

Complete Mitochondrial Genome for Lucilia cuprina dorsalis (Diptera: Calliphoridae) from the Northern Territory, Australia
 , , , and
, , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
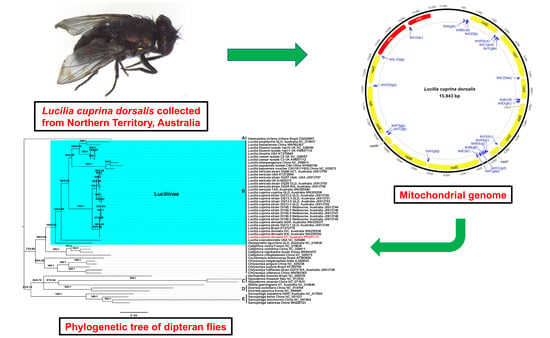
2.1. Sample Collection
2.2. DNA Extraction, Library Construction, and Sequencing
2.3. Mitochondrial Genome Assembly and Annotation
2.4. Genomic and Phylogenetic Analyses
3. Results
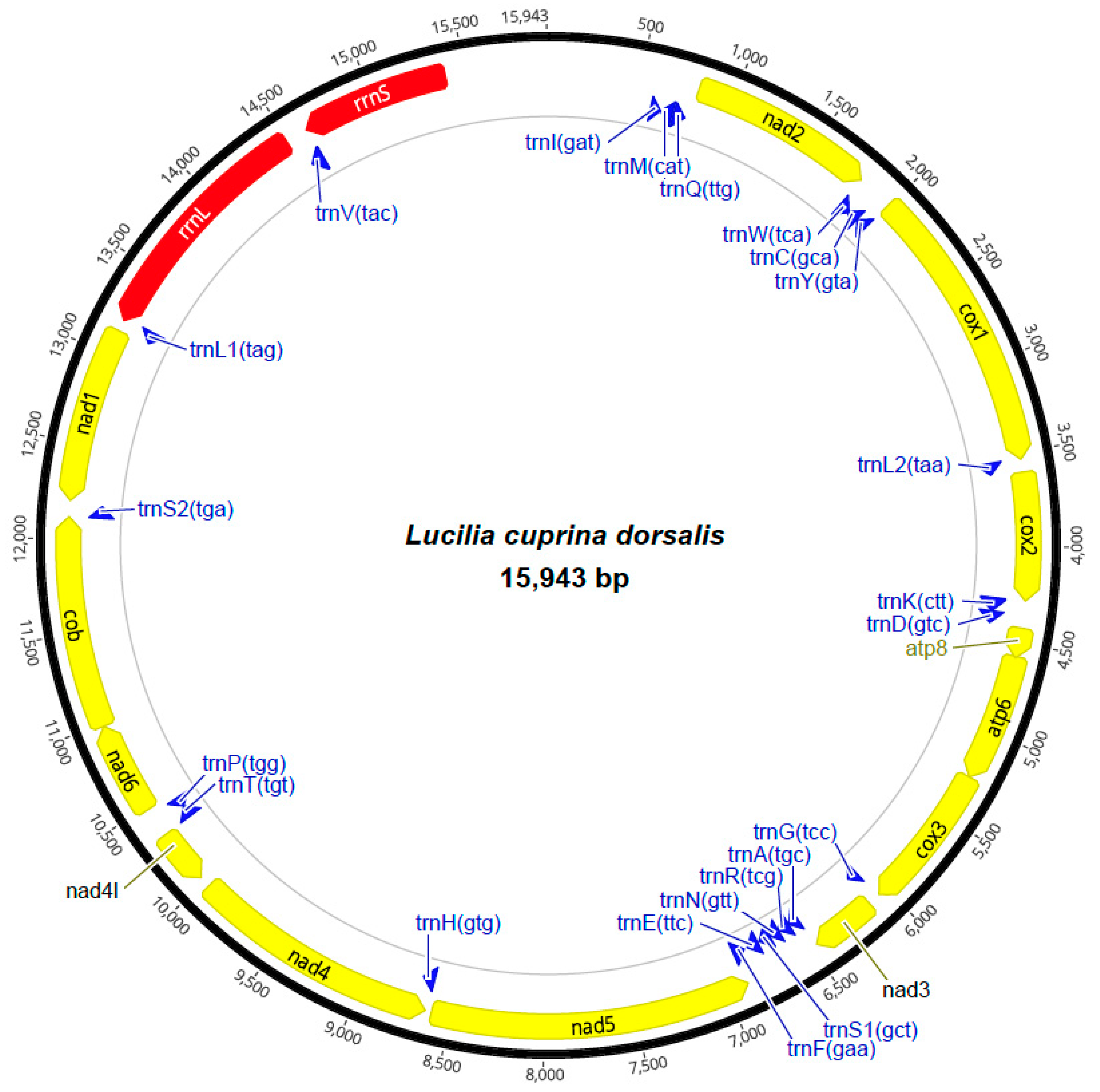
3.1. Mitochondrial Genome Organization and Base Composition Similar to Other Lucilia Species
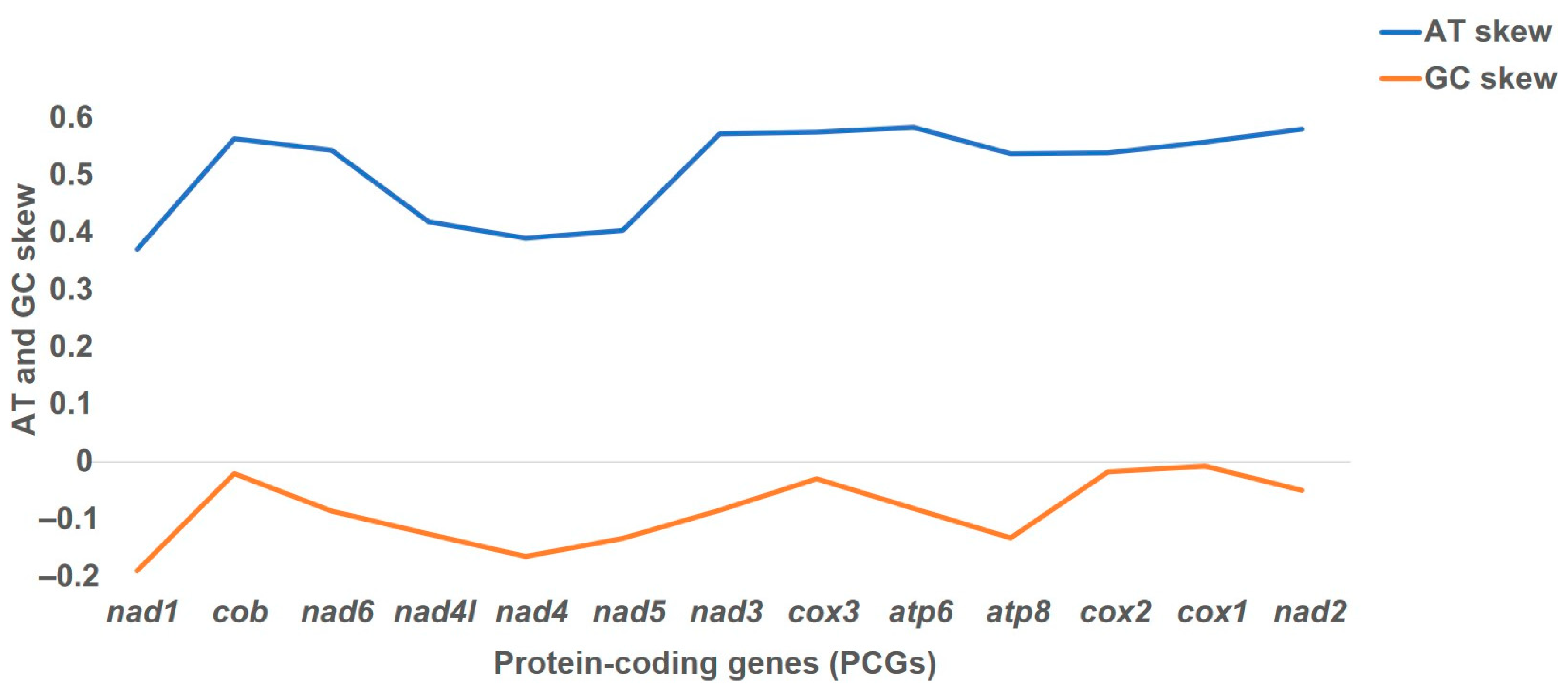
