Dissecting the Polygenic Basis of Cold Adaptation Using Genome-Wide Association of Traits and Environmental Data in Douglas-fir

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Whole-Genome Re-Sequencing
2.2. SNP Calling
2.3. Sample Collection Prior to Genotyping
2.4. DNA Extraction and SNP Genotyping
2.5. Population Structure
2.6. Isolation by Distance
2.7. Multivariate and Univariate Genome-Wide Association (GWAS) of Cold-Related Traits
2.8. Multivariate and Univariate Genotype-Environment Association Analyses (GEA)
2.9. Genome Scan for Detection of Selection Signatures
3. Results
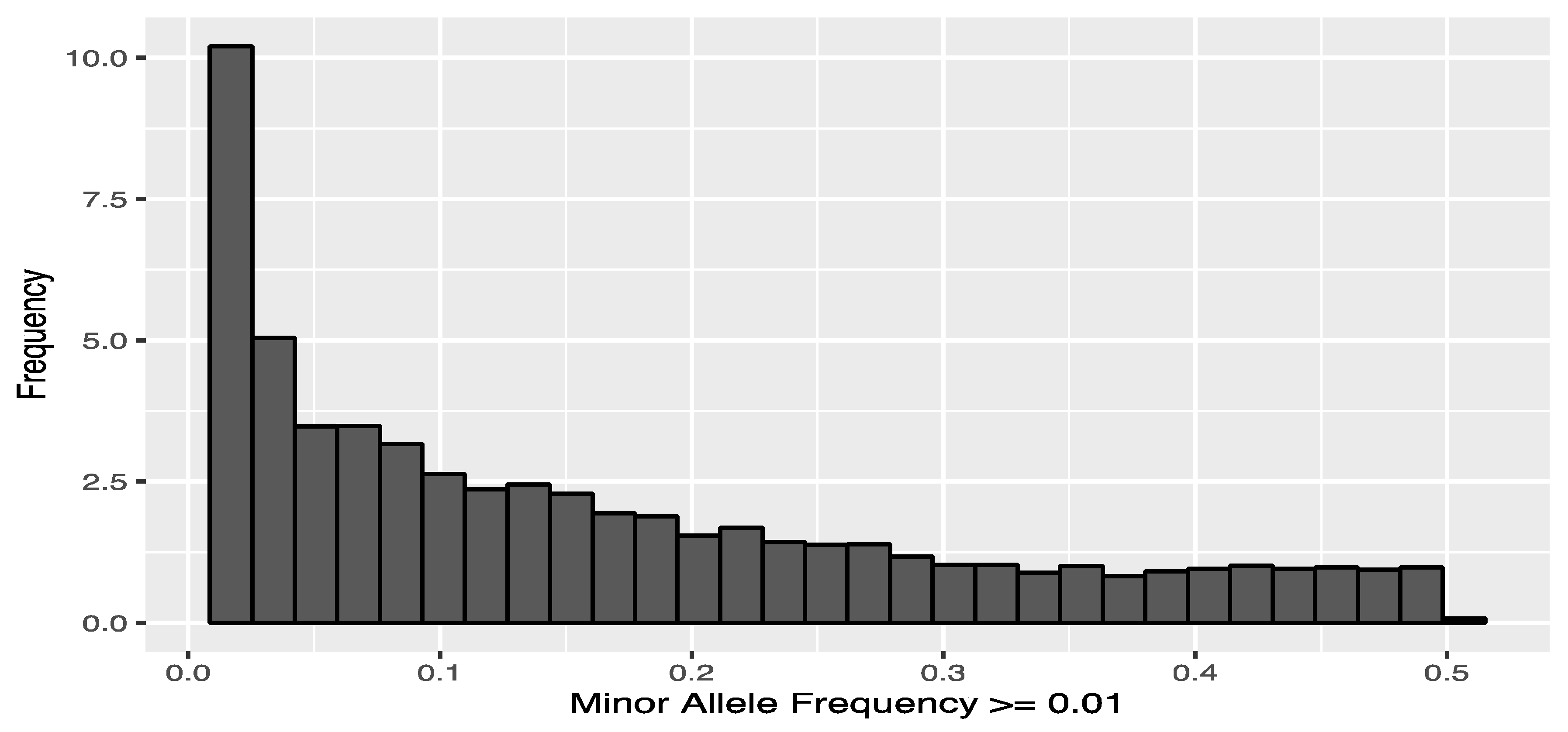
3.1. SNP Calling and SNP Genotyping
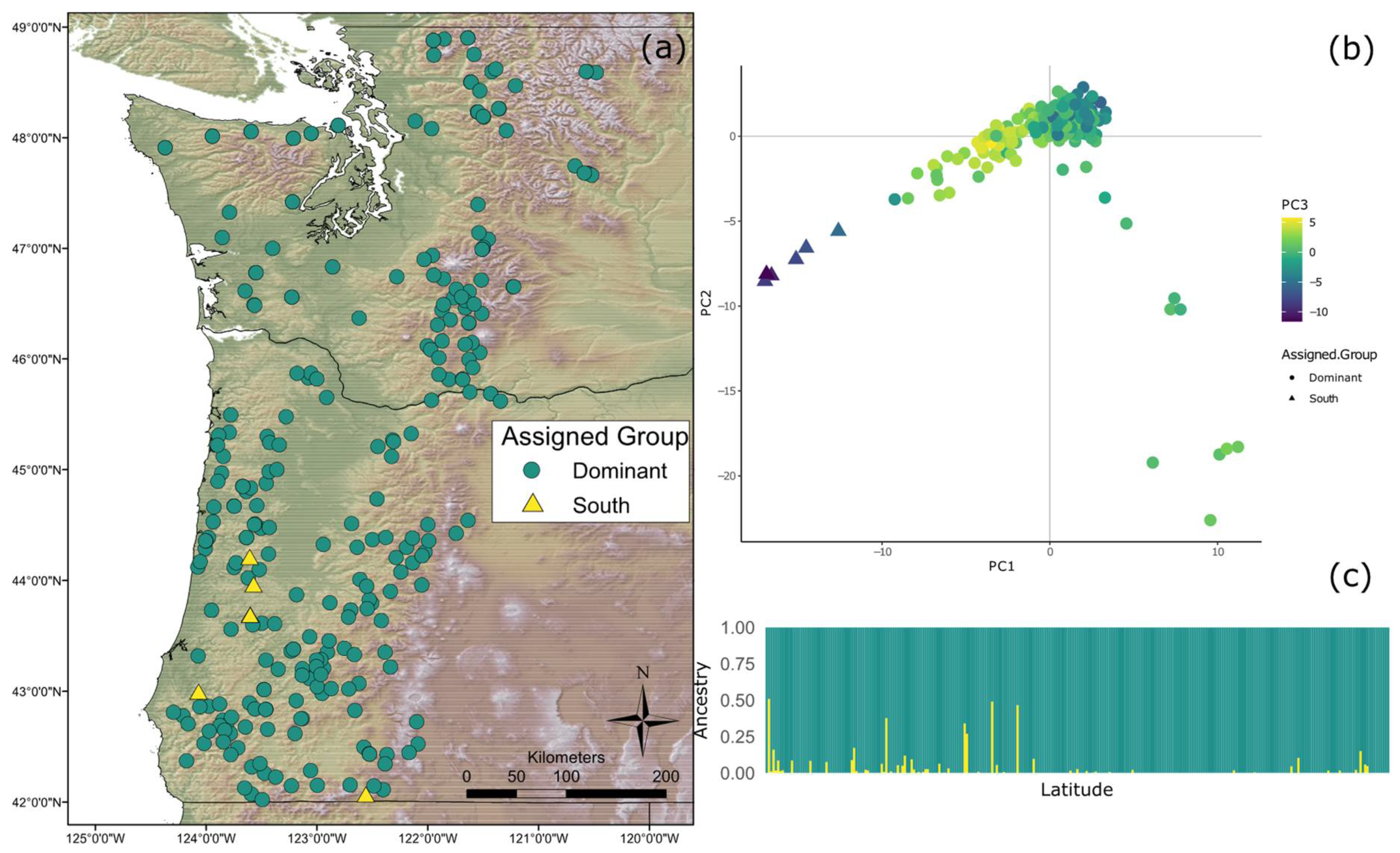
3.2. Population Structure
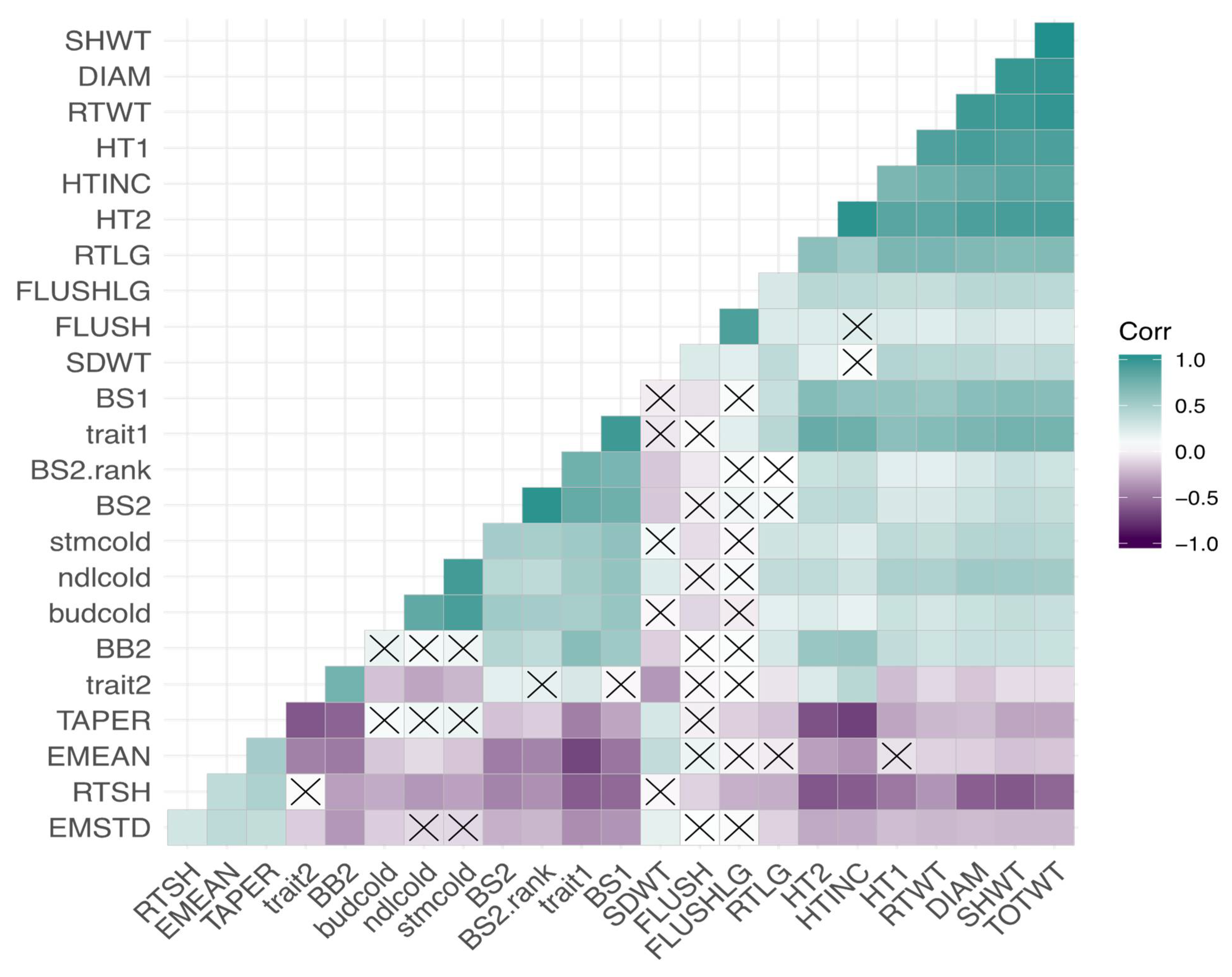
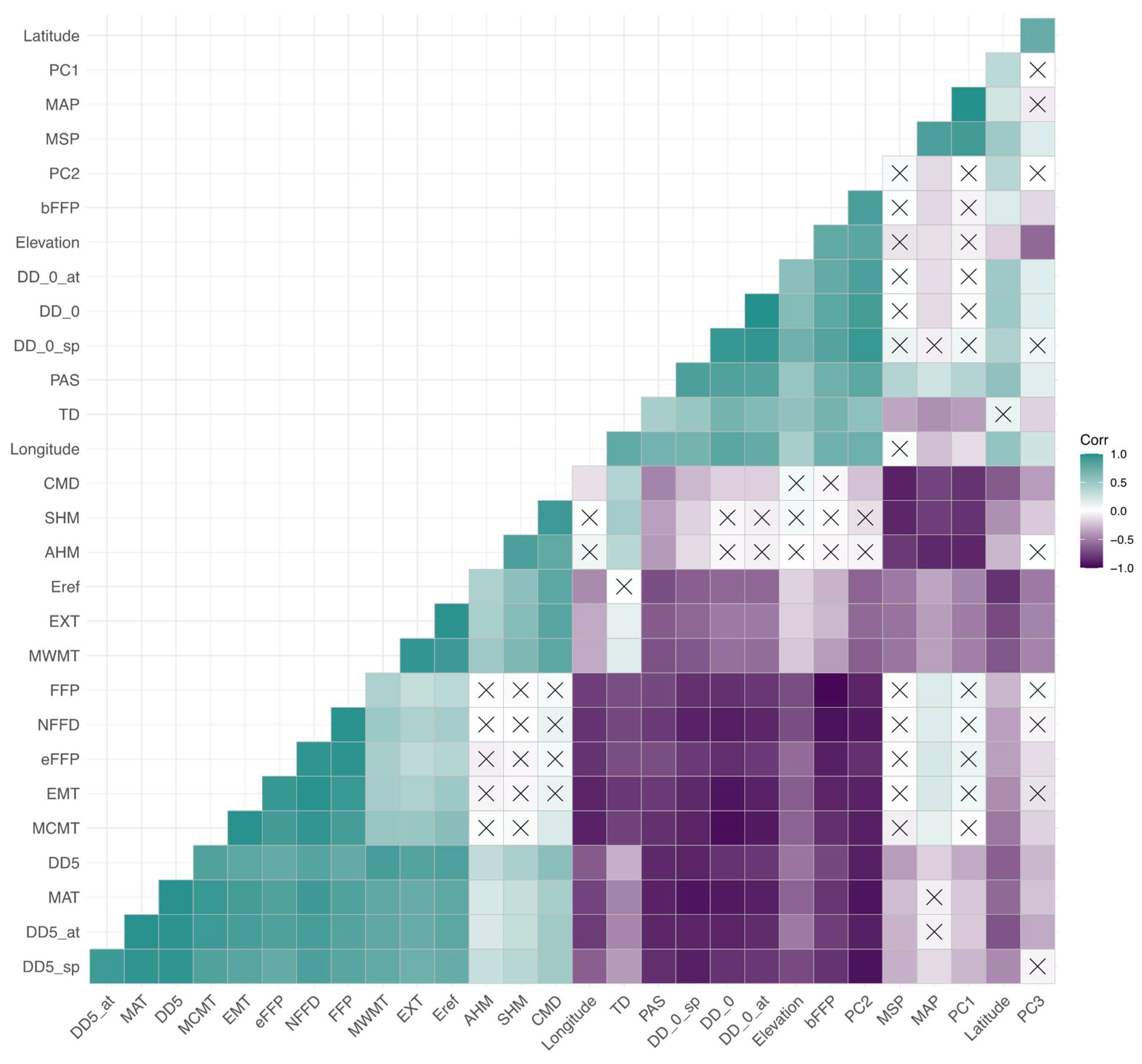
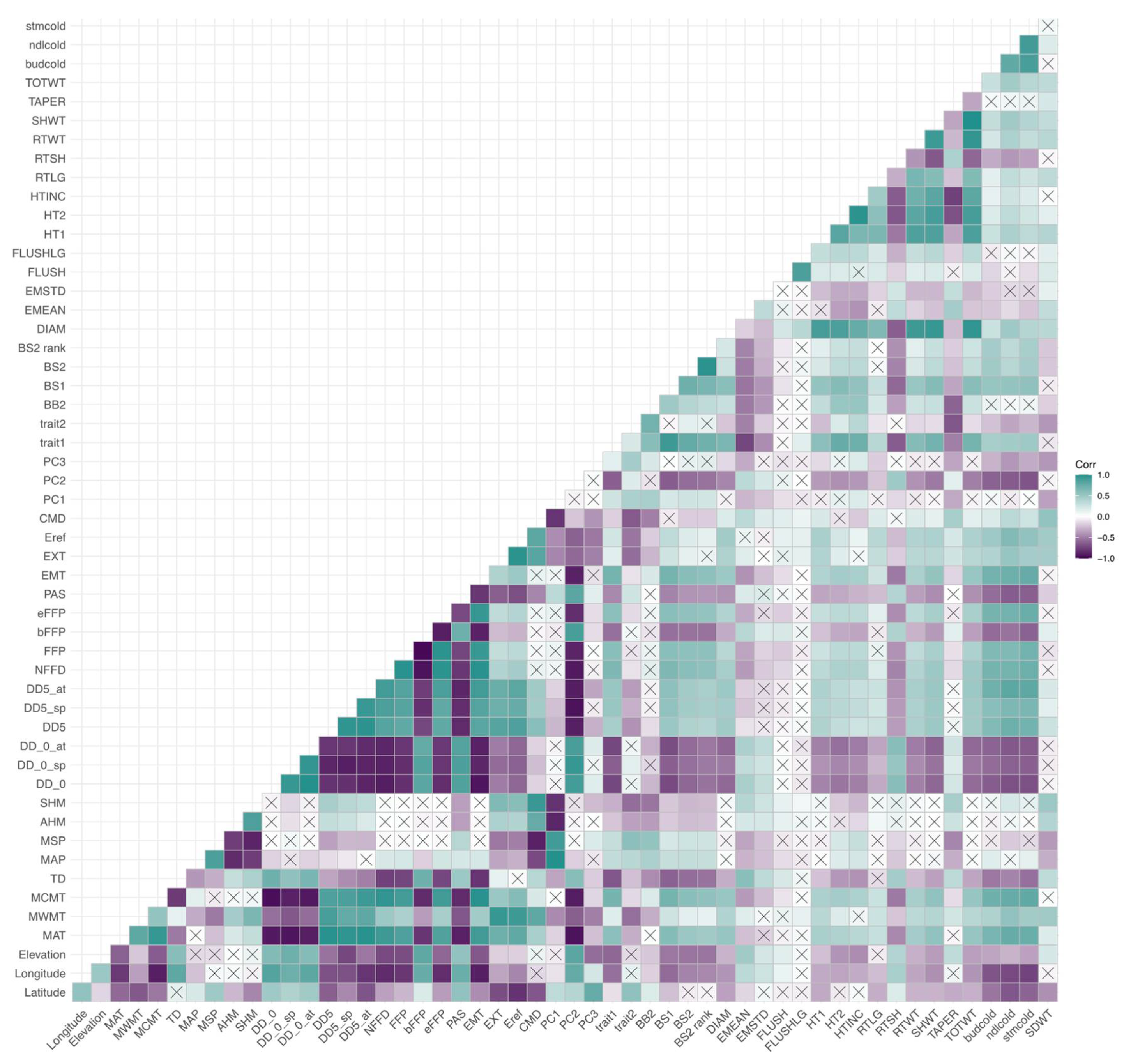
3.3. Correlations among Cold-Related Phenotypic Traits and Environmental Variables
3.4. Genome Scan for Detection of Selection Signatures
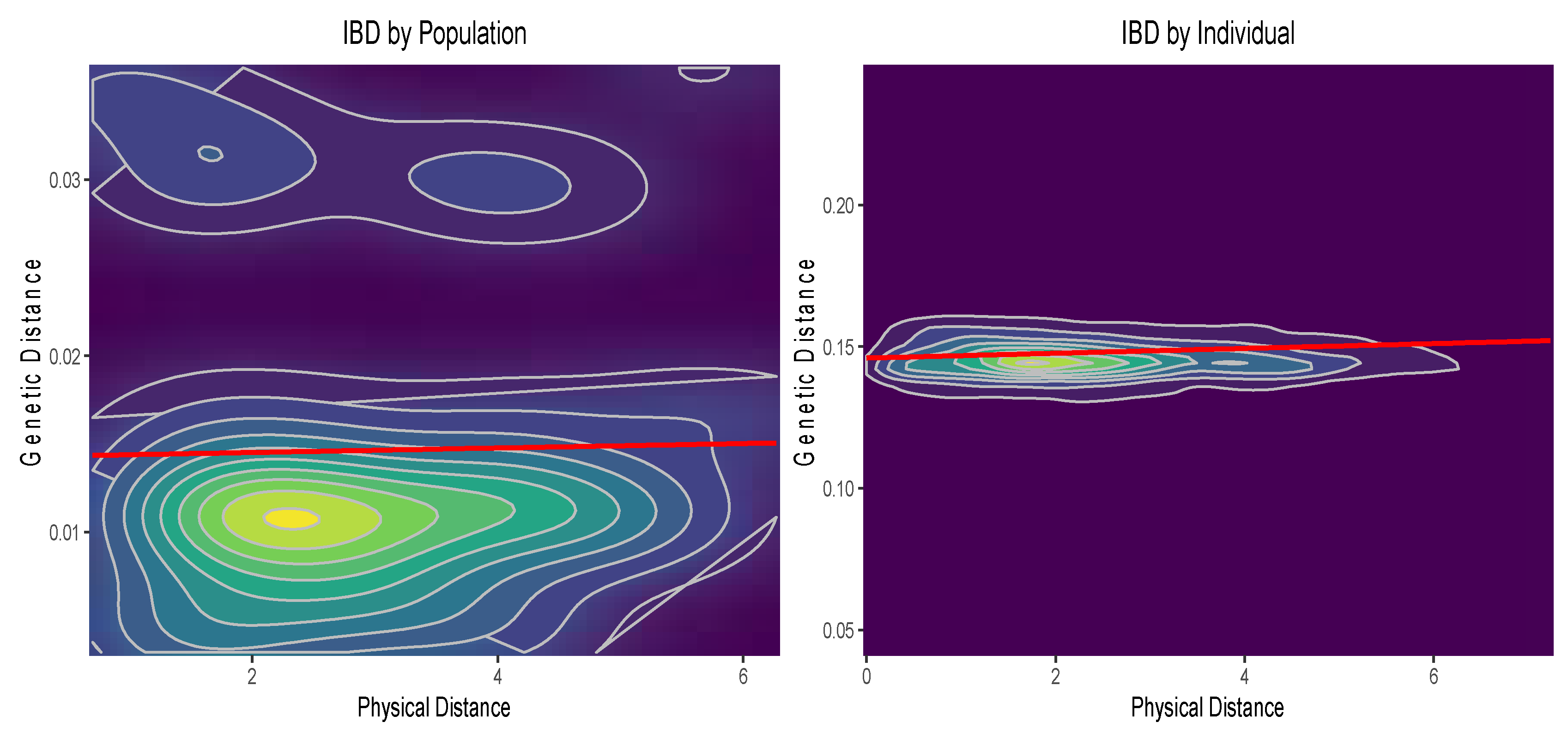
3.5. Isolation by Distance
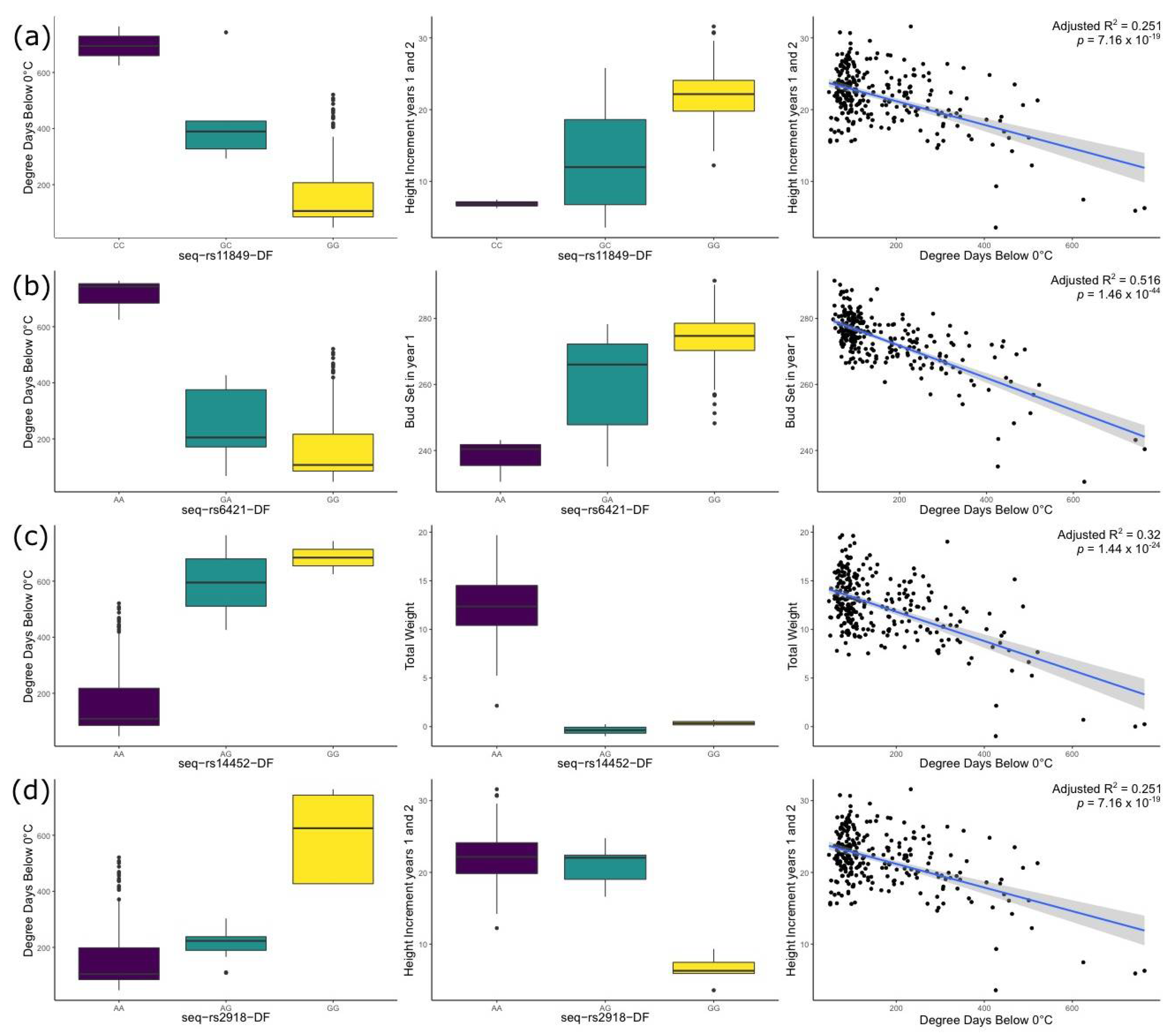
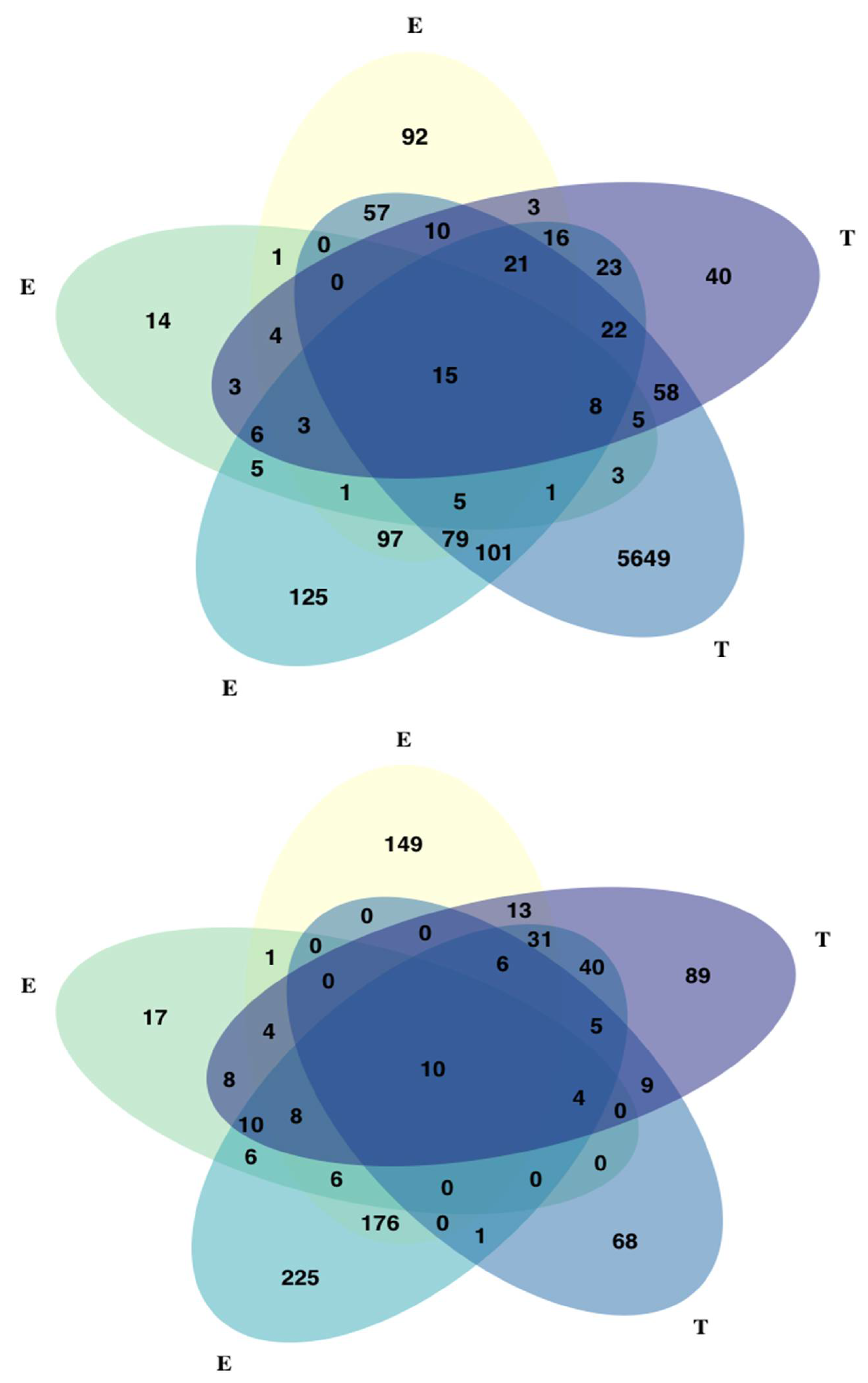
3.6. Univariate and Multivariate Genome-Wide Association Study of Cold-Related Traits
3.7. Univariate and Multivariate Genotype-Environment Association Analyses (GEA)
4. Discussion
4.1. Polygenic Basis of Cold Adaptation in Coastal Douglas-fir
4.2. Trade-Offs between Growth and Cold Hardiness
4.3. Functional Characterization of Genes Associated with Cold Adaptation
5. Conclusions and Predictions in the Face of Climate Change
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
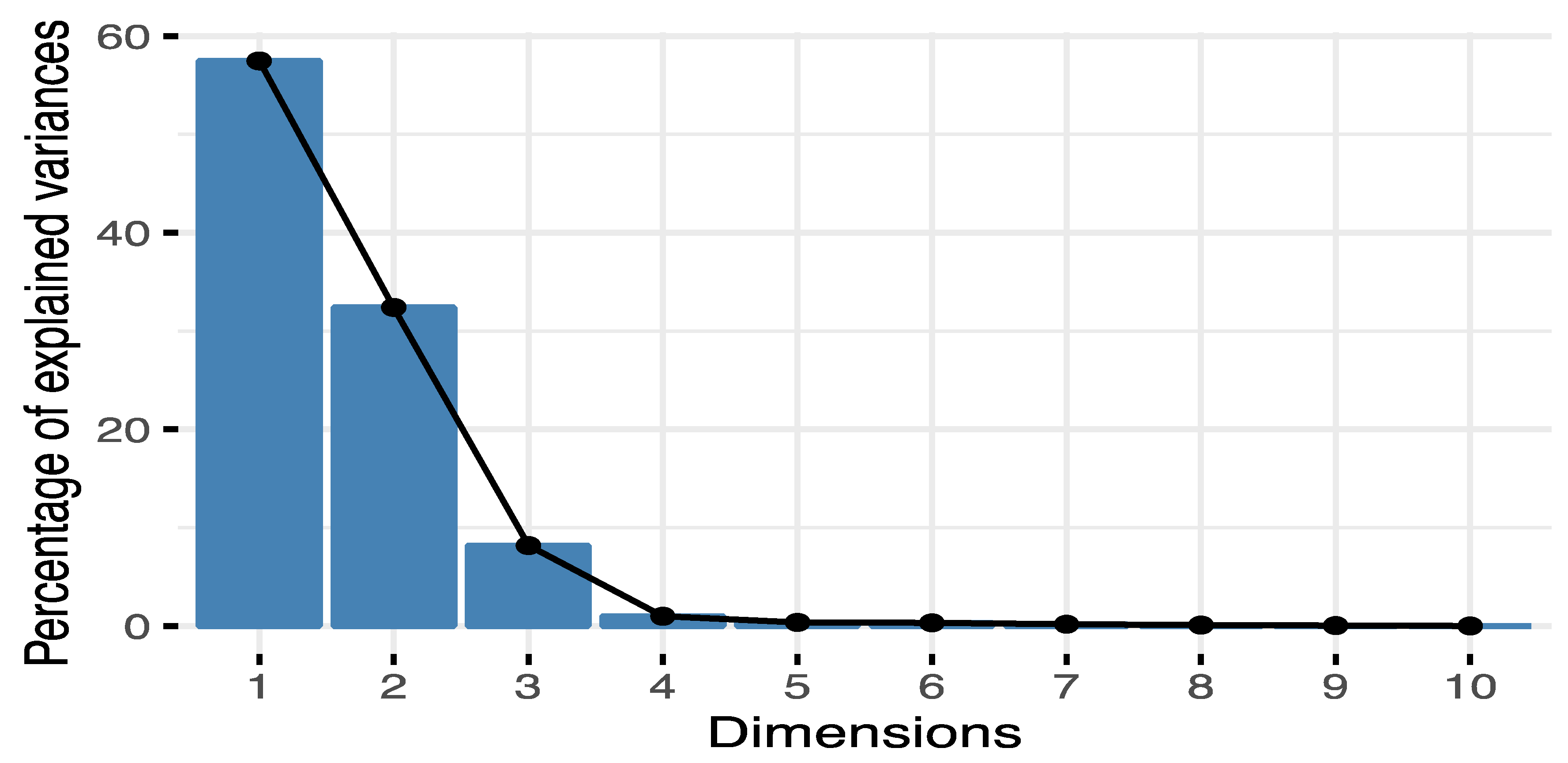{kind=link}
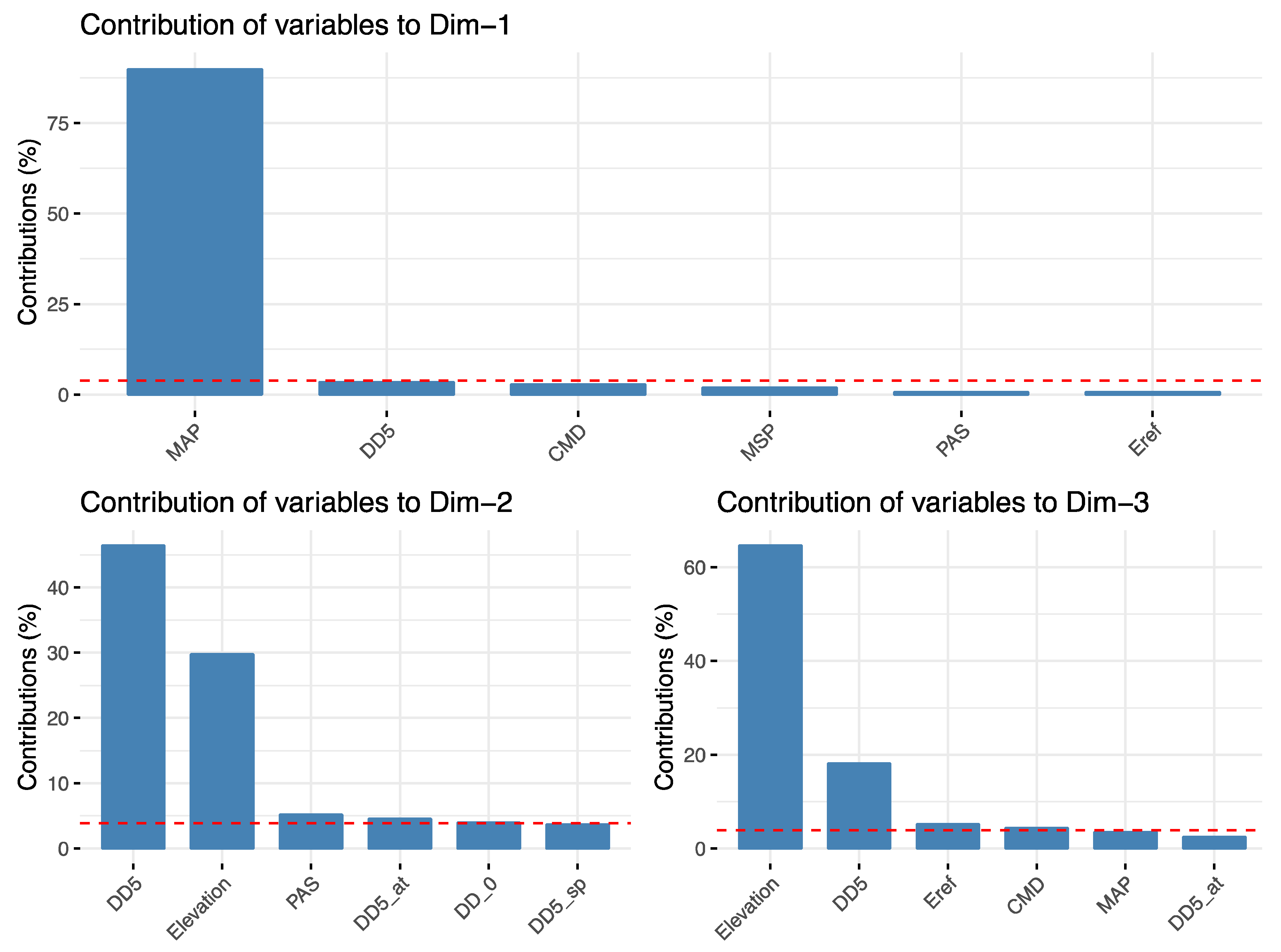{kind=link}
| Bio Process | Trait ID | Trait Description | Unit |
|---|---|---|---|
| Several | Trait1 | First canonical variate for traits | -- |
| Several | Trait2 | Second canonical variate for traits | -- |
| Phenology | BB2 | Budburst in year 2 | days since Jan 1 |
| Phenology | BS1 | Budset in year 1 | days since Jan 1 |
| Phenology | BS2 | Budset in year 2 | days since Jan 1 |
| Growth | DIAM | Diameter after year 2 | mm |
| Emergence | EMEAN | Rate of emergence | probits/day |
| Emergence | EMSTD | Standard deviation of emergence rate | probits/day |
| Emergence | FLUSH | Propensity to second flush | proportion |
| Emergence | FLUSHLG | Length of second flush | cm |
| Growth | HT1 | Height after year 1 | cm |
