Defining the Rhizobium leguminosarum Species Complex

 ,
,  , , , ,
, , , ,  , , ,
, , ,  , , , , ,
, , , , ,  ,
,  ,
,  , , , , ,
, , , , ,  , ,
, ,  and add
Show full author list
and add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. The Set of Genome Sequences
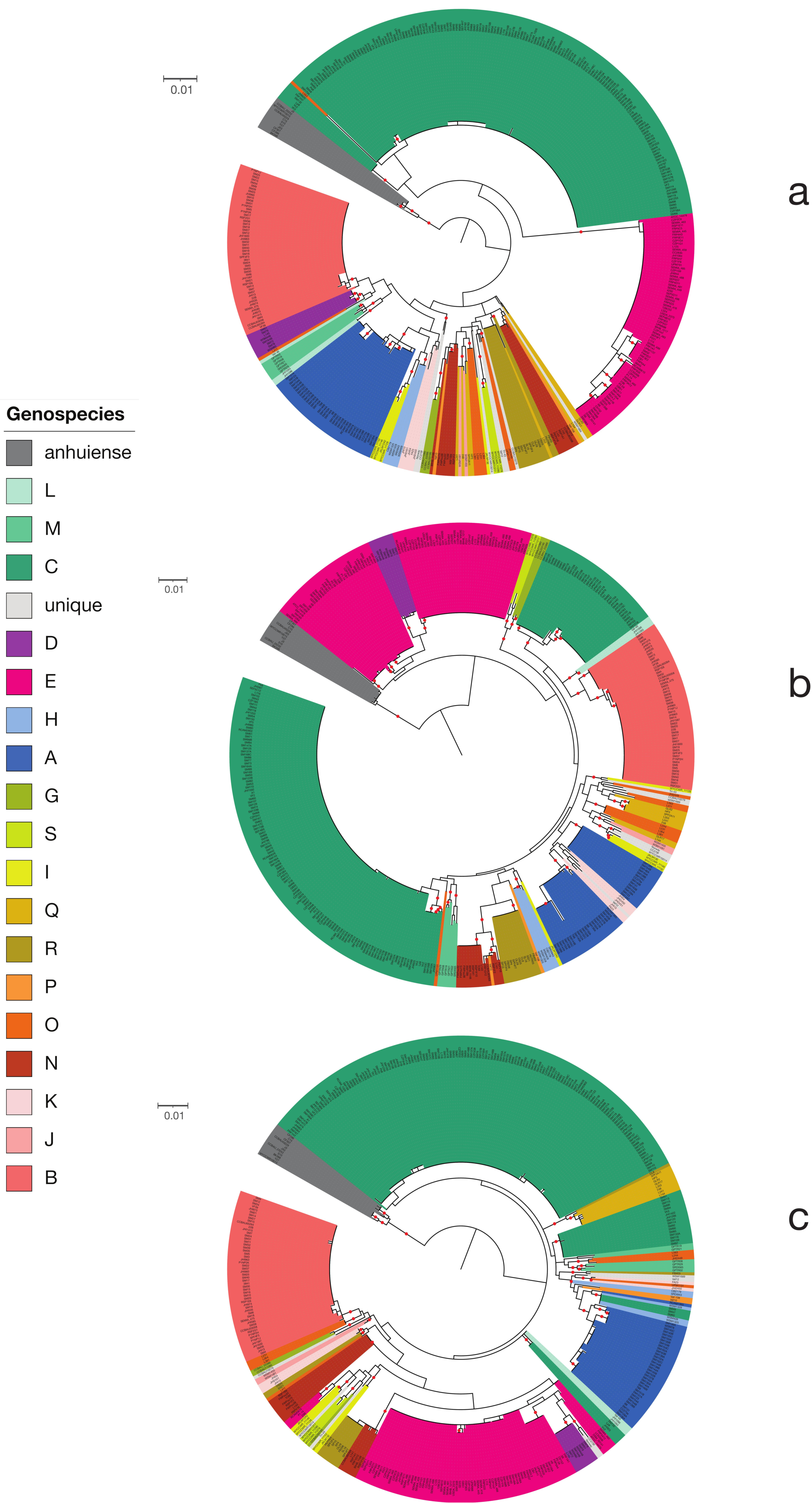
2.2. The Core Gene Phylogeny
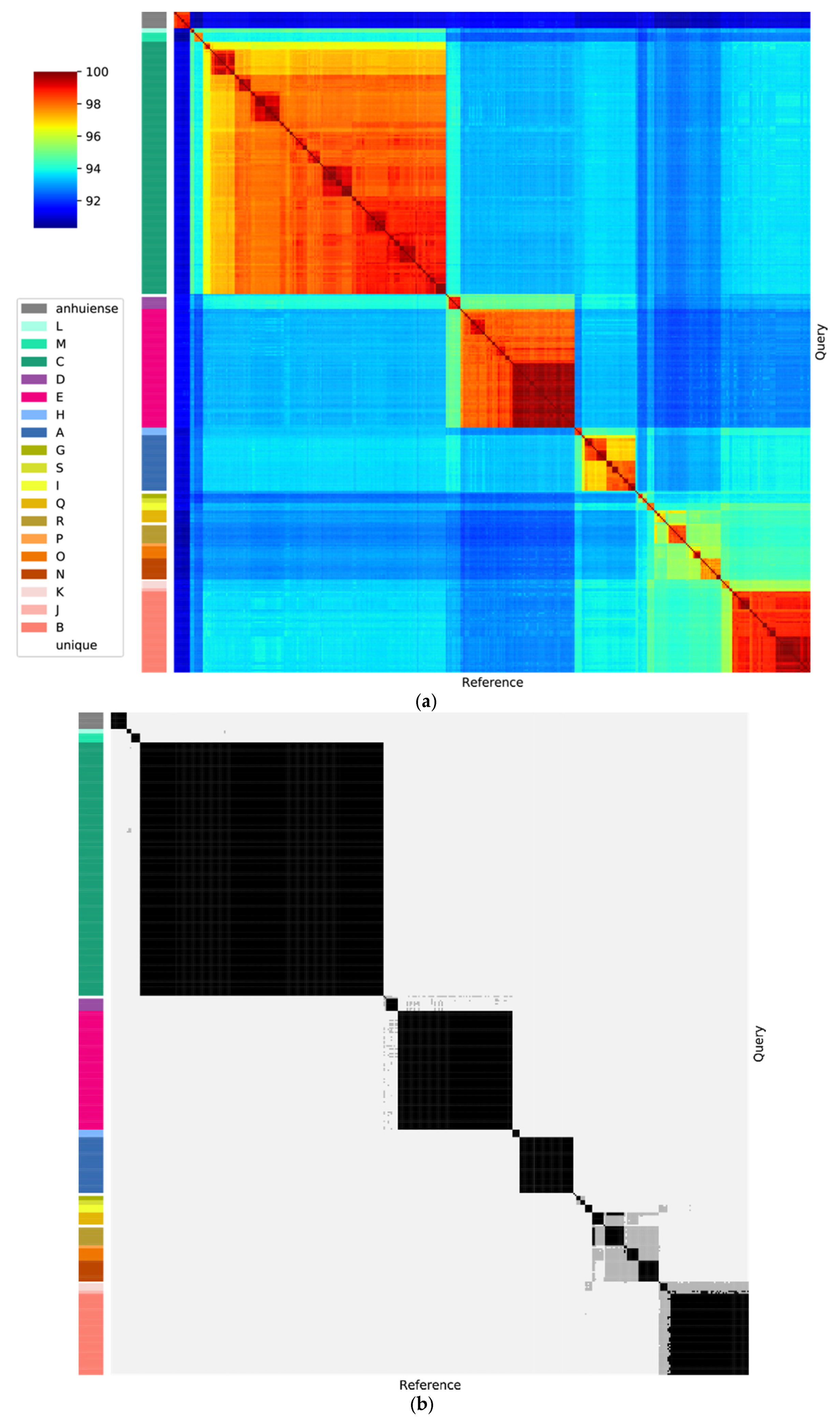
