
Comparative Mitogenome Analysis of Two Native Apple Snail Species (Ampullariidae, Pomacea) from Peruvian Amazon

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Annotation of Mitochondrial Genome and Predictions of Secondary Structures
2.4. Comparative Analysis
2.5. Phylogenetic Analysis
3. Results and Discussion
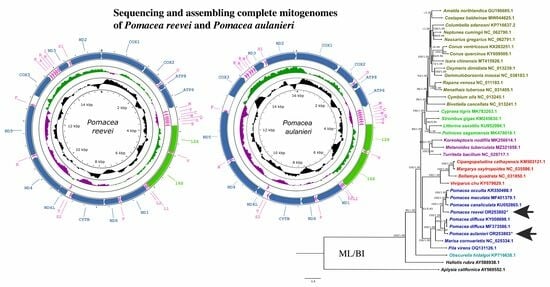
3.1. Organization and Structure of the Mitogenomes
3.2. Nucleotide Composition
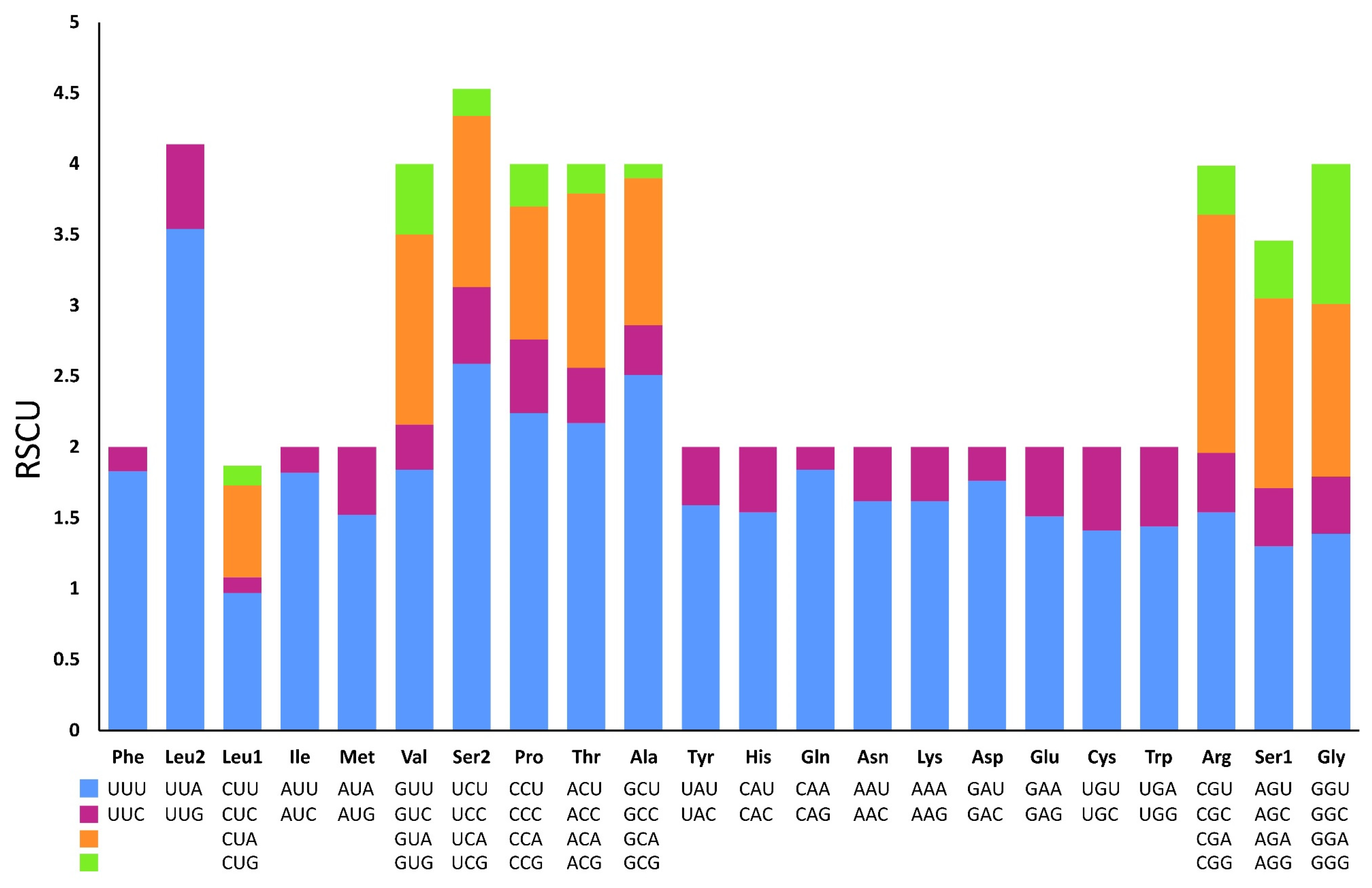
3.3. Protein-Coding Genes and Codon Usage
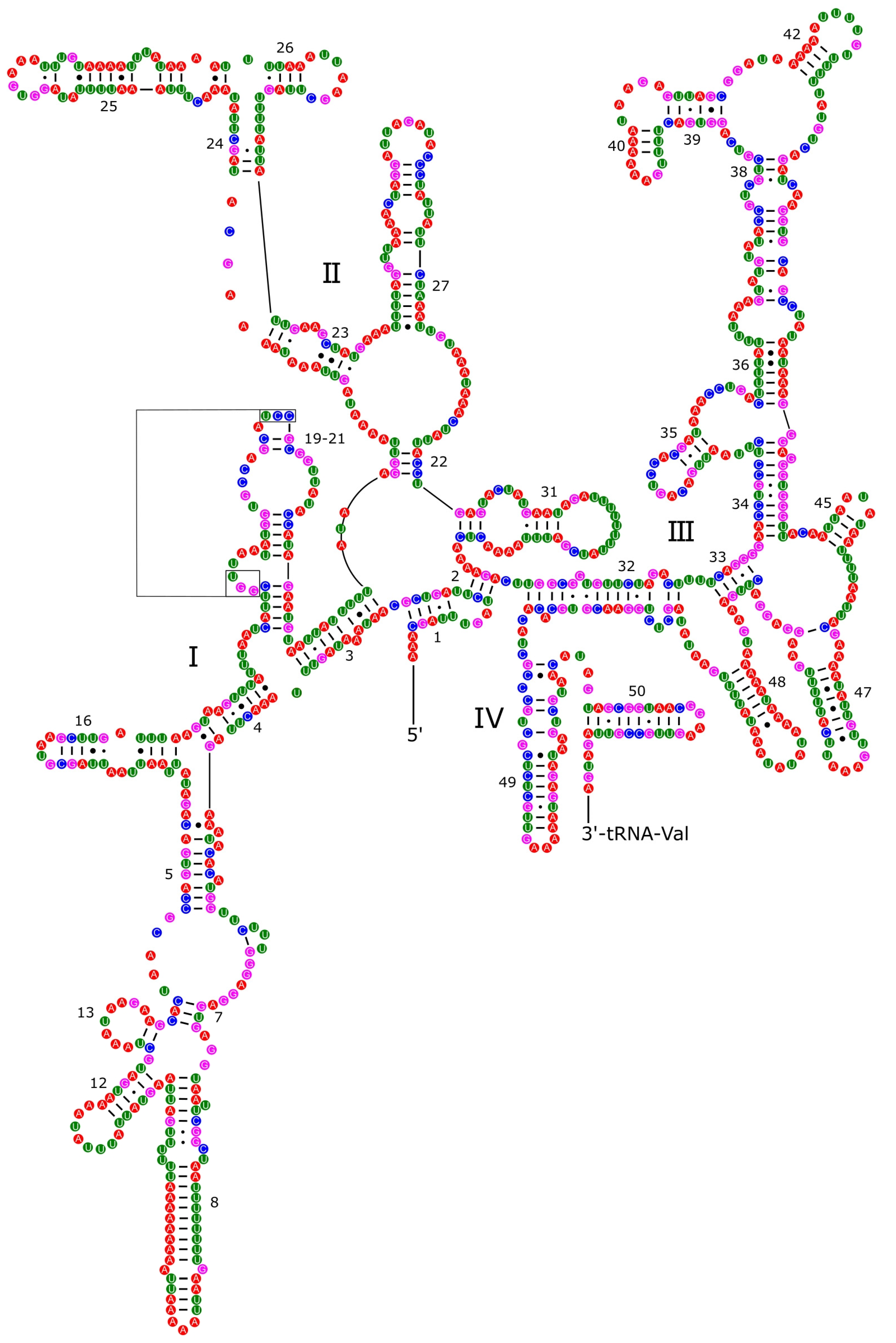
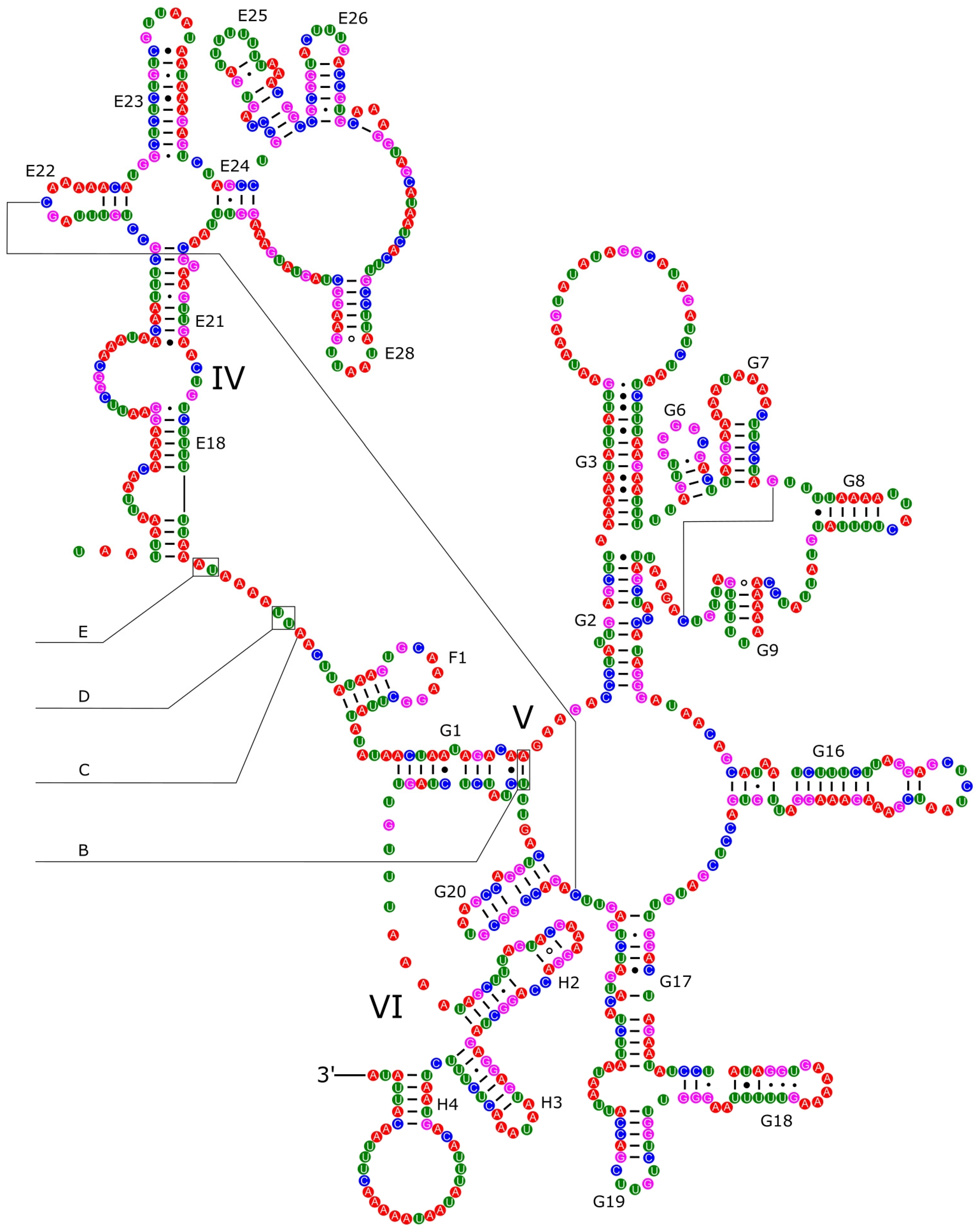
3.4. Ribosomal and Transfer RNA Genes
3.5. Control Region
3.6. Genetic Variation among Pomacea
3.7. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes, K.A.; Cowie, R.H.; Jørgensen, A.; Schultheiß, R.; Albrecht, C.; Thiengo, S.C. Molluscan models in evolutionary biology: Apple snails (Gastropoda: Ampullariidae) as a system for addressing fundamental questions. Am. Malacol. Bull. 2009, 27, 47–58. [Google Scholar] [CrossRef]
- Cowie, R.H.; Hayes, K.A.; Strong, E.E.; Thiengo, S.C. Nonnative apple snails: Systematics, distribution, invasion history and reasons for introduction. In Biology and Management of Invasive Apple Snails; Joshi, R.C., Cowie, R.H., Sebastian, L.S., Eds.; Philippine Rice Research Institute (PhilRice), Filipinas: Muñoz, Philippines, 2017; pp. 3–32. [Google Scholar]
- Rawlings, T.A.; Hayes, K.A.; Cowie, R.H.; Collins, T.M. The identity, distribution, and impacts of non-native apple snails in the continental United States. BMC Evol. Biol. 2007, 7, 97. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, Y.; Oulang, S.; Wu, X. Phylogeographic patterns and demographic history of Pomacea canaliculata and Pomacea maculata from different countries (Ampullariidae, Gastropoda, Mollusca). Nat. Conserv. 