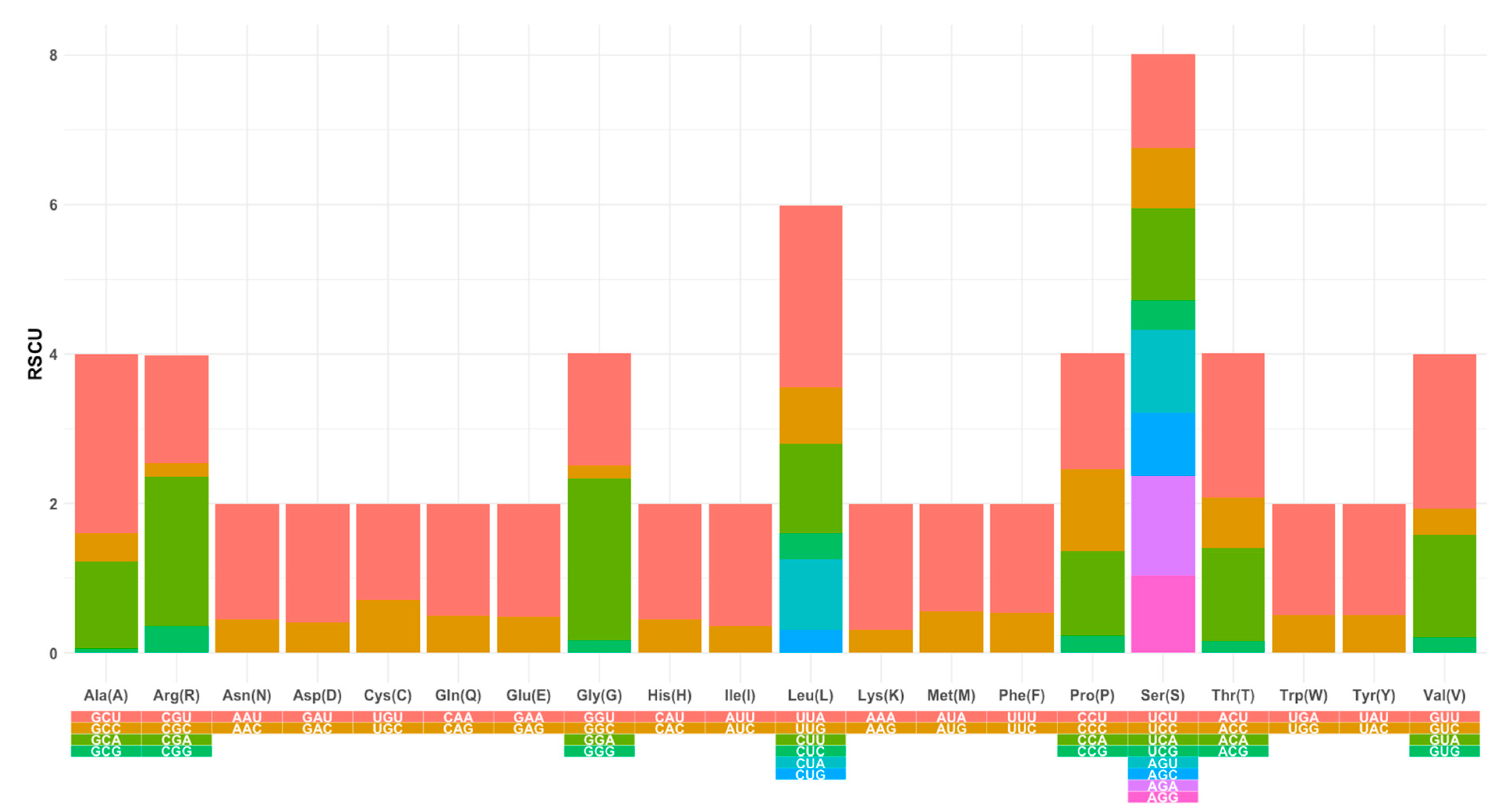
3.2. Protein-Coding Genes (PCGs) Are AT-Biased, and Codon Usage Is Dominant among Serine and Leucine Amino Acids
3.3. Transfer RNAs and Ribosomal RNAs Are AT-Rich
3.4. Nucleotide Polymorphisms Were Detected within Protein-Coding Genes (PCGs)
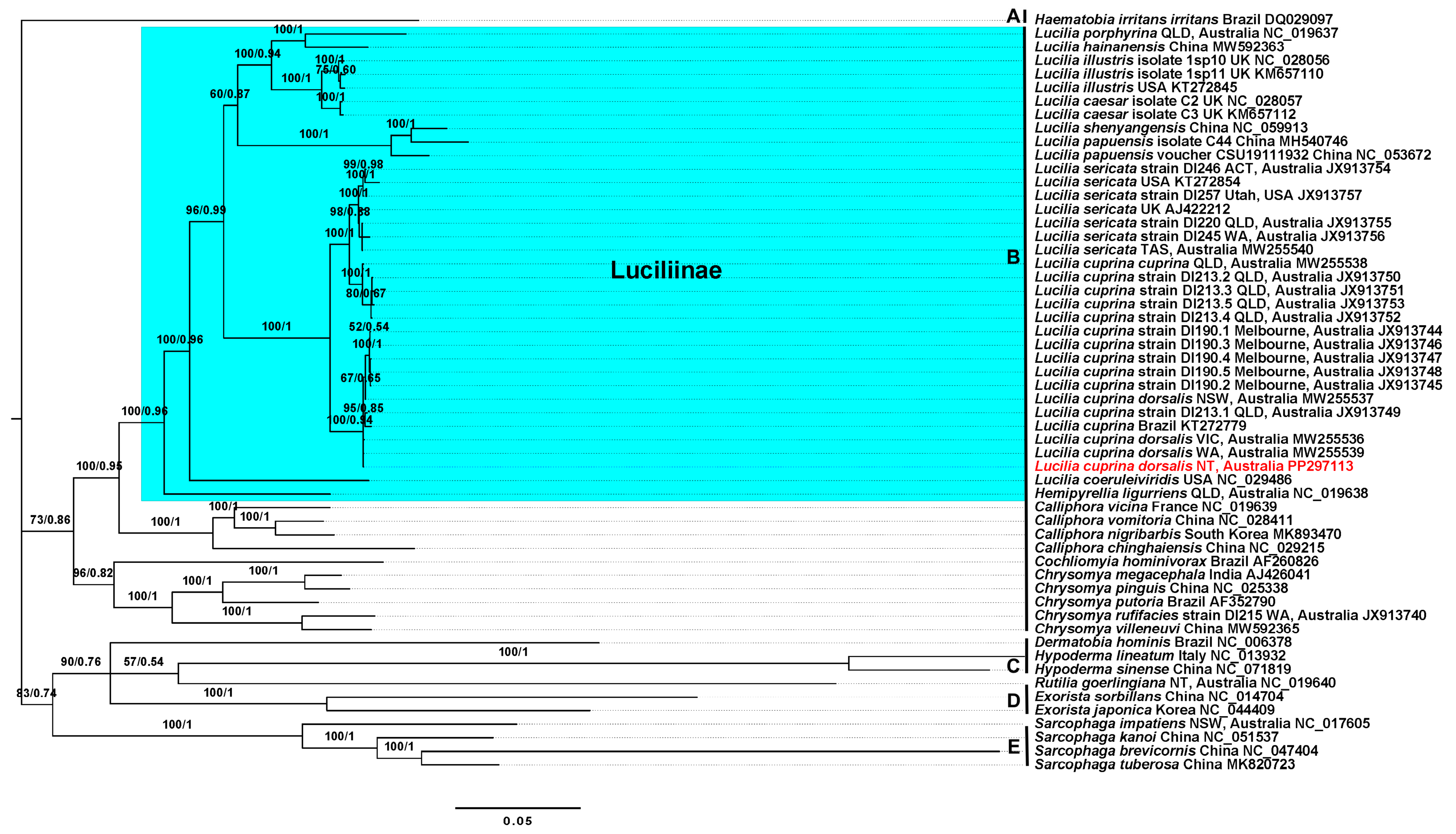
3.5. Phylogenetic Analyses Support Existing Dipteran Clades
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anstead, C.A.; Korhonen, P.K.; Young, N.D.; Hall, R.S.; Jex, A.R.; Murali, S.C.; Hughes, D.S.; Lee, S.F.; Perry, T.; Stroehlein, A.J.; et al. Lucilia cuprina genome unlocks parasitic fly biology to underpin future interventions. Nat. Commun. 