| Growth | HT2 | Height after year 2 | cm |
| Growth | HTINC | Height increment between years 1 & 2 | cm |
| Growth | RTLG | Root length | cm |
| Growth | RTSH | Root:shoot ratio | g/g |
| Growth | RTWT | Root weight | g |
| Growth | SHWT | Shoot weight | g |
| Growth | SDWT | Weight of 100 seeds | g |
| Growth | TAPER | Taper | mm/cm |
| Growth | TOTWT | Total weight | g |
| Cold hardiness | budcold | Cold damage of buds | %/10 |
| Cold hardiness | ndlcold | Cold damage of needles | %/10 |
| Cold hardiness | stmcold | Cold damage of stems | %/10 |
| Variable ID | Variable Name | Unit | Variable Description |
|---|---|---|---|
| Latitude | Latitude | degrees | From GIS after mapping parents. |
| Longitude | Longitude | degrees | From GIS after mapping parents. |
| Elevation | Elevation | m | From DEM after mapping parents. |
| MAT | Mean annual temperature | degrees C | Mean annual temperature |
| MWMT | Mean warmest month temperature | degrees C | Mean warmest month temperature |
| MCMT | Mean coldest month temperature | degrees C | Mean coldest month temperature |
| TD | Temperature difference between MWMT and MCMT, or continentality | degrees C | Temperature difference between MWMT and MCMT, or continentality |
| MAP | Mean annual precipitation | mm | Mean annual precipitation |
| MSP | May to September precipitation | mm | May to September precipitation |
| AHM | Annual heat-moisture index | (MAT+1-)/(MAP/1000) | |
| SHM | Summer heat-moisture index | (MWMT)/(MSP/1000) | |
| DD_0 | Degree-days below zero | days | chilling degree-days |
| DD_0_sp | Degree-days below zero in spring | days | chilling degree-days in spring |
| DD_0_at | Degree-days below zero in autumn | days | chilling degree-days in autumn |
| DD5 | Degree-days above 5 °C | degrees C | growing degree-days |
| DD5_sp | Degree-days above 5 °C in spring | degrees C | growing degree-days in spring |
| DD5_at | Degree-days above 5 °C in autumn | degrees C | growing degree-days in autumn |
| NFFD | Number of frost-free days | days | Number of frost-free days |
| FFP | Frost-free period | days | Frost-free period |
| bFFP | The day of the year FFP begins | day of year | The day of the year FFP begins |
| eFFP | The day of the year on which FFP ends | day of year | The day of the year on which FFP ends |
| PAS | Precipitation as snow | mm | Precipitation as snow |
| EMT | Extreme minimum temperature | degrees C | Lowest temperature over 30 years |