2.3. Average Nucleotide Identity (ANI)
2.4. Analysis of 16S and Nodulation Genes
2.5. Housekeeping Genes for Genospecies Assignment
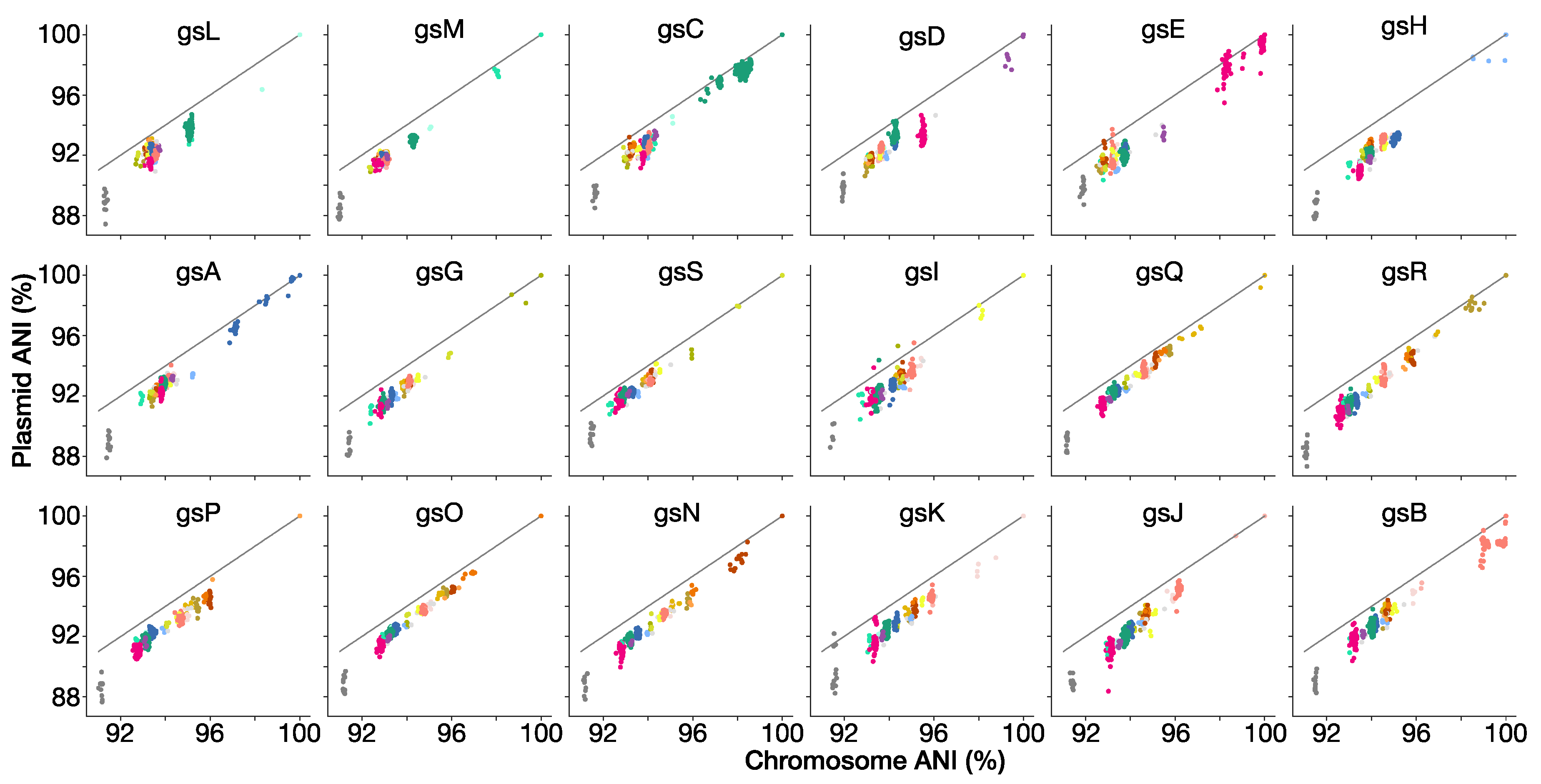
2.6. ANI of Chromosomal and Plasmid Compartments
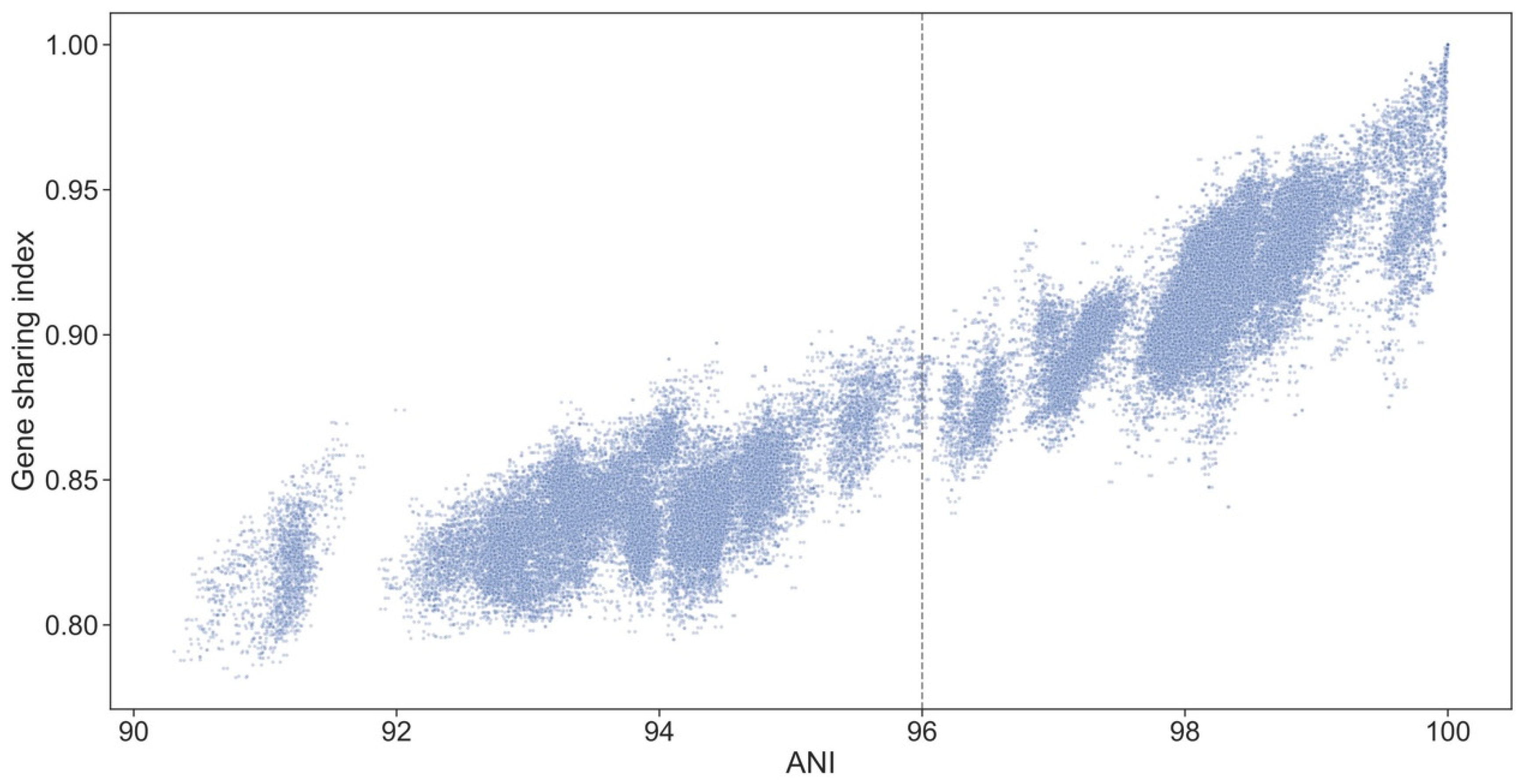