2019, 36, 71–92. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, M.; Zhang, Z.; Liu, Y.; Deng, Z.; Gong, X. Genetic Diversity of Two Globally Invasive Snails in Asia and Americas in Relation with Agricultural Habitats and Climate Factors. Diversity 2022, 14, 1069. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Zhu, S.; Xu, H.; Liu, Y.; Chen, L. The complete mitochondrial genome of Pomacea canaliculata (Gastropoda: Ampullariidae). Mitochondrial DNA Part A 2016, 27, 884–885. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.E.; Deng, Z.; Luo, H.; Guo, J.; He, S.; Luo, M.; Zhao, B. The complete mitochondrial genome of the golden apple snail Pomacea canaliculata (Gastropoda: Ampullariidae). Mitochondrial DNA Part B 2016, 1, 45–47. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, S.; Song, F.; Li, H.; Liu, J.; Liu, G.; Yu, X.P. The mitochondrial genome of Pomacea maculata (Gastropoda: Ampullariidae). Mitochondrial DNA Part A 2015, 27, 2895–2896. [Google Scholar] [CrossRef]
- Liu, S.; Yang, Q.; He, C.; Yu, X. The complete mitochondrial genome of Pomacea diffusa (Gastropoda: Ampullariidae). Mitochondrial DNA Part B 2017, 2, 491–492. [Google Scholar] [CrossRef]
- Guo, J.; Yang, H.; Zhang, C.; Xue, H.; Xia, Y.; Zhang, J.E. Complete mitochondrial genome of the apple snail Pomacea diffusa (Gastropoda, Ampullariidae) with phylogenetic consideration. Mitochondrial DNA Part B 2017, 2, 865–867. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.E.; Xia, J.; Yang, J.; Guo, J.; Deng, Z.; Luo, M. Comparative characterization of the complete mitochondrial genomes of the three apple snails (Gastropoda: Ampullariidae) and the phylogenetic analyses. Int. J. Mol. Sci. 2018, 19, 3646. