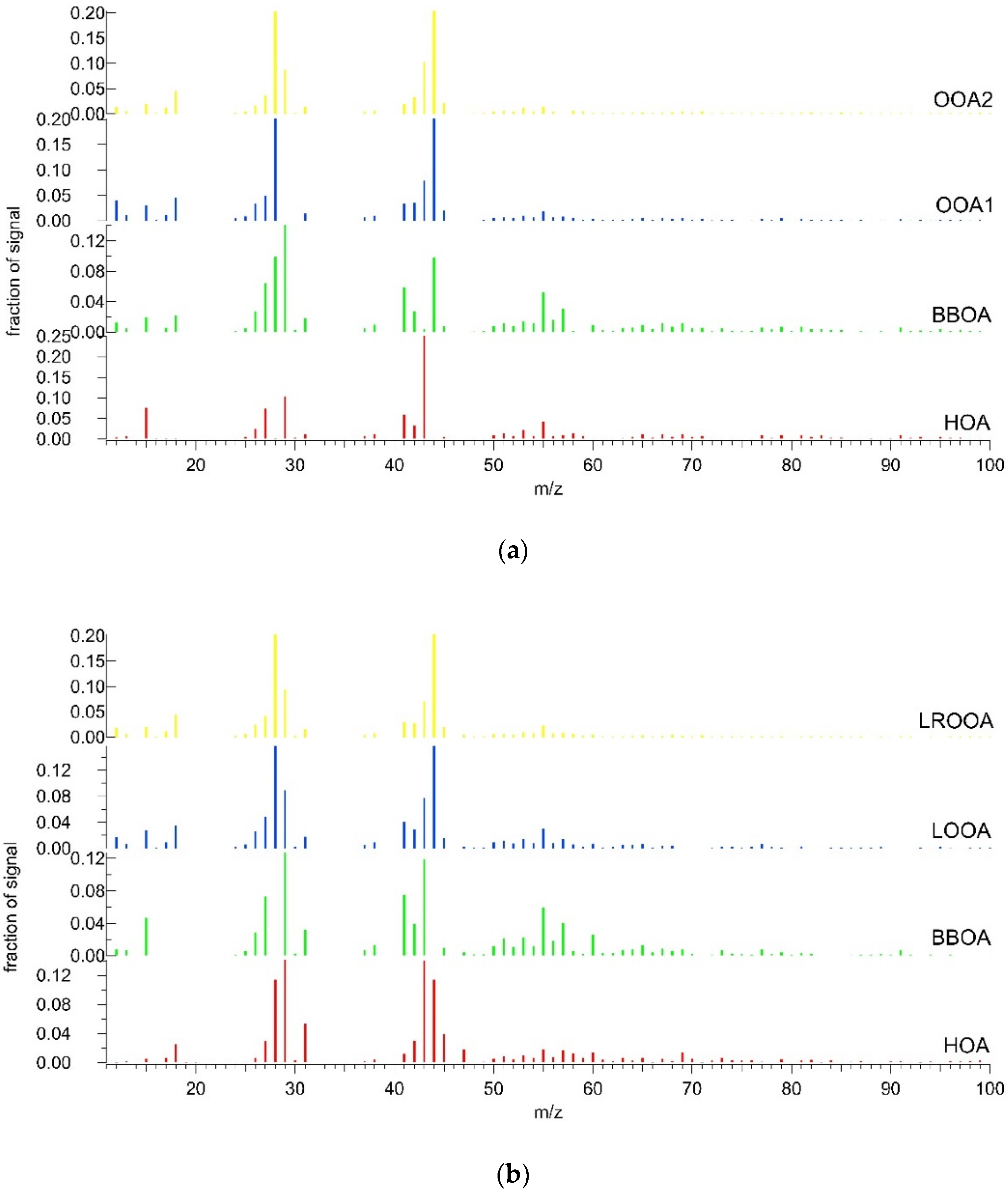
3.2. OA Source Apportionment
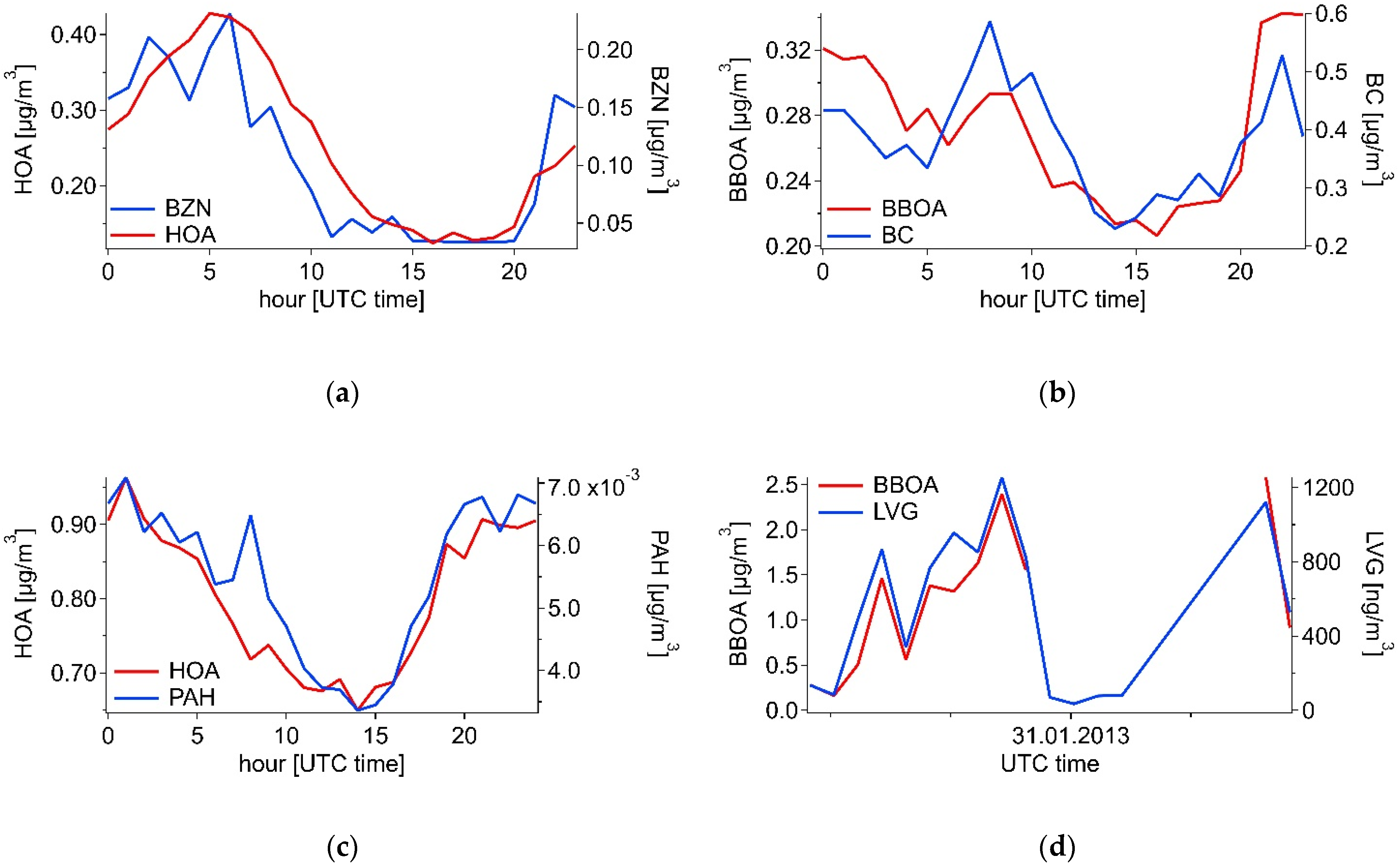
3.2.1. Summer
3.2.2. Winter
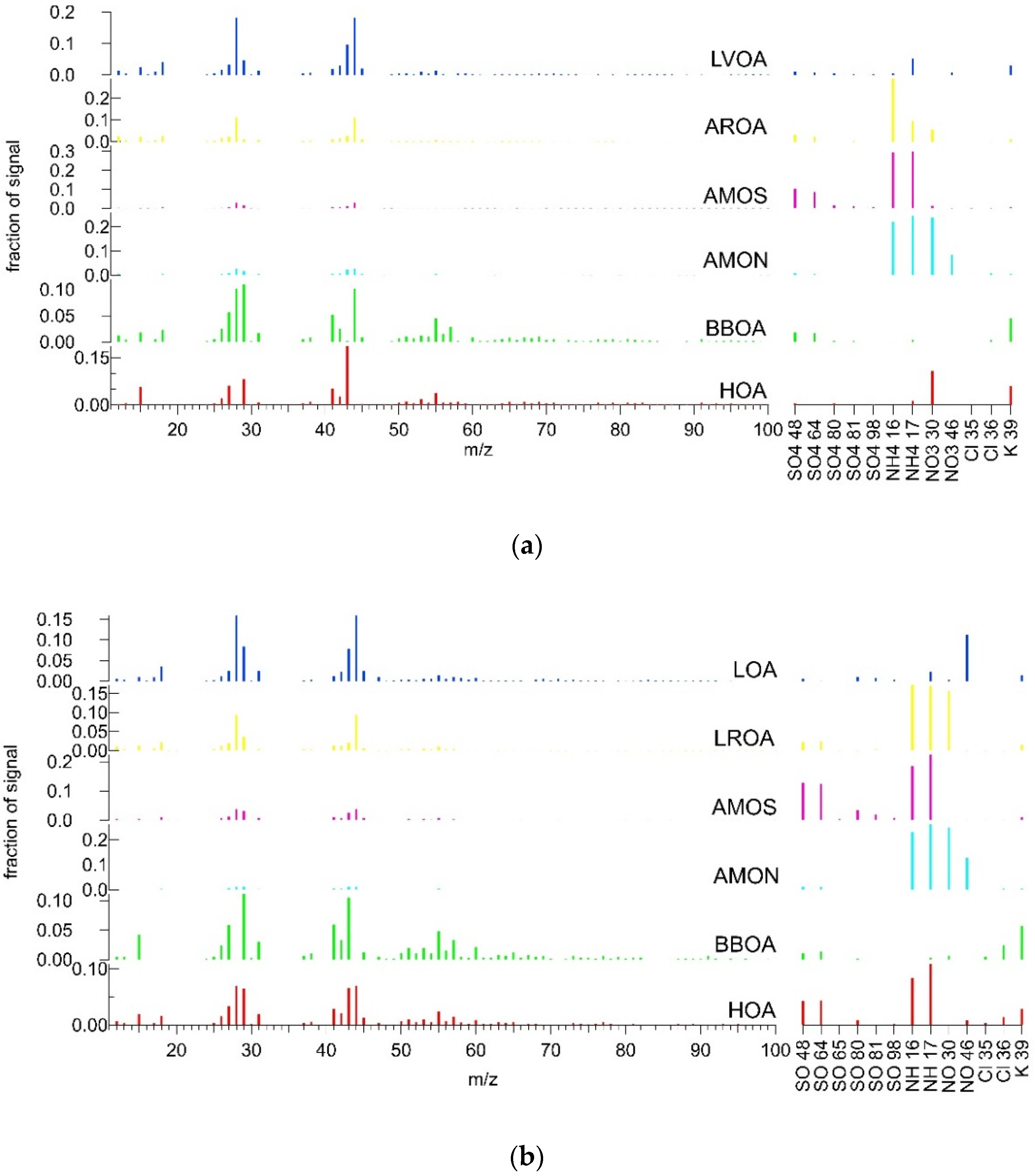
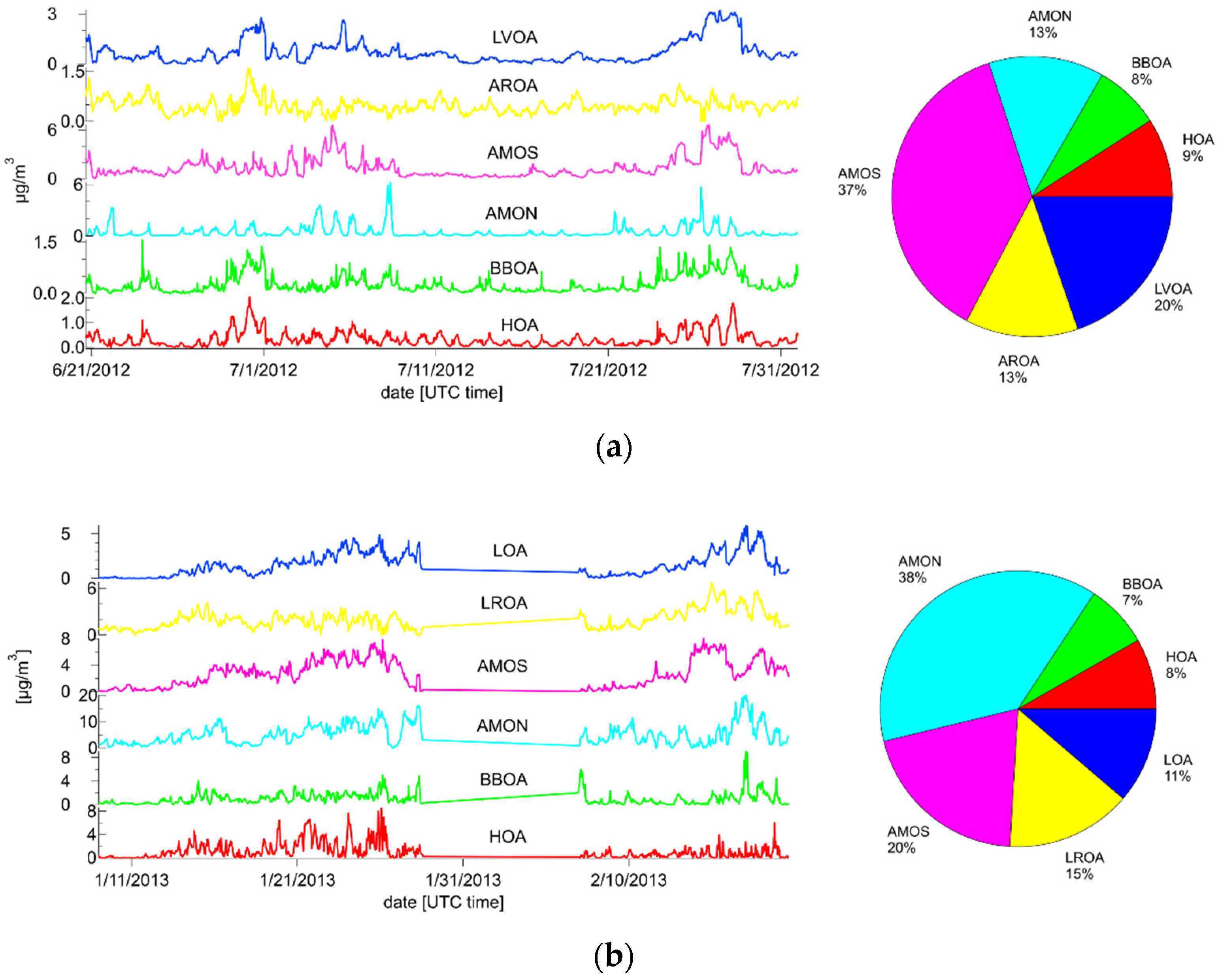
3.3. Combined Spectra Source Apportionment
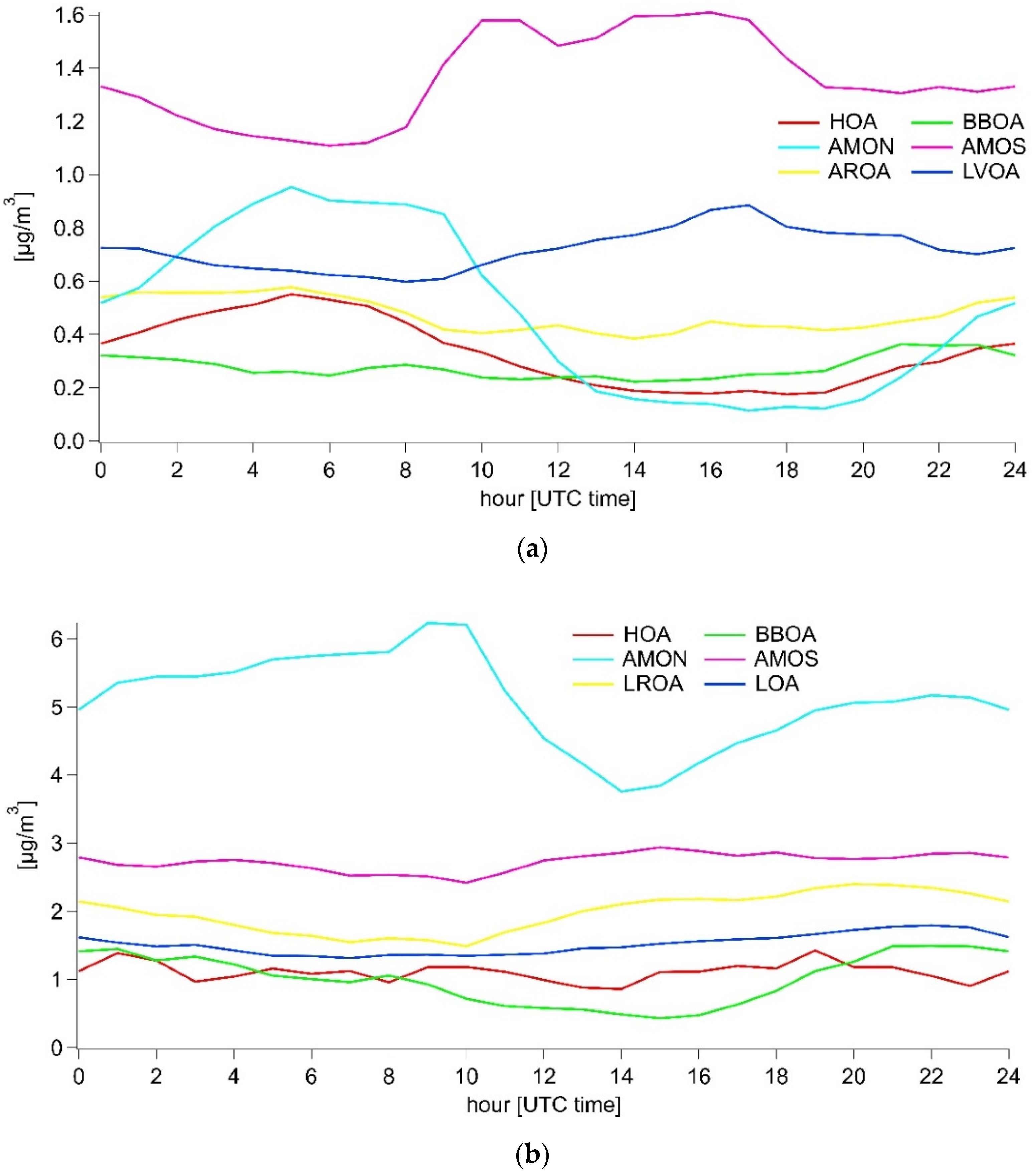
3.3.1. Summer
3.3.2. Winter
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Atmospheric Aerosol |
| AIM | Automated Immission Monitoring |
| AMON | Ammonium Nitrate |
| AMOS | Ammonium Sulphate |
| AMS | Aerosol Mass Spectrometer |
| AROA | Ammonia Rich Oxygenated Aerosol |
| ASL | Above Sea Level |
| BBOA | Biomass Burning Organic Aerosol |
| BC | Black Carbon |
| CE | Collection Efficiency |
| CHMI | Czech Hydrometeorological Institute |
| CMB | Chemical Mass Balance |
| COA | Cooking Organic Aerosol |
| HOA | Hydrocarbon-like Organic Aerosol |
| IA | Inorganic Aerosol |
| LOA | Local Oxygenated Aerosol |
| LOOA | Local Oxygenated Organic Aerosol |
| LROA | Long Range Oxygenated Aerosol |
| LROOA | Long Range Oxygenated Organic Aerosol |
| LVOA | Low Volatile Oxygenated Aerosol |
| ME-2 | Multilinear Engine |
| NR | Non-refractory |
| OA | Organic Aerosol |
| OOA1 | Oxygenated Organic Aerosol 1 |
| OOA2 | Oxygenated Organic Aerosol 2 |
| PAHs | Polycyclic Aromatic Hydrocarbons |
| PCA | Principal Component Analysis |
| PMF | Positive Matrix Factorization |
| PMX | Particulate Matter with diameter smaller than X |
| POA | Primary Organic Aerosol |
| R2 | Coefficient of determination |
| SOA | Secondary Organic Aerosol |
| UMR | Unit Mass Resolution |
References
- Engling, G.; Gelencsér, A. Atmospheric Brown Clouds: From Local Air Pollution to Climate Change. Elements 2010, 6, 223–228. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2014: Mitigation of Climate Change; IPCC: Geneva, Switzerland, 2014. [Google Scholar]
- Singh, A.; Dey, S. Influence of aerosol composition on visibility in megacity Delhi. Atmos. Environ. 2012, 62, 367–373. [Google Scholar] [CrossRef]
- Tuckermann, R.; Cammenga, H.K. The surface tension of aqueous solutions of some atmospheric water-soluble organic compounds. Atmos. Environ. 2004, 38, 6135–6138. [Google Scholar] [CrossRef]
- Pan, C.; Zhu, B.; Fang, C.; Kang, H.; Kang, Z.; Chen, H.; Liu, D.; Hou, X. The Fast Response of the Atmospheric Water Cycle to Anthropogenic Black Carbon Aerosols during Summer in East Asia. J. Clim. 2021, 34, 3049–3065. [Google Scholar] [CrossRef]
- Pope, C.A., 3rd; Dockery, D.W. Health Effects of Fine Particulate Air Pollution: Lines that Connect. J. Air Waste Manag. Assoc. 2006, 56, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Butt, E.W.; Rap, A.; Schmidt, A.; Scott, C.E.; Pringle, K.J.; Reddington, C.L.; Richards, N.A.D.; Woodhouse, M.T.; Ramirez-Villegas, J.; Yang, H.; et al. The impact of residential combustion emissions on atmospheric aerosol, human health, and climate. Atmos. Chem. Phys. Discuss. 2016, 16, 873–905. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.H.; Zhang, Q.; Kroll, J.H.; Decarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of Organic Aerosols in the Atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Jayne, J.T.; Leard, D.C.; Zhang, X.; Davidovits, P.; Smith, K.A.; Kolb, C.E.; Worsnop, D.R. Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles. Aerosol Sci. Technol. 2000, 33, 49–70. [Google Scholar] [CrossRef]