2.7. Identification and Analysis of Ortholog Sets
3. Results and Discussion
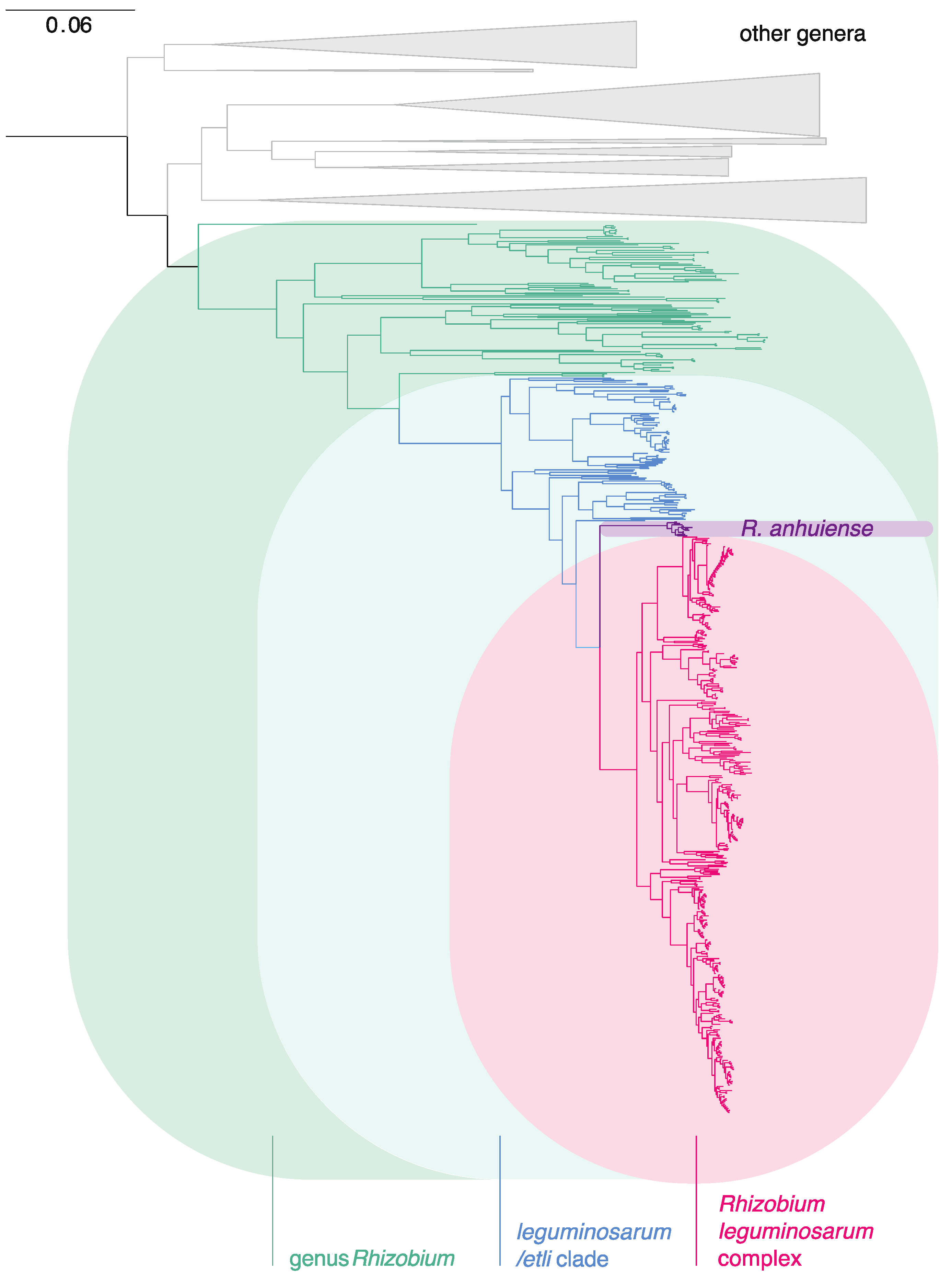
3.1. The Genus Rhizobium and Related Genera