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, S.; Song, F.; Liu, G.F.; Yu, X.P. Comparative mitogenome analysis on species of four apple snails (Ampullariidae: Pomacea). Int. J. Biol. Macromol. 2018, 118, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.A.; Cowie, R.H.; Thiengo, S.C. A global phylogeny of apple snails: Gondwanan origin, generic relationships, and the influence of outgroup choice (Caenogastropoda: Ampullariidae). Biol. J. Linn. Soc. 2009, 98, 61–76. [Google Scholar] [CrossRef]
- Hayes, K.A.; Cowie, R.H.; Thiengo, S.C.; Strong, E.E. Comparing apples with apples: Clarifying the identities of two highly invasive Neotropical Ampullariidae (Caenogastropoda). Zool. J. Linn. Soc. 2012, 166, 723–753. [Google Scholar] [CrossRef]
- Yang, Q.; Yu, X. A new species of apple snail in the genus Pomacea (Gastropoda: Caenogastropoda: Ampullariidae). Zool. Stud. 2019, 58, 13. [Google Scholar] [CrossRef]
- Ramírez, R.; Paredes, C.; Arenas, J. Moluscos del Perú. Revista de Biología Tropical 2003, 51 (Suppl. 3), 225–284. [Google Scholar]
- Ramírez, R.; Solis, M.; Ampuero, A.; Morín, J.; Jimenez-Vasquez, V.; Ramirez, J.; Congrains, C.; Temoche, H.; Shiga, Y.B. Identificación molecular y relaciones evolutivas de Pomacea nobilis, base para la autenticación específica del churo negro de la Amazonia peruana. Revista Peruana de Biología 2020, 27, 139–148. [Google Scholar] [CrossRef]
- Ramírez, R.; Ramírez, J.L.; Rivera, F.; Justino, S.; Solis, M.; Morín, J.; Ampuero, A.; Mendivil, A.; Congrains, C. Do not judge a snail by its shell: Molecular identification of Pomacea (Ampullariidae) species, with particular reference to the Peruvian Amazonian giant apple snail. Archiv für Molluskenkunde 2022, 151, 7–17. [Google Scholar] [CrossRef]