2015, 6, 7344. [Google Scholar] [CrossRef]
- Tellam, R.; Bowles, V. Control of blowfly strike in sheep: Current strategies and future prospects. Int. J. Parasitol. 1997, 27, 261–273. [Google Scholar] [CrossRef]
- Lane, J.; Jubb, T.; Shephard, R.; Webb-Ware, J.; Fordyce, G. Priority List of Endemic Diseases for the Red Meat Industries. 2015. Available online: https://era.daf.qld.gov.au/id/eprint/5030/1/B.AHE.0010_Final_Report_Priority%20list%20of%20endemic%20diseases%20for%20the%20red%20meat%20industries.pdf (accessed on 20 February 2024).
- Montossi, F.; Font-i-Furnols, M.; del Campo, M.; San Julián, R.; Brito, G.; Sañudo, C. Sustainable sheep production and consumer preference trends: Compatibilities, contradictions, and unresolved dilemmas. Meat Sci. 2013, 95, 772–789. [Google Scholar] [CrossRef]
- Yan, G.; Liu, S.; Schlink, A.C.; Flematti, G.R.; Brodie, B.S.; Bohman, B.; Greeff, J.C.; Vercoe, P.E.; Hu, J.; Martin, G.B. Behavior and electrophysiological response of gravid and non-gravid Lucilia cuprina (Diptera: Calliphoridae) to carrion-associated compounds. J. Econ. Entomol. 2018, 111, 1958–1965. [Google Scholar] [CrossRef]
- Paul, A.G.; Ahmad, N.W.; Lee, H.; Ariff, A.M.; Saranum, M.; Naicker, A.S.; Osman, Z. Maggot debridement therapy with Lucilia cuprina: A comparison with conventional debridement in diabetic foot ulcers. Int. Wound J. 2009, 6, 39–46. [Google Scholar] [CrossRef]
- Howlett, B. Optimising Pollination of Macadamia and Avocado in Australia. Horticulture Innovation Australia, Final Report, Project: MT13060. 2017. Available online: https://www.horticulture.com.au/globalassets/laserfiche/assets/project-reports/mt13060/mt13060-final-report-complete.pdf (accessed on 20 February 2024).