| EXT | Extreme maximum temperature | degrees C | Highest temperature over 30 years |
| Eref | Hargreaves reference evaporation | mm | Hargreaves reference evaporation |
| CMD | Hargreaves climatic moisture deficit | mm | Hargreaves climatic moisture deficit |
| PC1 | Principal component 1 | First principal component of climate variables | |
| PC2 | Principal component 2 | Second principal component of climate variables | |
| PC3 | Principal component 3 | Third principal component of climate variables |








References
- Stinchcombe, J.R.; Hoekstra, H.E. Combining population genomics and quantitative genetics: Finding the genes underlying ecologically important traits. Heredity 2007, 100, 158–170. [Google Scholar] [CrossRef]
- Feder, M.E.; Mitchell-Olds, T. Evolutionary and ecological functional genomics. Nat. Rev. Genet. 2003, 4, 649–655. [Google Scholar] [CrossRef]
- Ghalambor, C.K.; McKay, J.K.; Carroll, S.P.; Reznick, D.N. Adaptive versus non-adaptive phenotypic plasticity and the potential for contemporary adaptation in new environments. Funct. Ecol. 2007, 21, 394–407. [Google Scholar] [CrossRef]
- Savolainen, O.; Pyhäjärvi, T.; Knürr, T. Gene Flow and Local Adaptation in Trees. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 595–619. [Google Scholar] [CrossRef]
- Le Corre, V.; Kremer, A. The genetic differentiation at quantitative trait loci under local adaptation. Mol. Ecol. 2012, 21, 1548–1566. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, A.R.; Wilhite, B.; Neale, D.B. Environmental Genome-Wide Association Reveals Climate Adaptation Is Shaped by Subtle to Moderate Allele Frequency Shifts in Loblolly Pine. Genome Biol. Evol. 2019, 11, 2976–2989. [Google Scholar] [CrossRef]
- Neale, D.B.; Kremer, A. Forest tree genomics: Growing resources and applications. Nat. Rev. Genet. 2011, 12, 111–122. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Di Rienzo, A. Adaptation—Not by sweeps alone. Nat. Rev. Genet. 2010, 11, 665–667. [Google Scholar] [CrossRef]
- Howe, G.T.; Aitken, S.N.; Neale, D.B.; Jermstad, K.D.; Wheeler, N.C.; Chen, T.H.H. From genotype to phenotype: Unraveling the complexities of cold adaptation in forest trees. Can. J. Bot. 2003, 81, 1247–1266. [Google Scholar] [CrossRef]
- Ensminger, I.; Yao-Yun Chang, C.; Bräutigam, K. Chapter Seven—Tree Responses to Environmental Cues. In Advances in Botanical Research; Plomion, C., Adam-Blondon, A.-F., Eds.; Land Plants—Trees; Academic Press: Cambridge, MA, USA, 2015; Volume 74, pp. 229–263. [Google Scholar]