- Jimenez, J.L.; Jayne, J.T.; Shi, Q.; Kolb, C.E.; Worsnop, D.R.; Yourshaw, I.; Seinfeld, J.H.; Flagan, R.C.; Zhang, X.; Smith, K.A.; et al. Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer. J. Geophys. Res. Space Phys. 2003, 108. [Google Scholar] [CrossRef]
- Allan, J.D.; Delia, A.E.; Coe, H.; Bower, K.; Alfarra, R.; Jimenez, J.L.; Middlebrook, A.; Drewnick, F.; Onasch, T.B.; Canagaratna, M.R.; et al. A generalised method for the extraction of chemically resolved mass spectra from Aerodyne aerosol mass spectrometer data. J. Aerosol Sci. 2004, 35, 909–922. [Google Scholar] [CrossRef]
- Watson, J.G.; Zhu, T.; Chow, J.C.; Engelbrecht, J.; Fujita, E.M.; E Wilson, W. Receptor modeling application framework for particle source apportionment. Chemosphere 2002, 49, 1093–1136. [Google Scholar] [CrossRef]
- Roweis, S.T.; Saul, L.K.; Roweis, S.T.; Saul, L.K. Linked references are available on JSTOR for this article: Nonlinear Nonlinear Dimensionality Dimensionality Reduction by Locally Linear Embedding. Science 2000, 290, 2323–2326. [Google Scholar] [CrossRef] [PubMed]
- Paatero, P.; Tapper, U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Paatero, P. Least squares formulation of robust non-negative factor analysis. Chemom. Intell. Lab. Syst. 1997, 37, 23–35. [Google Scholar] [CrossRef]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Ulbrich, I.M.; Ng, N.L.; Worsnop, D.R.; Sun, Y. Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: A review. Anal. Bioanal. Chem. 2011, 401, 3045–3067. [Google Scholar] [CrossRef]
- Canonaco, F.; Crippa, M.; Slowik, J.G.; Baltensperger, U.; Prévôt, A.S.H. SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data. Atmos. Meas. Tech. 2013, 6, 3649–3661. [Google Scholar] [CrossRef]
- Srivastava, D.; Daellenbach, K.; Zhang, Y.; Bonnaire, N.; Chazeau, B.; Perraudin, E.; Gros, V.; Lucarelli, F.; Villenave, E.; Prévôt, A.; et al. Comparison of five methodologies to apportion organic aerosol sources during a PM pollution event. Sci. Total Environ. 2020, 757, 143168. [Google Scholar] [CrossRef]
- Lanz, V.A.; Prévôt, A.S.H.; Alfarra, M.R.; Weimer, S.; Mohr, C.; DeCarlo, P.F.; Gianini, M.F.D.; Hueglin, C.; Schneider, J.; Favez, O.; et al. Characterization of aerosol chemical composition with aerosol mass spectrometry in Central Europe: An overview. Atmos. Chem. Phys. Discuss. 2010, 10, 10453–10471. [Google Scholar] [CrossRef]
- Mohr, C.; DeCarlo, P.F.; Heringa, M.F.; Chirico, R.; Slowik, J.G.; Richter, R.; Reche, C.; Alastuey, A.; Querol, X.; Seco, R.; et al. Identification and quantification of organic aerosol from cooking and other sources in Barcelona using aerosol mass spectrometer data. Atmos. Chem. Phys. Discuss. 2012, 12, 1649–1665. [Google Scholar] [CrossRef]