- Ampuero, A.; Ramirez, R. Description of two new species of apple snail (Ampullariidae: Pomacea) from Peruvian Amazonia. Zootaxa 2023, 5258, 76–98. [Google Scholar] [CrossRef]
- Sturm, C.F.; Mayhew, R.; Bales, B.R. Field and laboratory methods in malacology. In The Mollusks: A Guide to Their Study, Collection, and Preservation; Sturm, C.F., Pearce, T.A., Valdes, A., Eds.; American Malacological Society: Pittsburgh, PA, USA, 2006; pp. 9–31. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; dePamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, B.A.; Hoksza, D.; Nawrocki, E.P.; Ribas, C.E.; Madeira, F.; Cannone, J.J.; Gutell, R.; Maddala, A.; Meade, C.D.; Williams, L.D.; et al. R2DT is a framework for predicting and visualising RNA secondary structure using templates. Nat. Commun. 2021, 12, 3494. [Google Scholar] [CrossRef]
- Hickson, R.E.; Simon, C.; Cooper, A.; Spicer, G.S.; Sullivan, J.; Penny, D. Conserved Sequence Motifs, Alignment, and Secondary Structure for the Third Domain of Animal 12S rRNA. Mol. Biol. Evol. 1996, 13, 150–169. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, C.; Holznagel, W.E.; Schnare, M.N.; Gutell, R.R. Phylogenetic Analysis of Molluscan Mitochondrial LSU rDNA Sequences and Secondary Structures. Mol. Phylogenet. Evol. 2000, 15, 83–102. [Google Scholar] [CrossRef]
- Wuys, J.; De Rijk, P.; Van de Peer, Y.; Winkelmans, T.; De Watcher, R. The European large subunit ribosomal RNA database. Nucleic Acids Res. 2001, 29, 175–177. [Google Scholar]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–359. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA Sequence Polymorphism Analysis of Large Dataset. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Nguyen, L.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 30 May 2022).