- Holliday, J.A.; Ralph, S.G.; White, R.; Bohlmann, J.; Aitken, S.N. Global monitoring of autumn gene expression within and among phenotypically divergent populations of Sitka spruce (Picea sitchensis). New Phytol. 2008, 178, 103–122. [Google Scholar] [CrossRef]
- Morgenstern, E.K. Geographic Variation in Forest Trees: Genetic Basis and Application of Knowledge in Silviculture; UBC Press: Vancouver, BC, Canada, 1996; ISBN 978-0-7748-0560-5. [Google Scholar]
- Whitlock, M.C.; Lotterhos, K.E. Reliable Detection of Loci Responsible for Local Adaptation: Inference of a Null Model through Trimming the Distribution of FST. Am. Nat. 2015, 186, S24–S36. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, S. Local adaptation by alleles of small effect. Am. Nat. 2015, 186 (S1), S74–S89. [Google Scholar] [CrossRef] [PubMed]
- Pulkrab, K.; Sloup, M.; Zeman, M. Economic Impact of Douglas-fir (Pseudotsuga menziesii [Mirb.] Franco) production in the Czech Republic. J. For. Sci. 2014, 60, 297–306. [Google Scholar] [CrossRef]
- Curtis, R.O.; Carey, A.B. Timber Supply in the Pacific Northwest: Managing for Economic and Ecological Values in Douglas-fir Forest. J. For. 1996, 94, 35–37. [Google Scholar]
- St. Clair, J.B. Genetic variation in fall cold hardiness in coastal Douglas-fir in western Oregon and Washington. Can. J. Bot. 2006, 84, 1110–1121. [Google Scholar] [CrossRef]
- Bansal, S.; Harrington, C.A.; Gould, P.J.; St. Clair, J.B. Climate-related genetic variation in drought-resistance of Douglas-fir (Pseudotsuga menziesii). Glob. Chang. Biol. 2015, 21, 947–958. [Google Scholar] [CrossRef]
- Bansal, S.; Harrington, C.A.; St. Clair, J.B. Tolerance to multiple climate stressors: A case study of Douglas-fir drought and cold hardiness. Ecol. Evol. 2016, 6, 2074–2083. [Google Scholar] [CrossRef]
- Wei, X.-X.; Beaulieu, J.; Khasa, D.P.; Vargas-Hernández, J.; López-Upton, J.; Jaquish, B.; Bousquet, J.; Vargas-Hernández, J.J. Range-wide chloroplast and mitochondrial DNA imprints reveal multiple lineages and complex biogeographic history for Douglas-fir. Tree Genet. Genomes 2011, 7, 1025–1040. [Google Scholar] [CrossRef]
- Ford, K.R.; Harrington, C.A.; Bansal, S.; Gould, P.J.; St. Clair, J.B. Will changes in phenology track climate change? A study of growth initiation timing in coast Douglas-fir. Glob. Chang. Biol. 2016, 22, 3712–3723. [Google Scholar] [CrossRef]