- Crippa, M.; Canonaco, F.; Lanz, V.A.; Äijälä, M.; Allan, J.D.; Carbone, S.; Capes, G.; Ceburnis, D.; Dall’Osto, M.; Day, D.A.; et al. Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach. Atmos. Chem. Phys. Discuss. 2014, 14, 6159–6176. [Google Scholar] [CrossRef]
- Ots, R.; Vieno, M.; Allan, J.D.; Reis, S.; Nemitz, E.; Young, D.E.; Coe, H.; Di Marco, C.; Detournay, A.; Mackenzie, I.A.; et al. Model simulations of cooking organic aerosol (COA) over the UK using estimates of emissions based on measurements at two sites in London. Atmos. Chem. Phys. Discuss. 2016, 16, 13773–13789. [Google Scholar] [CrossRef]
- Tobler, A.K.; Canonaco, F.; Skiba, A.; Močnik, G.; Pragati, R.; Styszko, K.; Nęcki, J.; Slowik, J.G.; Baltensperger, U.; Prevot, A.S.H.; et al. Characterization of non-refractory (NR) PM1 and source apportionment of organic aerosol in Kraków, Poland. Atmos. Chem. Phys. 2021, 21, 14893–14906. [Google Scholar] [CrossRef]
- Poulain, L.; Fahlbusch, B.; Spindler, G.; Müller, K.; van Pinxteren, D.; Wu, Z.; Iinuma, Y.; Birmili, W.; Wiedensohler, A.; Herrmann, H. Source apportionment and impact of long-range transport on carbonaceous aerosol particles in central Germany during HCCT-2010. Atmos. Chem. Phys. Discuss. 2021, 21, 3667–3684. [Google Scholar] [CrossRef]
- Schwarz, J.; Pokorná, P.; Rychlík, Š.; Škáchová, H.; Vlček, O.; Smolík, J.; Ždímal, V.; Hůnová, I. Assessment of air pollution origin based on year-long parallel measurement of PM2.5 and PM10 at two suburban sites in Prague, Czech Republic. Sci. Total Environ. 2019, 664, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Pokorná, P.; Hovorka, J.; Hopke, P.K. Elemental composition and source identification of very fine aerosol particles in a European air pollution hot-spot. Atmos. Pollut. Res. 2016, 7, 671–679. [Google Scholar] [CrossRef]
- Leoni, C.; Pokorná, P.; Hovorka, J.; Masiol, M.; Topinka, J.; Zhao, Y.; Krumal, K.; Cliff, S.; Mikuška, P.; Hopke, P.K. Source apportionment of aerosol particles at a European air pollution hot spot using particle number size distributions and chemical composition. Environ. Pollut. 2018, 234, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Vodička, P.; Schwarz, J.; Ždímal, V. Analysis of one year’s OC/EC data at a Prague suburban site with 2-h time resolution. Atmos. Environ. 2013, 77, 865–872. [Google Scholar] [CrossRef]
- Vodička, P.; Kawamura, K.; Schwarz, J.; Ždímal, V. Seasonal changes in stable carbon isotopic composition in the bulk aerosol and gas phases at a suburban site in Prague. Sci. Total Environ. 2021, 803, 149767. [Google Scholar] [CrossRef] [PubMed]
- Kubelová, L.; Vodička, P.; Schwarz, J.; Cusack, M.; Makeš, O.; Ondráček, J.; Ždímal, V. A study of summer and winter highly time-resolved submicron aerosol composition measured at a suburban site in Prague. Atmos. Environ. 2015, 118, 45–57. [Google Scholar] [CrossRef]