- Wang, M.; Qiu, J. Complete mitochondrial genome of the giant ramshorn snail Marisa cornuarietis (Gastropoda: Ampullariidae). Mitochondrial DNA 2014, 27, 1734–1735. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.K.; Stevenson, B.J.; Ownby, J.P.; Cady, M.T.; Watkins, M.; Olivera, B.M. The mitochondrial genome of Conus textile, coxI-coxII intergenic sequences and Conoidean evolution. Mol. Phylogenetics Evol. 2008, 46, 215–223. [Google Scholar] [CrossRef]
- Rawlings, T.A.; Macinnis, M.J.; Bieler, R.; Boore, J.L.; Collins, T.M. Sessile snails, dynamic genomes: Gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genom. 2010, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Peng, C.; Chen, Q.; Zhang, J.; Shi, Q. Mitochondrial genome sequencing of a vermivorous cone snail Conus quercinus supports the correlative analysis between phylogenetic relationships and dietary types of Conus species. PLoS ONE 2018, 13, e0193053. [Google Scholar] [CrossRef]
- Sun, S.; Li, Q.; Kong, L.; Yu, H. Multiple reversals of strand asymmetry in molluscs mitochondrial genomes, and consequences for phylogenetic inference. Mol. Phylogenetics Evol. 2018, 118, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved PCR primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Medina, M.; Collins, T.M.; Walsh, P.J. Mtdna Ribosomal Gene Phylogeny of Sea Hares in the Genus Aplysia (Gastropoda, Opisthobranchia, Anaspidea): Implications for Comparative Neurobiology. Syst. Biol. 2001, 50, 7676–7688. [Google Scholar] [CrossRef]
- Vogler, R.E.; Beltramino, A.A.; Strong, E.E.; Rumi, A.; Peso, J.G. Insights into the Evolutionary History of an Extinct South American Freshwater Snail Based on Historical DNA. PLoS ONE 2016, 11, e0169191. [Google Scholar] [CrossRef]
- Guzmán, L.B.; Vogler, R.E.; Beltramino, A.A. The mitochondrial genome of the semi-slug Omalonyx unguis (Gastropoda: Succineidae) and the phylogenetic relationships within Stylommatophora. PLoS ONE 2021, 16, e0253724. [Google Scholar] [CrossRef]
- Smith, S.D.; Bond, J.E. An analysis of the secondary structure of the mitochondrial large subunit rRNA gene (16S) in spiders and its implications for phylogenetic reconstruction. J. Arachnol. 2003, 31, 44–54. [Google Scholar] [CrossRef]
- Soroka, M. Characteristics of mitochondrial DNA of unionid bivalves (Mollusca: Bivalvia: Unionidae). II. Comparison of complete sequences of maternally inherited mitochondrial genomes of Sinanodonta woodiana and Unio pictorum. Folia Malacol. 2010, 18, 189–209. [Google Scholar] [CrossRef][Green Version]
- Brauer, A.; Kurz, A.; Stockwell, T.; Baden-Tillson, H.; Heidler, J.; Wittig, I.; Kauferstein, S.; Mebs, D.; Stöcklin, R.; Remm, M. The Mitochondrial Genome of the Venomous Cone Snail Conus consors. PLoS ONE 2012, 7, e51528. [Google Scholar] [CrossRef]
- Maynard, B.T.; Kerr, L.J.; Mckiernan, J.M.; Jansen, E.S.; Hanna, P.J. Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (Leach). Mar. Biotechnol. 2005, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Ramón-Laca, A.; Gallego, R.; Nichols, K.M. Affordable de novo generation of fish mitogenomes using amplification-free enrichment of mitochondrial DNA and deep sequencing of long fragments. Mol. Ecol. Resour. 2023, 23, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Castellana, S.; Vicario, S.; Saccone, C. Evolutionary Patterns of the Mitochondrial Genome in Metazoa: Exploring the Role of Mutation and Selection in Mitochondrial Protein–Coding Genes. Genome Biol. Evol. 2011, 3, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Peretolchina, T.E.; Sitnikova, T.Y.; Sherbakov, D.Y. The complete mitochondrial genomes of four Baikal molluscs from the endemic family Baicaliidae (Caenogastropoda: Truncatelloida). J. Molluscan Stud. 2020, 86, 201–209. [Google Scholar] [CrossRef]