- Ford, K.R.; Harrington, C.A.; St. Clair, J.B. Photoperiod cues and patterns of genetic variation limit phenological responses to climate change in warm parts of species’ range: Modeling diameter-growth cessation in coast Douglas-fir. Glob. Chang. Biol. 2017, 23, 3348–3362. [Google Scholar] [CrossRef]
- Cronn, R.C.; Dolan, P.C.; Jogdeo, S.; Wegrzyn, J.L.; Neale, D.B.; St. Clair, J.B.; Denver, D.R. Transcription through the eye of a needle: Daily and annual cyclic gene expression variation in Douglas-fir needles. BMC Genom. 2017, 18, 558. [Google Scholar] [CrossRef] [PubMed]
- Krutovsky, K.V.; Troggio, M.; Brown, G.R.; Jermstad, K.D.; Neale, D.B. Comparative Mapping in the Pinaceae. Genetics 2004, 168, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, N.C.; Jermstad, K.D.; Krutovsky, K.V.; Aitken, S.N.; Howe, G.T.; Krakowski, J.; Neale, D.B. Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. IV. Cold-hardiness QTL verification and candidate gene mapping. Mol. Breed. 2005, 15, 145–156. [Google Scholar] [CrossRef]
- Eckert, A.J.; Bower, A.D.; Wegrzyn, J.L.; Pande, B.; Jermstad, K.D.; Krutovsky, K.V.; St. Clair, J.B.; Neale, D.B. Association Genetics of Coastal Douglas fir (Pseudotsuga menziesii var. menziesii, Pinaceae). I. Cold-Hardiness Related Traits. Genetics 2009, 182, 1289–1302. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-fir Genome Sequence Reveals Specialization of the Photosynthetic Apparatus in Pinaceae. G3 Genes Genomes Genet. 2017, 7, 3157–3167. [Google Scholar] [CrossRef]
- TreeGenes Database. Douglas fir Genome. Available online: https://treegenesdb.org/FTP/Genomes/Psme/v1.0 (accessed on 16 November 2020).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- St. Clair, J.B.; Mandel, N.L.; Vance-Borland, K.W. Genecology of Douglas fir in Western Oregon and Washington. Ann. Bot. 2005, 96, 1199–1214. [Google Scholar] [CrossRef]
- Vangestel, C.; Eckert, A.J.; Wegrzyn, J.L.; Clair, J.B.S.; Neale, D.B. Linking phenotype, genotype and environment to unravel genetic components underlying cold hardiness in coastal Douglas-fir (Pseudotsuga menziesii var. menziesii). Tree Genet. Genomes 2018, 14, 10. [Google Scholar] [CrossRef]
- TreeGenes Database. Douglas fir Genome Annotations. Available online: https://treegenesdb.org/FTP/Genomes/Psme/v1.0/annotation/Psme.1_0.entap_annotations.tsv.gz (accessed on 16 November 2020).