- Horník, Š.; Sýkora, J.; Pokorná, P.; Vodička, P.; Schwarz, J.; Ždímal, V. Detailed NMR analysis of water-soluble organic compounds in size-resolved particulate matter seasonally collected at a suburban site in Prague. Atmos. Environ. 2021, 267, 118757. [Google Scholar] [CrossRef]
- Drewnick, F.; Hings, S.S.; Decarlo, P.; Jayne, J.T.; Gonin, M.; Fuhrer, K.; Weimer, S.; Jimenez, J.L.; Demerjian, K.L.; Borrmann, S.; et al. A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS)—Instrument Description and First Field Deployment. Aerosol Sci. Technol. 2005, 39, 637–658. [Google Scholar] [CrossRef]
- Zíková, N.; Vodička, P.; Ludwig, W.; Hitzenberger, R.; Schwarz, J. On the use of the field Sunset semi-continuous analyzer to measure equivalent black carbon concentrations. Aerosol Sci. Technol. 2016, 50, 284–296. [Google Scholar] [CrossRef][Green Version]
- Zhang, Z.-S.; Engling, G.; Chan, C.-Y.; Yang, Y.-H.; Lin, M.; Shi, S.; He, J.; Li, Y.-D.; Wang, X.-M. Determination of isoprene-derived secondary organic aerosol tracers (2-methyltetrols) by HPAEC-PAD: Results from size-resolved aerosols in a tropical rainforest. Atmos. Environ. 2013, 70, 468–476. [Google Scholar] [CrossRef]
- Dzepina, K.; Arey, J.; Marr, L.C.; Worsnop, D.R.; Salcedo, D.; Zhang, Q.; Onasch, T.B.; Molina, L.T.; Molina, M.J.; Jimenez, J.L. Detection of particle-phase polycyclic aromatic hydrocarbons in Mexico City using an aerosol mass spectrometer. Int. J. Mass Spectrom. 2007, 263, 152–170. [Google Scholar] [CrossRef]
- Lanz, V.A.; Alfarra, M.R.; Baltensperger, U.; Buchmann, B.; Hueglin, C.; Prévôt, A.S.H. Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra. Atmos. Chem. Phys. Discuss. 2007, 7, 1503–1522. [Google Scholar] [CrossRef]
- Fröhlich, R.; Cubison, M.J.; Slowik, J.G.; Bukowiecki, N.; Canonaco, F.; Croteau, P.L.; Gysel, M.; Henne, S.; Herrmann, E.; Jayne, J.T.; et al. Fourteen months of on-line measurements of the non-refractory submicron aerosol at the Jungfraujoch (3580 m a.s.l.)—Chemical composition, origins and organic aerosol sources. Atmos. Chem. Phys. Discuss. 2015, 15, 18225–18284. [Google Scholar] [CrossRef]
- Bressi, M.; Cavalli, F.; Putaud, J.; Fröhlich, R.; Petit, J.-E.; Aas, W.; Äijälä, M.; Alastuey, A.; Allan, J.; Aurela, M.; et al. A European aerosol phenomenology—7: High-time resolution chemical characteristics of submicron particulate matter across Europe. Atmos. Environ. X 2021, 10, 100108. [Google Scholar] [CrossRef]
- Canonaco, F.; Tobler, A.; Chen, G.; Sosedova, Y.; Slowik, J.G.; Bozzetti, C.; Daellenbach, K.R.; El Haddad, I.; Crippa, M.; Huang, R.-J.; et al. A new method for long-term source apportionment with time-dependent factor profiles and uncertainty assessment using SoFi Pro: Application to 1 year of organic aerosol data. Atmos. Meas. Tech. 2021, 14, 923–943. [Google Scholar] [CrossRef]