- Kapoor, S.; Young, N.D.; Yang, Y.T.; Batterham, P.; Gasser, R.B.; Bowles, V.M.; Anstead, C.A.; Perry, T. Mitochondrial genomic investigation reveals a clear association between species and genotypes of Lucilia and geographic origin in Australia. Parasites Vectors 2023, 16, 1–13. [Google Scholar] [CrossRef]
- Nelson, L.A.; Lambkin, C.L.; Batterham, P.; Wallman, J.F.; Dowton, M.; Whiting, M.F.; Yeates, D.K.; Cameron, S.L. Beyond barcoding: A mitochondrial genomics approach to molecular phylogenetics and diagnostics of blowflies (Diptera: Calliphoridae). Gene 2012, 511, 131–142. [Google Scholar] [CrossRef]
- Williams, K.; Villet, M.H. Ancient and modern hybridization between Lucilia sericata and L. cuprina (Diptera: Calliphoridae). Eur. J. Entomol. 2013, 110, 187–196. [Google Scholar] [CrossRef]
- Sandeman, R.M.; Levot, G.W.; Heath, A.C.G.; James, P.J.; Greeff, J.C.; Scott, M.J.; Batterham, P.; Bowles, V.M. Control of the sheep blowfly in Australia and New Zealand–are we there yet? Int. J. Parasitol. 2014, 44, 879–891. [Google Scholar]
- Stevens, J.R.; Wall, R. Species, sub-species and hybrid populations of the blowflies Lucilia cuprina and Lucilia sericata (Diptera: Calliphoridae). Proc. Biol. Sci. 1996, 263, 1335–1341. [Google Scholar]
- Sheep and Goats. Available online: https://nt.gov.au/industry/agriculture/livestock-and-animals/sheep-and-goats (accessed on 1 March 2024).
- Hall, R.N.; Huang, N.; Roberts, J.; Strive, T. Carrion flies as sentinels for monitoring lagovirus activity in Australia. Transbound. Emerg. Dis. 2019, 66, 2025–2032. [Google Scholar] [CrossRef]
- Holloway, B.A. Morphological characters to identify adult Lucilia sericata (Meigen, 1826) and L. cuprina (Wiedemann, 1830) (Diptera: Calliphoridae). N. Z. J. Zool. 1991, 18, 413–420. [Google Scholar]
- Wallman, J.F.; Leys, R.; Hogendoorn, K. Molecular systematics of Australian carrion-breeding blowflies (Diptera: Calliphoridae) based on mitochondrial DNA. Invertebr. Syst. 2005, 19, 1–15. [Google Scholar] [CrossRef]
- Williams, K.A.; Villet, M.H. Morphological identification of Lucilia sericata, Lucilia cuprina and their hybrids (Diptera, Calliphoridae). ZooKeys 2014, 420, 69–85. [Google Scholar]
- Marshall, S.A.; Whitworth, T.; Roscoe, L. Blow flies (Diptera: Calliphoridae) of eastern Canada with a key to Calliphoridae subfamilies and genera of eastern North America, and a key to the eastern Canadian species of Calliphorinae, Luciliinae and Chrysomyiinae. Can. J. Arthropod Identif. 2011, 11, 1–93. [Google Scholar]
- Green, M.R.; Sambrook, J. Isolation of High-Molecular-Weight DNA using organic solvents. Cold Spring Harb. Protoc. 2017, 2017, pdb-prot093450. [Google Scholar] [CrossRef]
- Stevens, J.R.; Wall, R. The use of random amplified polymorphic DNA (RAPD) analysis for studies of genetic variation in populations of the blowfly Lucilia sericata (Diptera: Calliphoridae) in southern England. Bull. Entomol. Res. 1995, 85, 549–555. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 April 2023).