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Francis, R.M. Pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Pembleton, L.W.; Cogan, N.O.I.; Forster, J.W. St AMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 2013, 13, 946–952. [Google Scholar] [CrossRef]
- Chessel, D.; Dufour, A.B.; Thioulouse, J. The Ade4 Package—I: One-Table Methods. R News 2004, 4, 5–10. [Google Scholar]
- White, T.L.; Hodge, G.R. Predicting Breeding Values with Applications in Forest Tree Improvement; Springer: Berlin/Heidelberg, Germany, 1989. [Google Scholar]
- Zhou, X.; Carbonetto, P.; Stephens, M. Polygenic Modeling with Bayesian Sparse Linear Mixed Models. PLoS Genet. 2013, 9, e1003264. [Google Scholar] [CrossRef]
- Comeault, A.A.; Soria-Carrasco, V.; Gompert, Z.; Farkas, T.E.; Buerkle, C.A.; Parchman, T.L.; Nosil, P. Genome-Wide Association Mapping of Phenotypic Traits Subject to a Range of Intensities of Natural Selection in Timema cristinae. Am. Nat. 2014, 183, 711–727. [Google Scholar] [CrossRef]
- Boyles, R.E.; Cooper, E.A.; Myers, M.T.; Brenton, Z.; Rauh, B.L.; Morris, G.P.; Kresovich, S. Genome-Wide Association Studies of Grain Yield Components in Diverse Sorghum Germplasm. Plant Genome 2016, 9, 1–17. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Wang, T.; Hamann, A.; Spittlehouse, D.; Carroll, C. Locally Downscaled and Spatially Customizable Climate Data for Historical and Future Periods for North America. PLoS ONE 2016, 11, e0156720. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Neilson, R.P.; Phillips, D.L. A Statistical-Topographic Model for Mapping Climatological Precipitation over Mountainous Terrain. J. Appl. Meteor. 1994, 33, 140–158. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2019. Available online: http://www.sortie-nd.org/lme/R%20Packages/vegan.pdf (accessed on 10 December 2019).
- Günther, T.; Coop, G. Robust Identification of Local Adaptation from Allele Frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef]
- Luu, K.; Bazin, E.; Blum, M.G.B. Pcadapt: An Rpackage to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 2017, 17, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure: Program note. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Aitken, S.N.; Adams, W.T. Spring Cold Hardiness under Strong Genetic Control in Oregon Populations of Pseudotsuga menziesii var. menziesii. Can. J. For. Res. 1997, 27, 1773–1780. [Google Scholar] [CrossRef][Green Version]
- Sun, Y.-Q.; Zhao, W.; Xu, C.-Q.; Xu, Y.; El-Kassaby, Y.A.; De La Torre, A.R.; Mao, J.-F. Genetic Variation Related to High Elevation Adaptation Revealed by Common Garden Experiments in Pinus yunnanensis. Front. Genet. 2020, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- Csilléry, K.; Lalagüe, H.; Vendramin, G.G.; González-Martínez, S.C.; Fady, B.; Oddou-Muratorio, S. Detecting short spatial scale local adaptation and epistatic selection in climate-related candidate genes in European beech (Fagus sylvatica) populations. Mol. Ecol. 2014, 23, 4696–4708. [Google Scholar] [CrossRef] [PubMed]
- Hornoy, B.; Pavy, N.; Gérardi, S.; Beaulieu, J.; Bousquet, J. Genetic Adaptation to Climate in White Spruce Involves Small to Moderate Allele Frequency Shifts in Functionally Diverse Genes. Genome Biol. Evol. 2015, 7, 3269–3285. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.J.; Wegrzyn, J.L.; Pande, B.; Jermstad, K.D.; Lee, J.M.; Liechty, J.D.; Tearse, B.R.; Krutovsky, K.V.; Neale, D.B. Multilocus Patterns of Nucleotide Diversity and Divergence Reveal Positive Selection at Candidate Genes Related to Cold Hardiness in Coastal Douglas fir (Pseudotsuga menziesii var. menziesii). Genetics 2009, 183, 289–298. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Puiu, D.; Crepeau, M.W.; Stevens, K.; Salzberg, S.L.; Langley, C.H.; Neale, D.B. Genomic architecture of complex traits in loblolly pine. New Phytol. 2018, 221, 1789–1801. [Google Scholar] [CrossRef]
- Weiss, M.; Sniezko, R.A.; Puiu, D.; Crepeau, M.W.; Stevens, K.; Salzberg, S.L.; Langley, C.H.; Neale, D.B.; De La Torre, A.R. Genomic basis of white pine blister rust quantitative disease resistance and its relationship with qualitative resistance. Plant J. 2020, 104, 365–376. [Google Scholar] [CrossRef]
- Prunier, J.; Giguère, I.; Ryan, N.; Guy, R.; Soolanayakanahally, R.; Isabel, N.; Mackay, J.; Porth, I. Gene copy number variations involved in balsam poplar (Populus balsamifera L.) adaptive variations. Mol. Ecol. 2018, 28, 1476–1490. [Google Scholar] [CrossRef]
- Frascaroli, E.; Revilla, P. Genomics of Cold Tolerance in Maize. In The Maize Genome; Bennetzen, J., Flint-Garcia, S., Hirsch, C., Tuberosa, R., Eds.; Compendium of Plant Genomes; Springer: Berlin/Heidelberg, Germany, 2018; pp. 287–303. ISBN 978-3-319-97427-9. [Google Scholar]
- Zhang, M.; Suren, H.; Holliday, J.A. Phenotypic and Genomic Local Adaptation across Latitude and Altitude in Populus trichocarpa. Genome Biol. Evol. 2019, 11, 2256–2272. [Google Scholar] [CrossRef]
- Yeaman, S.; Hodgins, K.A.; Lotterhos, K.E.; Suren, H.; Nadeau, S.; Degner, J.C.; Nurkowski, K.A.; Smets, P.; Wang, T.; Gray, L.K.; et al. Convergent local adaptation to climate in distantly related conifers. Science 2016, 353, 1431–1433. [Google Scholar] [CrossRef]
- Pyhäjärvi, T.; Kujala, S.T.; Savolainen, O. 275 years of forestry meets genomics in Pinus sylvestris. Evol. Appl. 2019, 13, 11–30. [Google Scholar] [CrossRef]
- Lotterhos, K.E.; Yeaman, S.; Degner, J.; Aitken, S.; Hodgins, K.A. Modularity of genes involved in local adaptation to climate despite physical linkage. Genome Biol. 2018, 19, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Vitasse, Y.; Lenz, A.; Kãrner, C. The interaction between freezing tolerance and phenology in temperate deciduous trees. Front. Plant Sci. 2014, 5, 541. [Google Scholar] [CrossRef] [PubMed]
- Mimura, M.; Aitken, S.N. Adaptive gradients and isolation-by-distance with postglacial migration in Picea sitchensis. Heredity 2007, 99, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, D.E.; McMahon, B.G.; Ostry, M.E. Population-dependent selection strategies needed for 2-year-old black cottonwood clones. Can. J. For. Res. 1994, 24, 1704–1710. [Google Scholar] [CrossRef]
- Hurme, P.; Repo, T.; Savolainen, O.; Pääkkönen, T. Climatic Adaptation of Bud Set and Frost Hardiness in Scots Pine (Pinus sylvestris). Can. J. For. Res. 1997, 27, 716–723. [Google Scholar] [CrossRef]
- Savolainen, O.; Bokma, F.; García-Gil, R.; Komulainen, P.; Repo, T. Genetic variation in cessation of growth and frost hardiness and consequences for adaptation of Pinus sylvestris to climatic changes. For. Ecol. Manag. 2004, 197, 79–89. [Google Scholar] [CrossRef]
- Goto, S.; Kajiya-Kanegae, H.; Ishizuka, W.; Hisamoto, Y.; Kudoh, H.; Yasugi, M.; Iwata, H.; Kitamura, K.; Ueno, S.; Nagano, A.J. Genetic mapping of local adaptation along the altitudinal gradient in Abies sachalinensis. Tree Genet. Genomes 2017, 13, 104. [Google Scholar] [CrossRef]
- Leites, L.; Rehfeldt, G.E.; Steiner, K.C. Adaptation to climate in five eastern North America broadleaf deciduous species: Growth clines and evidence of the growth-cold tolerance trade-off. Perspect. Plant Ecol. Evol. Syst. 2019, 37, 64–72. [Google Scholar] [CrossRef]
- Dauwe, R.; Holliday, J.A.; Aitken, S.N.; Mansfield, S.D. Metabolic dynamics during autumn cold acclimation within and among populations of Sitka spruce (Picea sitchensis). New Phytol. 2012, 194, 192–205. [Google Scholar] [CrossRef]
- Akula, R.; Ravishankar, G.A. Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. Behav. 2011, 6, 1720–1731. [Google Scholar] [CrossRef]