- Paatero, P. The Multilinear Engine—A Table-Driven, Least Squares Program for Solving Multilinear Problems, Including then-Way Parallel Factor Analysis Model. J. Comput. Graph. Stat. 1999, 8, 854–888. [Google Scholar] [CrossRef]
- Allan, J.; Jimenez, J.L.; Williams, P.; Alfarra, M.R.; Bower, K.; Jayne, J.T.; Coe, H.; Worsnop, D.R. Quantitative sampling using an Aerodyne aerosol mass spectrometer 1. Techniques of data interpretation and error analysis. J. Geophys. Res. Space Phys. 2003, 108, 4090. [Google Scholar] [CrossRef]
- Paatero, P.; Hopke, P.; Song, X.-H.; Ramadan, Z. Understanding and controlling rotations in factor analytic models. Chemom. Intell. Lab. Syst. 2002, 60, 253–264. [Google Scholar] [CrossRef]
- Ulbrich, I.M.; Canagaratna, M.R.; Zhang, Q.; Worsnop, D.R.; Jimenez, J.L. Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data. Atmos. Chem. Phys. Discuss. 2009, 9, 2891–2918. [Google Scholar] [CrossRef]
- Paatero, P.; Hopke, P.K. Discarding or downweighting high-noise variables in factor analytic models. Anal. Chim. Acta 2003, 490, 277–289. [Google Scholar] [CrossRef]
- Crippa, M.; DeCarlo, P.F.; Slowik, J.G.; Mohr, C.; Heringa, M.F.; Chirico, R.; Poulain, L.; Freutel, F.; Sciare, J.; Cozic, J.; et al. Wintertime aerosol chemical composition and source apportionment of the organic fraction in the metropolitan area of Paris. Atmos. Chem. Phys. Discuss. 2013, 13, 961–981. [Google Scholar] [CrossRef]
- Canagaratna, M.R.; Jayne, J.T.; Ghertner, D.A.; Herndon, S.; Shi, Q.; Jimenez, J.L.; Silva, P.; Williams, P.; Lanni, T.; Drewnick, F.; et al. Chase Studies of Particulate Emissions from in-use New York City Vehicles. Aerosol Sci. Technol. 2004, 38, 555–573. [Google Scholar] [CrossRef]
- Aiken, A.C.; DeCarlo, P.F.; Jimenez, J.L. Elemental Analysis of Organic Species with Electron Ionization High-Resolution Mass Spectrometry. Anal. Chem. 2007, 79, 8350–8358. [Google Scholar] [CrossRef] [PubMed]
- Alfarra, M.R.; Prevot, A.S.H.; Szidat, S.; Sandradewi, J.; Weimer, S.; Lanz, V.A.; Schreiber, D.; Mohr, M.; Baltensperger, U. Identification of the Mass Spectral Signature of Organic Aerosols from Wood Burning Emissions. Environ. Sci. Technol. 2007, 41, 5770–5777. [Google Scholar] [CrossRef]
- Ng, N.L.; Canagaratna, M.R.; Zhang, Q.; Jimenez, J.L.; Tian, J.; Ulbrich, I.M.; Kroll, J.H.; Docherty, K.S.; Chhabra, P.S.; Bahreini, R.; et al. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2010, 10, 4625–4641. [Google Scholar] [CrossRef]
- Crippa, M.; Canonaco, F.; Slowik, J.G.; El Haddad, I.; DeCarlo, P.F.; Mohr, C.; Heringa, M.F.; Chirico, R.; Marchand, N.; Temime-Roussel, B.; et al. Primary and secondary organic aerosol origin by combined gas-particle phase source apportionment. Atmos. Chem. Phys. Discuss. 2013, 13, 8411–8426. [Google Scholar] [CrossRef]
- Schwarz, J.; Cusack, M.; Karban, J.; Chalupníčková, E.; Havránek, V.; Smolík, J.; Ždímal, V. PM2.5 chemical composition at a rural background site in Central Europe, including correlation and air mass back trajectory analysis. Atmos. Res. 2016, 176–177, 108–120. [Google Scholar] [CrossRef]