- Fonseca, M.M.; Lopes-Lima, M.; Eackles, M.S.; King, T.L.; Froufe, E. The female and male mitochondrial genomes of Unio delphinus and the phylogeny of freshwater mussels (Bivalvia: Unionida). Mitochondrial DNA B 2016, 1, 954–957. [Google Scholar] [CrossRef]
- Osca, D.; Templado, J.; Zardoya, R. Caenogastropod mitogenomics. Mol. Phylogenetics Evol. 2015, 93, 118–128. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Position | Size (bp) | Intergenic Length | Anticodon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|
| COI | H | 1–1536 | 1536 | 18 | - | ATG | TAA |
| COII | H | 1555–2241 | 687 | 14 | - | ATG | TAA |
| tRNA-Asp | H | 2256–2323 | 68 | 0 | GTC | - | - |
| ATP8 | H | 2324–2482 | 159 | 8 | - | ATG | TAA |
| ATP6 | H | 2491–3189 | 699 | 33 | - | ATG | TAG |
| tRNA-Met | L | 3223–3288 | 66 | 39 | CAT | - | - |
| tRNA-Tyr | L | 3328–3393 | 66 | 38 | GTA | - | - |
| tRNA-Cys | L | 3432–3493 | 62 | 3 | GCA | - | - |
| tRNA-Trp | L | 3497–3562 | 66 | 8 | TCA | - | - |
| tRNA-Gln | L | 3571–3633 | 63 | 5 | TTG | - | - |
| tRNA-Gly | L | 3639–3704 | 66 | 26 | TCC | - | - |
| tRNA-Glu | L | 3731–3797 | 67 | 0 | TTC | - | - |
| 12S rRNA | H | 3798–4706 | 909 | 0 | - | - | - |
| tRNA-Val | H | 4707–4772 | 66 | 0 | TAC | - | - |
| 16S rRNA | H | 4773–6112 | 1340 | 0 | - | - | - |
| tRNA-Leu1 | H | 6113–6175 | 63 | 0 | TAG | - | - |
| tRNA-Leu2 | H | 6176–6241 | 66 | 0 | TAA | - | - |
| NAD1 | H | 6242–7180 | 939 | 13 | - | ATG | TAA |
| tRNA-Pro | H | 7194–7260 | 67 | 0 | TGG | - | - |
| NAD6 | H | 7261–7755 | 495 | 10 | - | ATG | TAA |
| CytB | H | 7766–8905 | 1140 | 3 | - | ATG | TAA |
| tRNA-Ser2 | H | 8909–8973 | 65 | 35 | TGA | - | - |
| tRNA-Thr | L | 9009–9074 | 66 | 7 | TGT | - | - |
| NAD4L | H | 9082–9378 | 297 | −7 | - | ATG | TAA |
| NAD4 | H | 9372–10,745 | 1374 | 38 | - | ATG | TAA |
| tRNA-His | H | 10,784–10,849 | 66 | 0 | GTG | - | - |
| NAD5 | H | 10,850–12,556 | 1707 | −2 | - | ATG | TAA |
| tRNA-Phe | H | 12,555–12,622 | 68 | 0 | GAA | - | - |
| CR | - | 12,623–12,914 | 292 | 0 | - | - | - |
| COIII | H | 12,915–13,694 | 780 | 27 | - | ATG | TAA |
| tRNA-Lys | H | 13,722–13,787 | 66 | 17 | TTT | - | - |
| tRNA-Ala | H | 13,805–13,872 | 68 | 21 | TGC | - | - |
| tRNA-Arg | H | 13,894–13,963 | 70 | 2 | TCG | - | - |
| tRNA-Asn | H | 13,966–14,038 | 73 | 15 | GTT | - | - |
| tRNA-Ile | H | 14,054–14,123 | 70 | 0 | GAT | - | - |
| NAD3 | H | 14,124–14,477 | 354 | 40 | - | ATG | TAA |
| tRNA-Ser1 | H | 14,518–14,584 | 67 | 0 | GCT | - | - |
| vNAD2 | H | 14,585–15,658 | 1074 | 2 | - | ATG | TAA |
| Gene | Strand | Position | Size (bp) | Intergenic Length | Anticodon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|
| COI | H | 1–1536 | 1536 | 35 | - | ATG | TAA |
| COII | H | 1572–2258 | 687 | 27 | - | ATG | TAA |
| tRNA-Asp | H | 2286–2353 | 68 | 0 | GTC | - | - |