- Bathe, U.; Tissier, A. Cytochrome P450 enzymes: A driving force of plant diterpene diversity. Phytochemistry 2019, 161, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.; Vuorinen, K.; Brosché, M. Interaction points in plant stress signaling pathways. Physiol. Plant. 2017, 162, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Joshi, D.; Yadav, P.K.; Gupta, A.K.; Bhatt, T.K. Role of Ubiquitin-Mediated Degradation System in Plant Biology. Front. Plant Sci. 2016, 7, 806. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xue, H.-W. The ubiquitin-proteasome system in plant responses to environments. Plant Cell Environ. 2019, 42, 2931–2944. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Fartyal, D.; Agarwal, A.; Shukla, T.; James, D.; Kaul, T.; Negi, Y.K.; Arora, S.; Reddy, M.K. Abiotic Stress Tolerance in Plants: Myriad Roles of Ascorbate Peroxidase. Front. Plant Sci. 2017, 8, 581. [Google Scholar] [CrossRef]
- Mageroy, M.H.; Parent, G.; Germanos, G.; Giguère, I.; Delvas, N.; Maaroufi, H.; Bauce, É.; Bohlmann, J.; Mackay, J. Expression of the β-glucosidase gene Pgβglu-1 underpins natural resistance of white spruce against spruce budworm. Plant J. 2014, 81, 68–80. [Google Scholar] [CrossRef]
- Porth, I.; White, R.; Jaquish, B.; Ritland, K. Partial correlation analysis of transcriptomes helps detangle the growth and defense network in spruce. New Phytol. 2018, 218, 1349–1359. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Piot, A.; Liu, B.; Wilhite, B.; Weiss, M.; Porth, I. Functional and morphological evolution in gymnosperms: A portrait of implicated gene families. Evol. Appl. 2020, 13, 210–227. [Google Scholar] [CrossRef]
- Bansal, S.; St. Clair, J.B.; Harrington, C.A.; Gould, P.J. Impact of climate change on cold hardiness of Douglas-fir (Pseudotsuga menziesii): Environmental and genetic considerations. Glob. Chang. Biol. 2015, 21, 3814–3826. [Google Scholar] [CrossRef]
- Hansen, J.E.; Sato, M.; Ruedy, R. Perception of climate change. Proc. Natl. Acad. Sci. USA 2012, 109, E2415–E2423. [Google Scholar] [CrossRef]
- Moreau, G.; Chagnon, C.; Auty, D.; Caspersen, J.; Achim, A. Impacts of Climatic Variation on the Growth of Black Spruce Across the Forest-Tundra Ecotone: Positive Effects of Warm Growing Seasons and Heat Waves Are Offset by Late Spring Frosts. Front. For. Glob. Chang. 2020, 3. [Google Scholar] [CrossRef]
- Loarie, S.R.; Duffy, P.B.; Hamilton, H.; Asner, G.P.; Field, C.B.; Ackerly, D.D. The velocity of climate change. Nat. Cell Biol. 2009, 462, 1052–1055. [Google Scholar] [CrossRef] [PubMed]



| Trait | PVE | PGE | SNPs | BSLMM | MLM |
|---|---|---|---|---|---|
| trait1 | 0.75 ± 0.17 | 0.52 ± 0.16 | 3 ± 2 | 23 | 106 |
| trait2 | 0.88 ± 0.13 | 0.24 ± 0.2 | 78 ± 71 | 1617 | 0 |
| BB2 | 0.65 ± 0.19 | 0.38 ± 0.19 | 3 ± 2 | 15 | 25 |
| BS1 | 0.6 ± 0.17 | 0.56 ± 0.18 | 3 ± 2 | 11 | 70 |
| BS2 | 0.68 ± 0.21 | 0.13 ± 0.16 | 34 ± 40 | 52 | 0 |
| DIAM | 0.8 ± 0.16 | 0.37 ± 0.14 | 2 ± 1 | 11 | 75 |
| EMEAN | 0.86 ± 0.16 | 0.21 ± 0.1 | 3 ± 4 | 18 | 54 |
| EMSTD | 0.47 ± 0.2 | 0.45 ± 0.21 | 3 ± 2 | 8 | 48 |
| FLUSH | 0.39 ± 0.25 | 0.2 ± 0.24 | 17 ± 24 | 18 | 1 |
| FLUSHLG | 0.26 ± 0.19 | 0.27 ± 0.27 | 25 ± 38 | 26 | 0 |
| HT1 | 0.64 ± 0.2 | 0.41 ± 0.18 | 2 ± 1 | 6 | 46 |
| HT2 | 0.54 ± 0.16 | 0.64 ± 0.19 | 2 ± 1 | 14 | 77 |
| HTINC | 0.52 ± 0.19 | 0.5 ± 0.22 | 3 ± 2 | 15 | 54 |
| RTLG | 0.68 ± 0.23 | 0.34 ± 0.2 | 10 ± 18 | 34 | 30 |
| RTSH | 0.48 ± 0.14 | 0.71 ± 0.18 | 2 ± 2 | 14 | 115 |
| RTWT | 0.76 ± 0.21 | 0.22 ± 0.16 | 3 ± 5 | 23 | 20 |
| SHWT | 0.66 ± 0.22 | 0.33 ± 0.2 | 3 ± 6 | 18 | 29 |
| TAPER | 0.69 ± 0.23 | 0.23 ± 0.21 | 38 ± 54 | 188 | 10 |
| TOTWT | 0.69 ± 0.21 | 0.31 ± 0.18 | 2 ± 2 | 22 | 30 |
| budcold | 0.65 ± 0.19 | 0.29 ± 0.25 | 99 ± 84 | 2453 | 1 |
| ndlcold | 0.93 ± 0.09 | 0.42 ± 0.24 | 96 ± 78 | 2857 | 2 |
| stmcold | 0.88 ± 0.13 | 0.15 ± 0.13 | 11 ± 16 | 34 | 4 |
| SDWT | 0.82 ± 0.19 | 0.13 ± 0.15 | 23 ± 35 | 58 | 2 |
| Gene | NCBI Accession | S | O | Trait | Env. | Processes/Pathways | Functional Annotation |
|---|---|---|---|---|---|---|---|
| PSME_32494 | XP_020083319.1 | 1 | N | cold | TD | Pentose phosphate pathway | glucose-6-phosphate 1-dehydrogenase 4 |
| PSME_35569 | XP_011080970.1 | 1 | N | cold | SHM | Cell wall pectin biosynthesis | xyloglucan-specific galacturonosyltransferase 1 |
| PSME_01956 | XP_023919718.1 | 1 | N | cold | bFFP | Starch and sucrose metab. | neutral trehalase-like |
| PSME_04717 | XP_023911511.1 | 2 | Y | grow cold | TD, PC1 | CH and aminoacid metab. | 4-aminobutyrate aminotransferase |
| PSME_39868 | XP_020187038.1 | 1 | N | cold | PAS, TD | Glycolysis, fatty acid degradation, tyrosine metab. | alcohol dehydrogenase-like 4 |
| PSME_34754 | XP_011622948.1 | 1 | Y | grow | TD | Glycolisis | plastidial pyruvate kinase 4 |
| PSME_37622 | XP_023873676.1 | 1 | N | cold | Lat | Glycolysis, phosphorylation of fructose-6-phosphate | ATP-dependent 6-phosphofructokinase 5 |
| PSME_46816 | MA_72344g0010 | 1 | N | cold | TD | sugar transporter | Bidirectional sugar transporter SWEET3-like |
| PSME_47335 | XP_006858646.2 | 1 | N | cold | TD | sugar transporter | CMP-sialic acid transporter 5 |
| PSME_39947 | XP_014501229.1 | 1 | N | grow phe | DD_0, TD | Fructose and mannose metabolism | mannan endo-1,4-β-mannosidase 7-like |
| PSME_39362 | RVW55203.1 | 4 | N | grow cold | EXT, Eref, TD | cellulose metabolism | cellulose synthase |
| PSME_40047 | XP_020534550.1 | 1 | N | cold | TD | cellulose metabolism | β-glucosidase 18 |
| PSME_20810 | MA_121907g0010 | 1 | N | cold | TD | CH and photosynthesis | plastocyanin-like |
| PSME_32128 | XP_020532303.1 | 1 | N | phe | MCMT | Glycerophospholipid metab. | lysophospholipid acyltransferase LPEAT2 |
| PSME_23694 | XP_011100368.1 | 1 | N | cold phe | TD | arginine, proline, β-alanine, pantothenate and CoA | polyamine oxidase 2 |
| PSME_30851 | XP_006836334.1 | 1 | Y | grow cold | DD5_at | arginine, proline, β-alanine, pantothenate and CoA | polyamine oxidase 2 |
| PSME_40791 | XP_004289831.1 | 1 | N | grow cold | DD_0_at | Cysteine and methionine metabolism | 5’-methylthioadenosine/S-adenosylhomocysteine nucleosidase |
| PSME_01345 | XP_023540379.1 | 1 | N | grow | TD | RNA metabolism | DEAD-box ATP-dependent RNA helicase 57 |
| PSME_39361 | XP_021895115.1 | 3 | N | grow cold | PAS, TD, DD_0 | Terpenoid backbone biosynthesis | 1-deoxy-D-xylulose 5-phosphate reductoisomerase |
| PSME_35875 | XP_023520455.1 | 1 | N | grow cold phe | TD | Ubiquinone and other terpenoid-quinone biosynthesis | NAD(P)H dehydrogenase (quinone) FQR1 |
| PSME_03065 | XP_023925599.1 | 1 | N | phe | PAS | Steroid and diterpenes biosynthesis | cytochrome P450 |
| PSME_13751 | XP_007209932.1 | 1 | N | cold | MWMT, TD | Steroid biosynthesis | geraniol 8-hydroxylase, cytochrome P450 |
| PSME_06220 | XP_018684598.1 | 1 | Y | cold | PAS | Glycosylation (terpenes and others biosynthesis) | UDP-rhamnose:rhamnosyltransferase 1 |
| PSME_42889 | XP_021819748.1 | 1 | N | cold phe | PC1 | Porphyrin and chlorophyll metabolism | phytochromobilin:ferredoxin oxidoreductase |
| PSME_41367 | XP_011080788.1 | 1 | N | cold | PAS | vitamin B6 metabolism | pyridoxal 5’-phosphate synthase subunit PDX1 |
| PSME_28922 | XP_007202107.1 | 1 | N | grow | DD_0, CMD | flavonoid biosynthesis | naringenin,2-oxoglutarate 3-dioxygenase |
| PSME_40932 | XP_023531135.1 | 2 | N | grow cold phe | DD_0, MSP, TD | growth and development | protein COBRA-like |
| PSME_04771 | XP_006433957.1 | 1 | N | grow cold phe | DD_0, EXT, TD | growth and development | EXORDIUM-like 2 |
| PSME_02459 | XP_023541193.1 | 1 | N | cold phe | TD | growth and development | EXORDIUM-like 3 |
| PSME_32867 | XP_020102207.1 | 1 | N | grow phe | DD_0, TD | growth and development | NAC domain-containing protein 68-like |