- Štefancová, L.; Schwarz, J.; Maenhaut, W.; Chi, X.; Smolik, J. Hygroscopic growth of atmospheric aerosol sampled in Prague 2008 using humidity controlled inlets. Atmos. Res. 2010, 98, 237–248. [Google Scholar] [CrossRef]
- Řimnáčová, D.; Ždímal, V.; Schwarz, J.; Smolík, J.; Řimnáč, M. Atmospheric aerosols in suburb of Prague: The dynamics of particle size distributions. Atmos. Res. 2011, 101, 539–552. [Google Scholar] [CrossRef]
- Kenagy, H.S.; Sparks, T.L.; Ebben, C.J.; Wooldridge, P.J.; Lopez-Hilfiker, F.D.; Lee, B.H.; Thornton, J.A.; McDuffie, E.E.; Fibiger, D.L.; Brown, S.S.; et al. NO x Lifetime and NO y Partitioning During WINTER. J. Geophys. Res. Atmos. 2018, 123, 9813–9827. [Google Scholar] [CrossRef]
- Schwarz, J.; Štefancová, L.; Maenhaut, W.; Smolík, J.; Ždímal, V. Mass and chemically speciated size distribution of Prague aerosol using an aerosol dryer—The influence of air mass origin. Sci. Total Environ. 2012, 437, 348–362. [Google Scholar] [CrossRef]
- McCulloch, A.; Aucott, M.L.; Benkovitz, C.M.; E Graedel, T.; Kleiman, G.; Midgley, P.M.; Li, Y.-F. Global emissions of hydrogen chloride and chloromethane from coal combustion, incineration and industrial activities: Reactive Chlorine Emissions Inventory. J. Geophys. Res. Space Phys. 1999, 104, 8391–8403. [Google Scholar] [CrossRef]
- Vodička, P.; Schwarz, J.; Cusack, M.; Ždímal, V. Detailed comparison of OC/EC aerosol at an urban and a rural Czech background site during summer and winter. Sci. Total Environ. 2015, 518-519, 424–433. [Google Scholar] [CrossRef]
- Petit, J.-E.; Favez, O.; Sciare, J.; Canonaco, F.; Croteau, P.; Močnik, G.; Jayne, J.; Worsnop, D.; Leoz-Garziandia, E. Submicron aerosol source apportionment of wintertime pollution in Paris, France by double positive matrix factorization (PMF2) using an aerosol chemical speciation monitor (ACSM) and a multi-wavelength Aethalometer. Atmos. Chem. Phys. Discuss. 2014, 14, 13773–13787. [Google Scholar] [CrossRef]
- Cubison, M.J.; Ortega, A.M.; Hayes, P.L.; Farmer, D.K.; Day, D.; Lechner, M.J.; Brune, W.H.; Apel, E.C.; Diskin, G.S.; Fisher, J.A.; et al. Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies. Atmos. Chem. Phys. 2011, 11, 12049–12064. [Google Scholar] [CrossRef]
- Bae, M.-S.; Lee, J.Y.; Kim, Y.-P.; Oak, M.-H.; Shin, J.-S.; Lee, K.-Y.; Lee, H.; Lee, S.Y.; Kim, Y.-J. Analytical Methods of Levoglucosan, a Tracer for Cellulose in Biomass Burning, by Four Different Techniques. Asian J. Atmos. Environ. 2012, 6, 53–66. [Google Scholar] [CrossRef]
- Durana, N.; Navazo, M.C.G.; Gómez, M.; Alonso, L.; Garcia, J.A.; Ilardia, J.; Gangoiti, G.; Iza, J. Long term hourly measurement of 62 non-methane hydrocarbons in an urban area: Main results and contribution of non-traffic sources. Atmos. Environ. 2006, 40, 2860–2872. [Google Scholar] [CrossRef]