| ATP8 | H | 2354–2512 | 159 | 30 | - | ATG | TAG |
| ATP6 | H | 2543–3241 | 699 | 28 | - | ATG | TAG |
| tRNA-Met | L | 3270–3332 | 63 | 21 | CAT | - | - |
| tRNA-Tyr | L | 3354–3418 | 65 | 22 | GTA | - | - |
| tRNA-Cys | L | 3441–3502 | 62 | 22 | GCA | - | - |
| tRNA-Trp | L | 3525–3590 | 66 | 25 | TCA | - | - |
| tRNA-Gln | L | 3616–3681 | 66 | 26 | TTG | - | - |
| tRNA-Gly | L | 3708–3774 | 67 | 34 | TCC | - | - |
| tRNA-Glu | L | 3809–3873 | 65 | 0 | TTC | - | - |
| 12S rRNA | H | 3874–4833 | 960 | 0 | - | - | - |
| tRNA-Val | H | 4834–4900 | 67 | 0 | TAC | - | - |
| 16S rRNA | H | 4901–6248 | 1348 | 0 | - | - | - |
| tRNA-Leu1 | H | 6249–6312 | 64 | 0 | TAG | - | - |
| tRNA-Leu2 | H | 6313–6381 | 69 | 0 | TAA | - | - |
| NAD1 | H | 6382–7326 | 945 | 9 | - | ATG | TAA |
| tRNA-Pro | H | 7336–7402 | 67 | −1 | TGG | - | - |
| NAD6 | H | 7402–7896 | 495 | 10 | - | ATG | TAA |
| CytB | H | 7907–9046 | 1140 | 15 | - | ATG | TAA |
| tRNA-Ser2 | H | 9062–9127 | 66 | 29 | TGA | - | - |
| tRNA-Thr | L | 9157–9227 | 71 | 10 | TGT | - | - |
| NAD4L | H | 9238–9534 | 297 | −7 | - | ATG | TAA |
| NAD4 | H | 9528–10,889 | 1362 | 10 | - | ATG | TAA |
| tRNA-His | H | 10,900–10,964 | 65 | 0 | GTG | - | - |
| NAD5 | H | 10,965–12,692 | 1728 | −21 | - | ATG | TAA |
| tRNA-Phe | H | 12,672–12,740 | 69 | 0 | GAA | - | - |
| CR | - | 12,741–13,264 | 524 | 0 | - | ||
| COIII | H | 13,265–14,044 | 780 | 65 | - | ATG | TAA |
| tRNA-Lys | H | 14,110–14,176 | 68 | 10 | TTT | - | - |
| tRNA-Ala | H | 14,187–14,260 | 74 | 45 | TGC | - | - |
| tRNA-Arg | H | 14,306–14,374 | 69 | 10 | TCG | - | - |
| tRNA-Asn | H | 14,385–14,455 | 71 | 27 | GTT | - | - |
| tRNA-Ile | H | 14,483–14,552 | 70 | 5 | GAT | - | - |
| NAD3 | H | 14,558–14,911 | 354 | 39 | - | ATG | TAG |
| tRNA-Ser1 | H | 14,951–15,020 | 70 | −1 | GCT | - | - |
| NAD2 | H | 15,020–16,093 | 1074 | 3 | - | ATG | TAG |
| Feature | Length | A% | T% | C% | G% | %A + T | %G + C | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| Whole mitogenome | |||||||||
| P. reevei | 15,660 | 30.10 | 40.88 | 13.14 | 15.87 | 70.98 | 29.02 | −0.152 | 0.094 |
| P. aulanieri | 16,096 | 29.48 | 39.37 | 14.83 | 16.32 | 68.85 | 31.15 | −0.144 | 0.048 |
| PCGs | |||||||||
| P. reevei | 11,241 | 27.52 | 42.45 | 13.95 | 16.08 | 69.98 | 30.02 | −0.213 | 0.071 |
| P. aulanieri | 11,256 | 26.87 | 40.70 | 15.67 | 16.76 | 67.57 | 32.43 | −0.205 | 0.033 |
| rRNAs | |||||||||
| P. reevei | 2251 | 37.23 | 35.41 | 11.28 | 16.08 | 72.63 | 27.37 | 0.025 | 0.175 |
| P. aulanieri | 2308 | 36.74 | 34.84 | 11.92 | 16.51 | 71.58 | 28.42 | 0.027 | 0.162 |
| tRNAs | |||||||||
| P. reevei | 1465 | 35.77 | 36.04 | 11.95 | 16.25 | 71.81 | 28.19 | −0.004 | 0.153 |
| P. aulanieri | 1485 | 34.28 | 35.35 | 12.93 | 17.44 | 69.63 | 30.37 | −0.015 | 0.149 |
| CR | |||||||||
| P. reevei | 292 | 36.64 | 40.75 | 9.25 | 13.36 | 77.40 | 22.60 | −0.053 | 0.182 |
| P. aulanieri | 524 | 33.40 | 38.17 | 16.60 | 11.83 | 71.56 | 28.44 | −0.067 | −0.168 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 311 | 1.83 | UCU(S) | 120 | 2.59 | UAU(Y) | 108 | 1.59 | UGU(C) | 31 | 1.41 |
| UUC(F) | 29 | 0.17 | UCC(S) | 25 | 0.54 | UAC(Y) | 28 | 0.41 | UGC(C) | 13 | 0.59 |
| UUA(L) | 367 | 3.54 | UCA(S) | 56 | 1.21 | UAA(*) | 12 | 1.85 | UGA(W) | 75 | 1.44 |