| PSME_31370 | MA_101849g0010 | 1 | N | grow | TD | reproductive development | NAC domain-containing 35-like (PF02365) |
| PSME_17369 | XP_016710129.1 | 1 | N | grow phe | DD_0, TD | growth and development | PREDICTED: LOB domain-containing protein 19 |
| PSME_47800 | XP_023516539.1 | 1 | N | grow phe | DD_0, TD | growth and development | probable inactive purple acid phosphatase 27 |
| PSME_40261 | PSS23947.1 | 1 | N | grow phe | DD_0_at, PC1 | growth and development | Purple acid phosphatase |
| PSME_30046 | BBC78345.1 | 1 | N | grow | DD_0, Eref | reproductive development | NEEDLY-like protein |
| PSME_37515 | XP_010242469.1 | 1 | N | phe | TD | reproductive development | LETM1 and EF-hand domain-containing protein 1 |
| PSME_47186 | XP_011627569.1 | 1 | N | grow | DD_0_at, PAS | growth | methionine aminopeptidase 1B |
| PSME_27219 | XP_012838463.1 | 1 | N | grow cold phe | DD_0, PC1, TD | cell cycle control | cyclin-dependent kinase C-2-like isoform X2 |
| PSME_38027 | XP_031497943.1 | 1 | N | grow cold | TD | cell cycle control | GAMETE EXPRESSED 1 |
| PSME_51103 | XP_022153266.1 | 1 | N | grow phe | DD_0, Lat, TD | Transcription regulation | CASP isoform X2 |
| PSME_30616 | XP_001754344.1 | 1 | N | grow phe | PC1, TD | Transcription regulation | TATA-box binding |
| PSME_18347 | MA_8626g0010 | 1 | N | phe | EXT | Transcription regulation | Tannin-related R2R3 MYB transcription |
| PSME_01485 | MA_4929994g0010 | 1 | N | cold | PC1 | Transcription regulation | AP2 domain transcription factor |
| PSME_34977 | MA_2589g0010 | 1 | N | cold | DD_0_at | Transcription regulation | Transcription termination factor |
| PSME_46703 | KHN01498.1 | 1 | N | grow | TD | Transcription, splicing factor SF1 | KH Domain-containing protein |
| PSME_37207 | MA_9457778g0010 | 1 | N | grow cold phe | DD_0_at, TD | antioxidative defense | peroxidase 5 (PF00141) |
| PSME_46413 | XP_021618646.1 | 1 | N | grow phe | PAS, TD | antioxidative defense | peroxidase 4 |
| PSME_40921 | MA_629244g0010 | 1 | N | grow | DD_0_at | Biotic stress | ABC transporter C family member 14-like |
| PSME_16294 | XP_021726759.1 | 1 | N | grow | TD | Biotic stress, plant-pathogen interaction | protein SGT1 homolog |
| PSME_47942 | MA_35233g0010 | 1 | N | grow phe | TD | Salt stress | cysteine rich repeat secretory 5 (PF01657) |
| PSME_32719 | XP_020268797.1 | 1 | N | grow | TD | abiotic stress | 17.8 kDa class I heat shock protein-like |
| PSME_47504 | ADB97926.1 | 1 | N | grow phe | DD_0, AHM, TD | cold stress | thaumatin-like L2 |
| PSME_33611 | XP_010920821.1 | 1 | N | grow cold | TD | biotic stress | protein PMR5 |
| PSME_02881 | OVA07401.1 | 1 | Y | grow phe | PC1, TD | abiotic stress | Leucine-rich repeat |
| PSME_03186 | MA_52212g0010 | 1 | N | cold phe | PAS | stress and calcium signaling | Probable calcium-binding CML25 |
| PSME_09147 | XP_021984819.1 | 1 | N | grow cold phe | DD_0_at, TD | biotic stress, signal transduction | serine/threonine-protein kinase AFC1-like |
| PSME_32617 | XP_011077233.1 | 1 | N | grow cold phe | PC1, TD | biotic stress, signal transduction | phosphoenolpyruvate carboxylase kinase 2 |
| PSME_42855 | XP_021297375.1 | 1 | N | cold | PAS, TD | biotic stress, signal transduction | protein kinase and PP2C-like domain-protein |
| PSME_08981 | XP_020099349.1 | 1 | N | grow cold | EXT | stress, vesicular transport | ras-related protein Rab7 |
| PSME_47475 | XP_021275390.1 | 1 | Y | phe | TD | stress, signal transduction | lanthionine synthetase component C (lanC)-like protein GCL1 |
| PSME_05154 | XP_007218270.1 | 1 | Y | grow | SHM | Biotic stress | carboxylesterase 15 |
| PSME_01336 | XP_010269100.1 | 1 | N | grow phe | DD_0_at, TD | Biotic stress | carboxylesterase 2 |
| PSME_34054 | MA_15220g0010 | 1 | Y | grow | TD | Biotic stress | Metallothiol transferase |
| PSME_00292 | XP_023900051.1 | 1 | N | grow phe | DD_0, MSP, TD | Ubiquitin mediated proteolisis | ubiquitin-40S ribosomal protein S27a |
| PSME_03290 | XP_023547970.1 | 1 | N | phe | PAS | Ubiquitin mediated proteolisis | U-box domain-containing protein 26 |
| PSME_02609 | XP_016487911.1 | 1 | N | grow cold phe | Long, TD | Ubiquitin mediated proteolisis | cullin-1-like |
| PSME_01721 | MA_83109g0010 | 1 | Y | grow | DD_0 | Ubiquitin mediated proteolisis | cullin-1 like |
| PSME_04517 | XP_023538234.1 | 1 | N | cold phe | SHM | Ubiquitin mediated proteolisis | SKP1-like protein 1B |
| PSME_37900 | MA_10432666g0010 | 1 | Y | grow cold | TD | Ubiquitin mediated proteolisis | U11 U12 small nuclear ribonucleo 25 kDa |
| PSME_15528 | XP_010250248.1 | 1 | N | cold | PC3 | Ubiquitin mediated proteolisis | ubiquitin carboxyl-terminal hydrolase 22 |
| PSME_02072 | XP_024392985.1 | 1 | N | grow | TD | Ubiquitin mediated proteolisis | E3 ubiquitin protein ligase RING1-like |
| PSME_30516 | AAX92710.1 | 1 | N | grow | PAS | Ubiquitin mediated proteolisis | SCF ubiquitin ligase |
| PSME_48521 | XP_022751818.1 | 1 | N | cold | TD | stress, signal transduction | histidine kinase 5-like |
| PSME_27170 | XP_021677291.1 | 1 | N | grow phe | PAS | stress, signalling and cellular processes | pollen-specific protein SF21-like isoform X2 |
| PSME_30964 | MA_279935g0010 | 1 | N | grow | DD_0_at, TD | Methylation and growth | Histone-lysine N-methyltransferase family member SUVH9 |
| PSME_15751 | XP_012079811.1 | 1 | N | cold | TD | Methylation and growth | putative methyltransferase DDB_G0268948 |
| PSME_40578 | KAF5183317.1 | 1 | N | cold | Elev | Merthylation | 23S rRNA (uracil(1939)-C(5))-methyltransferase RlmD |
| PSME_32636 | XP_021691879.1 | 1 | N | grow | PC2 | translation | 50S ribosomal protein L35, chloroplastic-like |
| PSME_02851 | XP_013468200.1 | 1 | N | phe | PAS, TD | translation | 60S ribosomal L12-like |
| PSME_36577 | XP_020101722.1 | 1 | N | phe | MAP | translation | probable GTP-binding protein OBGM, mitochondrial |
| PSME_46659 | RWR94859.1 | 1 | N | cold | PAS, TD | translation | eukaryotic translation initiation factor 5B |
| PSME_31058 | XP_020522223.1 | 1 | N | grow | MSP, TD | DNA replication and repair | crossover junction endonuclease MUS81 |
| PSME_01087 | XP_004229823.1 | 1 | N | grow phe | PAS, TD | DNA replication and repair | AT-hook motif nuclear-localized protein 22 |
| PSME_41853 | XP_010277286.1 | 1 | N | grow | PC1, TD | post-translational protein modification | PREDICTED: protein S-acyltransferase 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De La Torre, A.R.; Wilhite, B.; Puiu, D.; St. Clair, J.B.; Crepeau, M.W.; Salzberg, S.L.; Langley, C.H.; Allen, B.; Neale, D.B. Dissecting the Polygenic Basis of Cold Adaptation Using Genome-Wide Association of Traits and Environmental Data in Douglas-fir. Genes 2021, 12, 110. https://doi.org/10.3390/genes12010110
De La Torre AR, Wilhite B, Puiu D, St. Clair JB, Crepeau MW, Salzberg SL, Langley CH, Allen B, Neale DB. Dissecting the Polygenic Basis of Cold Adaptation Using Genome-Wide Association of Traits and Environmental Data in Douglas-fir. Genes. 2021; 12(1):110. https://doi.org/10.3390/genes12010110
Chicago/Turabian StyleDe La Torre, Amanda R., Benjamin Wilhite, Daniela Puiu, John Bradley St. Clair, Marc W. Crepeau, Steven L. Salzberg, Charles H. Langley, Brian Allen, and David B. Neale. 2021. "Dissecting the Polygenic Basis of Cold Adaptation Using Genome-Wide Association of Traits and Environmental Data in Douglas-fir" Genes 12, no. 1: 110. https://doi.org/10.3390/genes12010110
APA StyleDe La Torre, A. R., Wilhite, B., Puiu, D., St. Clair, J. B., Crepeau, M. W., Salzberg, S. L., Langley, C. H., Allen, B., & Neale, D. B. (2021). Dissecting the Polygenic Basis of Cold Adaptation Using Genome-Wide Association of Traits and Environmental Data in Douglas-fir. Genes, 12(1), 110. https://doi.org/10.3390/genes12010110