- Gkatzelis, G.I.; Coggon, M.M.; McDonald, B.C.; Peischl, J.; Aikin, K.C.; Gilman, J.B.; Trainer, M.; Warneke, C. Identifying Volatile Chemical Product Tracer Compounds in U.S. Cities. Environ. Sci. Technol. 2020, 55, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Villegas, E.; Priestley, M.; Ting, Y.-C.; Haslett, S.; Bannan, T.; Le Breton, M.; Williams, P.I.; Bacak, A.; Flynn, M.J.; Coe, H.; et al. Simultaneous aerosol mass spectrometry and chemical ionisation mass spectrometry measurements during a biomass burning event in the UK: Insights into nitrate chemistry. Atmos. Chem. Phys. Discuss. 2018, 18, 4093–4111. [Google Scholar] [CrossRef]
- Kaskaoutis, D.; Grivas, G.; Stavroulas, I.; Bougiatioti, A.; Liakakou, E.; Dumka, U.; Gerasopoulos, E.; Mihalopoulos, N. Apportionment of black and brown carbon spectral absorption sources in the urban environment of Athens, Greece, during winter. Sci. Total Environ. 2021, 801, 149739. [Google Scholar] [CrossRef]
- Leoz-Garziandia, E.; Carlier, P.; Tatry, V. Sampling and analysis of PAH and oxygenated PAH in diesel exhaust and ambient air. Int. Symp. Polycycl. Aromat. Compd. 2000, 20, 245–258. [Google Scholar] [CrossRef][Green Version]
- Latif, M.T.; Anuwar, N.Y.; Srithawirat, T.; Razak, I.S.; Ramli, N.A. Composition of Levoglucosan and Surfactants in Atmospheric Aerosols from Biomass Burning. Aerosol Air Qual. Res. 2011, 11, 837–845. [Google Scholar] [CrossRef]
- Farmer, D.K.; Matsunaga, A.; Docherty, K.S.; Surratt, J.; Seinfeld, J.H.; Ziemann, P.J.; Jimenez, J.L. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 6670–6675. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.D. An Aerosol Mass Spectrometer: Instrument Development, Data Analysis Techniques and Quantitative Atmospheric Particulate Measurements; UMIST: Manchester, UK, 2004. [Google Scholar]
- Heywood, J.B.; Sher, E.; Carel, R.; Plaut, S.E.; Plaut, P.O.; Stone, R.; Hochgreb, S.; Dulger, M.; Milton, B.; Pischinger, F.; et al. Handbook of Air Pollution from Internal Combustion Engines; Academic Press: San Diego, CA, USA, 1998; pp. xiii–xvii. [Google Scholar]
- Ng, N.L.; Canagaratna, M.R.; Jimenez, J.L.; Zhang, Q.; Ulbrich, I.M.; Worsnop, D.R. Real-Time Methods for Estimating Organic Component Mass Concentrations from Aerosol Mass Spectrometer Data. Environ. Sci. Technol. 2010, 45, 910–916. [Google Scholar] [CrossRef]
- Zhou, W.; Jiang, J.; Duan, L.; Hao, J. Evolution of Submicrometer Organic Aerosols during a Complete Residential Coal Combustion Process. Environ. Sci. Technol. 2016, 50, 7861–7869. [Google Scholar] [CrossRef]
- Horák, J.; Laciok, V.; Krpec, K.; Hopan, F.; Dej, M.; Kubesa, P.; Ryšavý, J.; Molchanov, O.; Kuboňová, L. Influence of the type and output of domestic hot-water boilers and wood moisture on the production of fine and ultrafine particulate matter. Atmos. Environ. 2020, 229, 117437. [Google Scholar] [CrossRef]
- Chan, K.M.; Wood, R. The seasonal cycle of planetary boundary layer depth determined using COSMIC radio occultation data. J. Geophys. Res. Atmos. 2013, 118, 12,422–12,434. [Google Scholar] [CrossRef]