| UUG(L) | 62 | 0.6 | UCG(S) | 9 | 0.19 | UAG(*) | 1 | 0.15 | UGG(W) | 29 | 0.56 |
| CUU(L) | 100 | 0.97 | CCU(P) | 74 | 2.24 | CAU(H) | 61 | 1.54 | CGU(R) | 22 | 1.54 |
| CUC(L) | 10 | 0.11 | CCC(P) | 17 | 0.52 | CAC(H) | 18 | 0.46 | CGC(R) | 6 | 0.42 |
| CUA(L) | 67 | 0.65 | CCA(P) | 31 | 0.94 | CAA(Q) | 69 | 1.84 | CGA(R) | 23 | 1.68 |
| CUG(L) | 14 | 0.14 | CCG(P) | 10 | 0.3 | CAG(Q) | 6 | 0.16 | CGG(R) | 5 | 0.35 |
| AUU(I) | 281 | 1.82 | ACU(T) | 83 | 2.17 | AAU(N) | 112 | 1.62 | AGU(S) | 60 | 1.3 |
| AUC(I) | 28 | 0.18 | ACC(T) | 16 | 0.39 | AAC(N) | 26 | 0.38 | AGC(S) | 19 | 0.41 |
| AUA(M) | 162 | 1.52 | ACA(T) | 47 | 1.23 | AAA(K) | 81 | 1.62 | AGA(S) | 62 | 1.34 |
| AUG(M) | 51 | 0.48 | ACG(T) | 8 | 0.21 | AAG(K) | 19 | 0.38 | AGG(S) | 19 | 0.41 |
| GUU(V) | 114 | 1.84 | GCU(A) | 146 | 2.51 | GAU(D) | 65 | 1.76 | GGU(G) | 81 | 1.39 |
| GUC(V) | 19 | 0.32 | GCC(A) | 19 | 0.35 | GAC(D) | 9 | 0.24 | GGC(G) | 23 | 0.4 |
| GUA(V) | 83 | 1.34 | GCA(A) | 60 | 1.04 | GAA(E) | 62 | 1.51 | GGA(G) | 70 | 1.22 |
| GUG(V) | 31 | 0.5 | GCG(A) | 6 | 0.1 | GAG(E) | 19 | 0.49 | GGG(G) | 57 | 0.99 |
| Feature | P. reevei | P. aulanieri | P. diffusa | P. canaliculata | P. maculata | P. occulta |
|---|---|---|---|---|---|---|
| Overlapping NAD5/tRNA-Phe | + | + | + | − | − | − |
| Overlapping NAD1/tRNA-Pro | − | − | − | + | + | + |
| ATP6 length (bp) | 699 | 699 | 714 | 699 | 699 | 699 |
| NAD1 length (bp) | 939 | 945 | 945 | 960 | 960 | 960 |
| NAD2 length (bp) | 1074 | 1074 | 1071 | 1062 | 1062 | 1065 |
| NAD4 length(bp) | 1374 | 1362 | 1362 | 1368 | 1368 | 1368 |
| NAD5 length (bp) | 1707 | 1728 | 1728 | 1710 | 1710 | 1710 |
| Start codon COIII | ATG | ATG | ATG | ATA | ATA | ATA |
| Range of tRNAs length (bp) | 12 | 13 | 15 | 7 | 7 | 7 |
| Tallo D en tRNA-Ser1 | − | + | + | − | − | − |
| Tallo D en tRNA-Ser2 | − | − | − | + | + | + |
| 12S rRNA length (bp) | 909 | 960 | 952 | 929 | 936 | 934 |
| 16S rRNA length (bp) | 1340 | 1348 | 1346 | 1334 | 1336 | 1331 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendivil, A.; Ramírez, R.; Morin, J.; Ramirez, J.L.; Siccha-Ramirez, R.; Britzke, R.; Rivera, F.; Ampuero, A.; Oliveros, N.; Congrains, C. Comparative Mitogenome Analysis of Two Native Apple Snail Species (Ampullariidae, Pomacea) from Peruvian Amazon. Genes 2023, 14, 1769. https://doi.org/10.3390/genes14091769
Mendivil A, Ramírez R, Morin J, Ramirez JL, Siccha-Ramirez R, Britzke R, Rivera F, Ampuero A, Oliveros N, Congrains C. Comparative Mitogenome Analysis of Two Native Apple Snail Species (Ampullariidae, Pomacea) from Peruvian Amazon. Genes. 2023; 14(9):1769. https://doi.org/10.3390/genes14091769
Chicago/Turabian StyleMendivil, Alejandro, Rina Ramírez, Jaime Morin, Jorge L. Ramirez, Raquel Siccha-Ramirez, Ricardo Britzke, Fátima Rivera, Andre Ampuero, Nilda Oliveros, and Carlos Congrains. 2023. "Comparative Mitogenome Analysis of Two Native Apple Snail Species (Ampullariidae, Pomacea) from Peruvian Amazon" Genes 14, no. 9: 1769. https://doi.org/10.3390/genes14091769
APA StyleMendivil, A., Ramírez, R., Morin, J., Ramirez, J. L., Siccha-Ramirez, R., Britzke, R., Rivera, F., Ampuero, A., Oliveros, N., & Congrains, C. (2023). Comparative Mitogenome Analysis of Two Native Apple Snail Species (Ampullariidae, Pomacea) from Peruvian Amazon. Genes, 14(9), 1769. https://doi.org/10.3390/genes14091769