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2007, 24, 172–175. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochrome b gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma Ki Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Schoofs, K.R.; Ahmadzai, U.K.; Goodwin, W. Analysis of the complete mitochondrial genomes of two forensically important blowfly species: Lucilia caesar and Lucilia illustris. Mitochondrial DNA Part B 2018, 3, 1114–1116. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- DeBry, R.W.; Timm, A.E.; Dahlem, G.A.; Stamper, T. mtDNA-based identification of Lucilia cuprina (Wiedemann) and Lucilia sericata (Meigen) (Diptera: Calliphoridae) in the continental United States. Forensic Sci. Int. 2010, 202, 102–109. [Google Scholar]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar]
- Junqueira, A.C.M.; Lessinger, A.C.; Torres, T.T.; da Silva, F.R.; Vettore, A.L.; Arruda, P.; Espin, A.M.L.A. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 2004, 339, 7–15. [Google Scholar] [CrossRef]
- Chen, T.; Li, X.; Wang, Y. The complete mitochondrial genome of Lucilia shenyangensis (Diptera: Calliphoridae). Mitochondrial DNA Part B 2021, 6, 2299–2301. [Google Scholar] [CrossRef]
- Stevens, J.R.; Wall, R.; Wells, J.D. Paraphyly in Hawaiian hybrid blowfly populations and the evolutionary history of anthropophilic species. Insect Mol. Biol. 2002, 11, 141–148. [Google Scholar]
- Negrisolo, E.; Babbucci, M.; Patarnello, T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genom. 2011, 12, 221. [Google Scholar] [CrossRef]
- Yan, L.; Pape, T.; Elgar, M.A.; Gao, Y.; Zhang, D. Evolutionary history of stomach bot flies in the light of mitogenomics. Syst. Entomol. 2019, 44, 797–809. [Google Scholar] [CrossRef]
- Li, X.-Y.; Yan, L.-P.; Pape, T.; Gao, Y.-Y.; Zhang, D. Evolutionary insights into bot flies (Insecta: Diptera: Oestridae) from comparative analysis of the mitochondrial genomes. Int. J. Biol. Macromol. 2020, 149, 371–380. [Google Scholar] [CrossRef]
- Chen, W.-H.; Lu, G.; Bork, P.; Hu, S.; Lercher, M.J. Energy efficiency trade-offs drive nucleotide usage in transcribed regions. Nat. Commun. 2016, 7, 11334. [Google Scholar] [CrossRef]
- Lessinger, A.C.; Junqueira, A.C.M.; Lemos, T.A.; Kemper, E.L.; Da Silva, F.R.; Vettore, A.L.; Arruda, P.; Azeredo-Espin, A.M.L. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol. Biol. 2000, 9, 521–529. [Google Scholar] [CrossRef]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Li, H. Characterization and phylogenetic implications of the complete mitochondrial genome of syrphidae. Genes 2019, 10, 563. [Google Scholar] [CrossRef]
- Stevens, J.R.; Wall, R. Genetic variation in populations of the blowflies Lucilia cuprina and Lucilia sericata (Diptera: Calliphoridae). Random amplified polymorphic DNA analysis and mitochondrial DNA sequences. Biochem. Syst. Ecol. 1997, 25, 81–97. [Google Scholar]
- Tourle, R.; Downie, D.A.; Villet, M.H. Flies in the ointment: A morphological and molecular comparison of Lucilia cuprina and Lucilia sericata (Diptera: Calliphoridae) in South Africa. Med. Veter. Entomol. 2009, 23, 6–14. [Google Scholar] [CrossRef]
- Stevens, J.R.; West, H.; Wall, R. Mitochondrial genomes of the sheep blowfly, Lucilia sericata, and the secondary blowfly, Chrysomya megacephala. Med. Vet. Entomol. 2008, 22, 89–91. [Google Scholar]
- Cameron, S.L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Anstead, C.A.; Perry, T.; Richards, S.; Korhonen, P.K.; Young, N.D.; Bowles, V.M.; Batterham, P.; Gasser, R.B. The battle against flystrike–past research and new prospects through genomics. Adv. Parasitol. 2017, 98, 227–281. [Google Scholar]
- Moving and Exporting Livestock. Available online: https://nt.gov.au/industry/agriculture/livestock-and-animals/moving-and-exporting-livestock (accessed on 1 March 2024).