- Surratt, J.D.; Kroll, J.H.; Kleindienst, T.E.; Edney, E.O.; Claeys, M.; Sorooshian, A.; Ng, N.L.; Offenberg, J.H.; Lewandowski, M.; Jaoui, M.; et al. Evidence for Organosulfates in Secondary Organic Aerosol. Environ. Sci. Technol. 2006, 41, 517–527. [Google Scholar] [CrossRef]
- Tiitta, P.; Leskinen, A.; Hao, L.; Yli-Pirilä, P.; Kortelainen, M.; Grigonyte, J.; Tissari, J.; Lamberg, H.; Hartikainen, A.; Kuuspalo, K.; et al. Transformation of logwood combustion emissions in a smog chamber: Formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging. Atmos. Chem. Phys. Discuss. 2016, 16, 13251–13269. [Google Scholar] [CrossRef]
- Sun, Y.L.; Zhang, Q.; Schwab, J.J.; Yang, T.; Ng, N.L.; Demerjian, K.L. Factor analysis of combined organic and inorganic aerosol mass spectra from high resolution aerosol mass spectrometer measurements. Atmos. Chem. Phys. Discuss. 2012, 12, 8537–8551. [Google Scholar] [CrossRef]
- Äijälä, M.; Daellenbach, K.R.; Canonaco, F.; Heikkinen, L.; Junninen, H.; Petäjä, T.; Kulmala, M.; Prevot, A.; Ehn, M. Constructing a data-driven receptor model for organic and inorganic aerosol—A synthesis analysis of eight mass spectrometric data sets from a boreal forest site. Atmos. Chem. Phys. Discuss. 2019, 19, 3645–3672. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Winter | Summer | ||||
|---|---|---|---|---|---|
| factor All/OA | R2 | Slope | factor All/OA | R2 | Slope |
| HOA/HOA | 0.89 | 0.52 | HOA/HOA | 1.00 | 0.77 |
| BBOA/BBOA | 1.00 | 0.85 | BBOA/BBOA | 0.98 | 0.90 |
| LOA/LOOA | 0.96 | 0.95 | AROA/OOA1 | 0.98 | 0.54 |
| LROA/LROOA | 0.99 | 0.45 | LVOA/OOA2 | 0.99 | 0.88 |
| Winter | Summer | ||
|---|---|---|---|
| factor All/OA | R2 | factor All/OA | R2 |
| HOA/HOA | 0.16 | HOA/HOA | 0.98 |
| BBOA/BBOA | 0.98 | BBOA/BBOA | 0.76 |
| LOA/LOOA | 0.10 | AROA/OOA1 | 0.73 |
| LROA/LROOA | 0.71 | LVOA/OOA2 | 0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makeš, O.; Schwarz, J.; Vodička, P.; Engling, G.; Ždímal, V. Determination of PM1 Sources at a Prague Background Site during the 2012–2013 Period Using PMF Analysis of Combined Aerosol Mass Spectra. Atmosphere 2022, 13, 20. https://doi.org/10.3390/atmos13010020
Makeš O, Schwarz J, Vodička P, Engling G, Ždímal V. Determination of PM1 Sources at a Prague Background Site during the 2012–2013 Period Using PMF Analysis of Combined Aerosol Mass Spectra. Atmosphere. 2022; 13(1):20. https://doi.org/10.3390/atmos13010020
Chicago/Turabian StyleMakeš, Otakar, Jaroslav Schwarz, Petr Vodička, Guenter Engling, and Vladimír Ždímal. 2022. "Determination of PM1 Sources at a Prague Background Site during the 2012–2013 Period Using PMF Analysis of Combined Aerosol Mass Spectra" Atmosphere 13, no. 1: 20. https://doi.org/10.3390/atmos13010020
APA StyleMakeš, O., Schwarz, J., Vodička, P., Engling, G., & Ždímal, V. (2022). Determination of PM1 Sources at a Prague Background Site during the 2012–2013 Period Using PMF Analysis of Combined Aerosol Mass Spectra. Atmosphere, 13(1), 20. https://doi.org/10.3390/atmos13010020