- Bambaradeniya, Y.T.B.; Karunaratne, W.I.P.; Tomberlin, J.K.; Goonerathne, I.; Kotakadeniya, R.B. Temperature and tissue type impact development of Lucilia cuprina (Diptera: Calliphoridae) in Sri Lanka. J. Med. Entomol. 2017, 55, 285–291. [Google Scholar] [CrossRef]
- Junqueira, A.C.M.; Azeredo-Espin, A.M.L.; Paulo, D.F.; Marinho, M.A.T.; Tomsho, L.P.; Drautz-Moses, D.I.; Purbojati, R.W.; Ratan, A.; Schuster, S.C. Large-scale mitogenomics enables insights into Schizophora (Diptera) radiation and population diversity. Sci. Rep. 2016, 6, 21762. [Google Scholar] [CrossRef]
- Arias-Robledo, G.; Wall, R.; Szpila, K.; Shpeley, D.; Whitworth, T.; Stark, T.; King, R.; Stevens, J. Ecological and geographical speciation in Lucilia bufonivora: The evolution of amphibian obligate parasitism. Int. J. Parasitol. Parasites Wildl. 2019, 10, 218–230. [Google Scholar] [CrossRef]
- Diakova, A.V.; Schepetov, D.M.; Oyun, N.Y.; Shatalkin, A.I.; Galinskaya, T.V. Assessing genetic and morphological variation in populations of Eastern European Lucilia sericata (Diptera: Calliphoridae). Eur. J. Entomol. 2018, 115, 192–197. [Google Scholar] [CrossRef]
- Wall, R.; French, N.P.; Morgan, K.L. Blowfly species composition in sheep myiasis in Britain. Med. Veter. Entomol. 1992, 6, 177–178. [Google Scholar] [CrossRef]
- Kotze, A.C.; James, P.J. Control of sheep flystrike: What’s been tried in the past and where to from here. Aust. Vet. J. 2022, 100, 1–19. [Google Scholar]
- Stevens, J.R.; Wall, R. The evolution of ectoparasitism in the genus Lucilia (Diptera: Calliphoridae). Int. J. Parasitol. 1997, 27, 51–59. [Google Scholar]
- Harvey, M.; Gaudieri, S.; Villet, M.; Dadour, I. A global study of forensically significant calliphorids: Implications for identification. Forensic Sci. Int. 2008, 177, 66–76. [Google Scholar] [CrossRef]
- Palevich, N.; Carvalho, L.; Maclean, P. The complete mitochondrial genome of the New Zealand parasitic blowfly Lucilia sericata (Insecta: Diptera: Calliphoridae). Mitochondrial DNA Part B 2021, 6, 1267–1269. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, S.; Yang, Y.T.; Hall, R.N.; Gasser, R.B.; Bowles, V.M.; Perry, T.; Anstead, C.A. Complete Mitochondrial Genome for Lucilia cuprina dorsalis (Diptera: Calliphoridae) from the Northern Territory, Australia. Genes 2024, 15, 506. https://doi.org/10.3390/genes15040506
Kapoor S, Yang YT, Hall RN, Gasser RB, Bowles VM, Perry T, Anstead CA. Complete Mitochondrial Genome for Lucilia cuprina dorsalis (Diptera: Calliphoridae) from the Northern Territory, Australia. Genes. 2024; 15(4):506. https://doi.org/10.3390/genes15040506
Chicago/Turabian StyleKapoor, Shilpa, Ying Ting Yang, Robyn N. Hall, Robin B. Gasser, Vernon M. Bowles, Trent Perry, and Clare A. Anstead. 2024. "Complete Mitochondrial Genome for Lucilia cuprina dorsalis (Diptera: Calliphoridae) from the Northern Territory, Australia" Genes 15, no. 4: 506. https://doi.org/10.3390/genes15040506
APA StyleKapoor, S., Yang, Y. T., Hall, R. N., Gasser, R. B., Bowles, V. M., Perry, T., & Anstead, C. A. (2024). Complete Mitochondrial Genome for Lucilia cuprina dorsalis (Diptera: Calliphoridae) from the Northern Territory, Australia. Genes, 15(4), 506. https://doi.org/10.3390/genes15040506