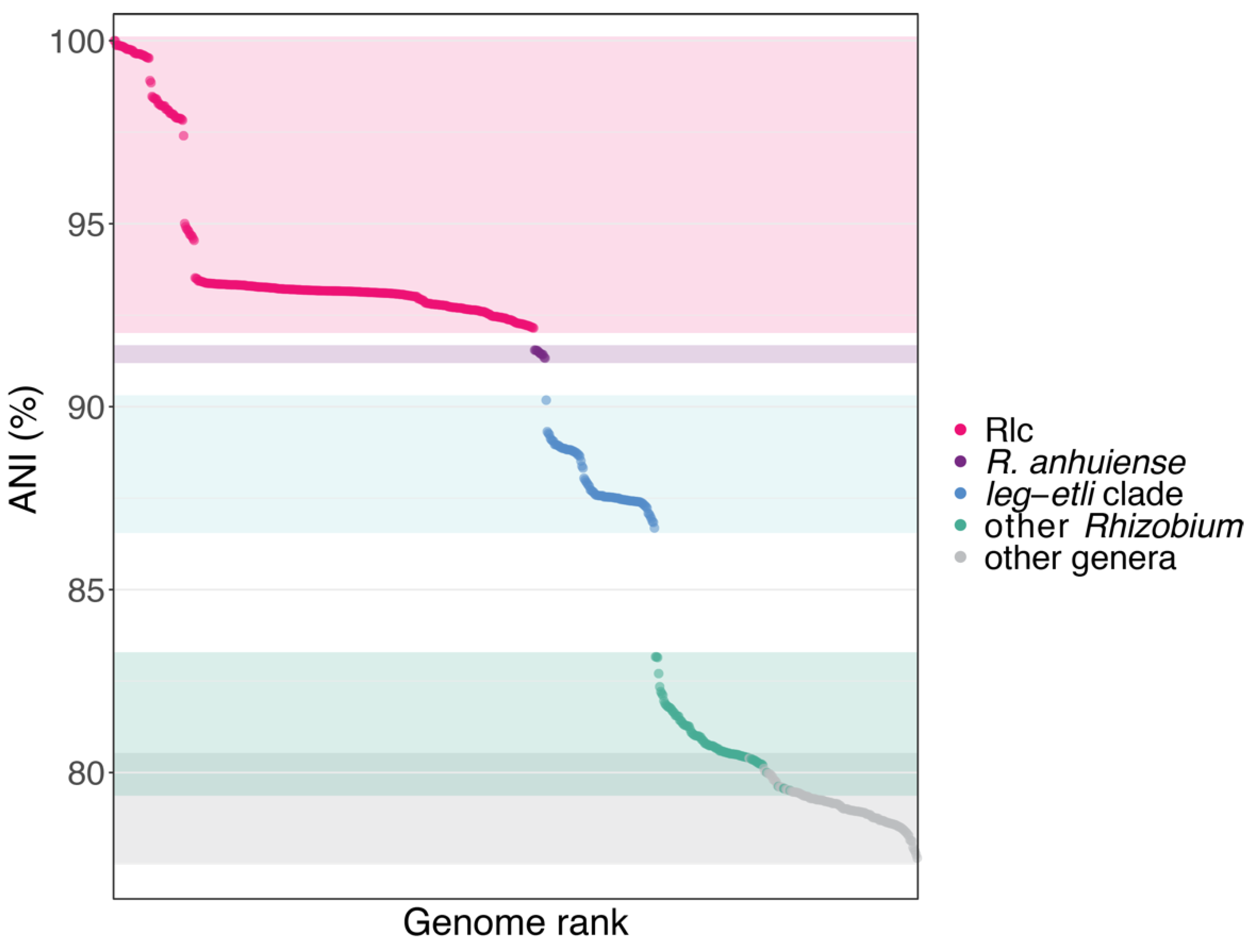
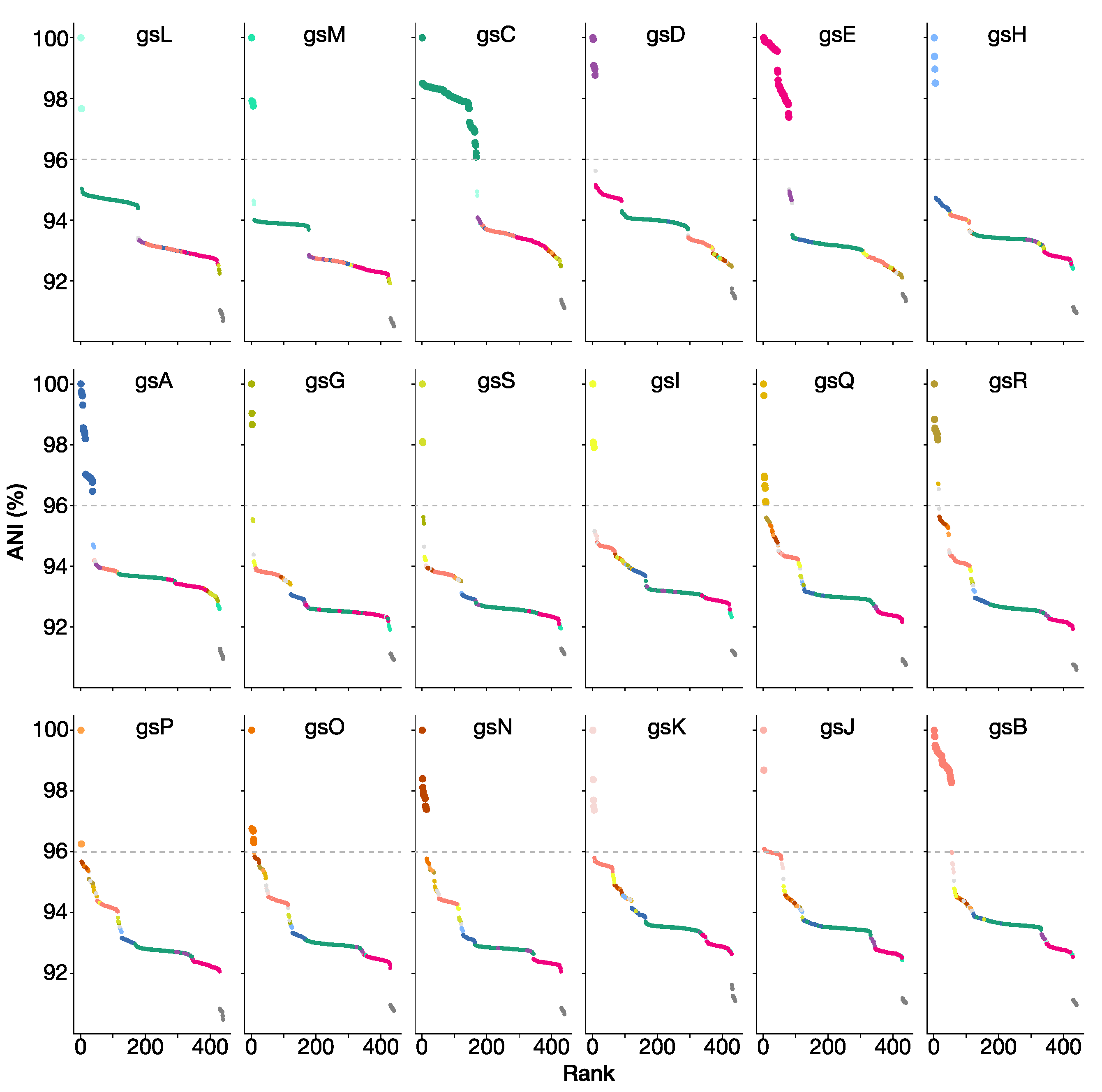
3.2. The Rlc has a Clear Boundary
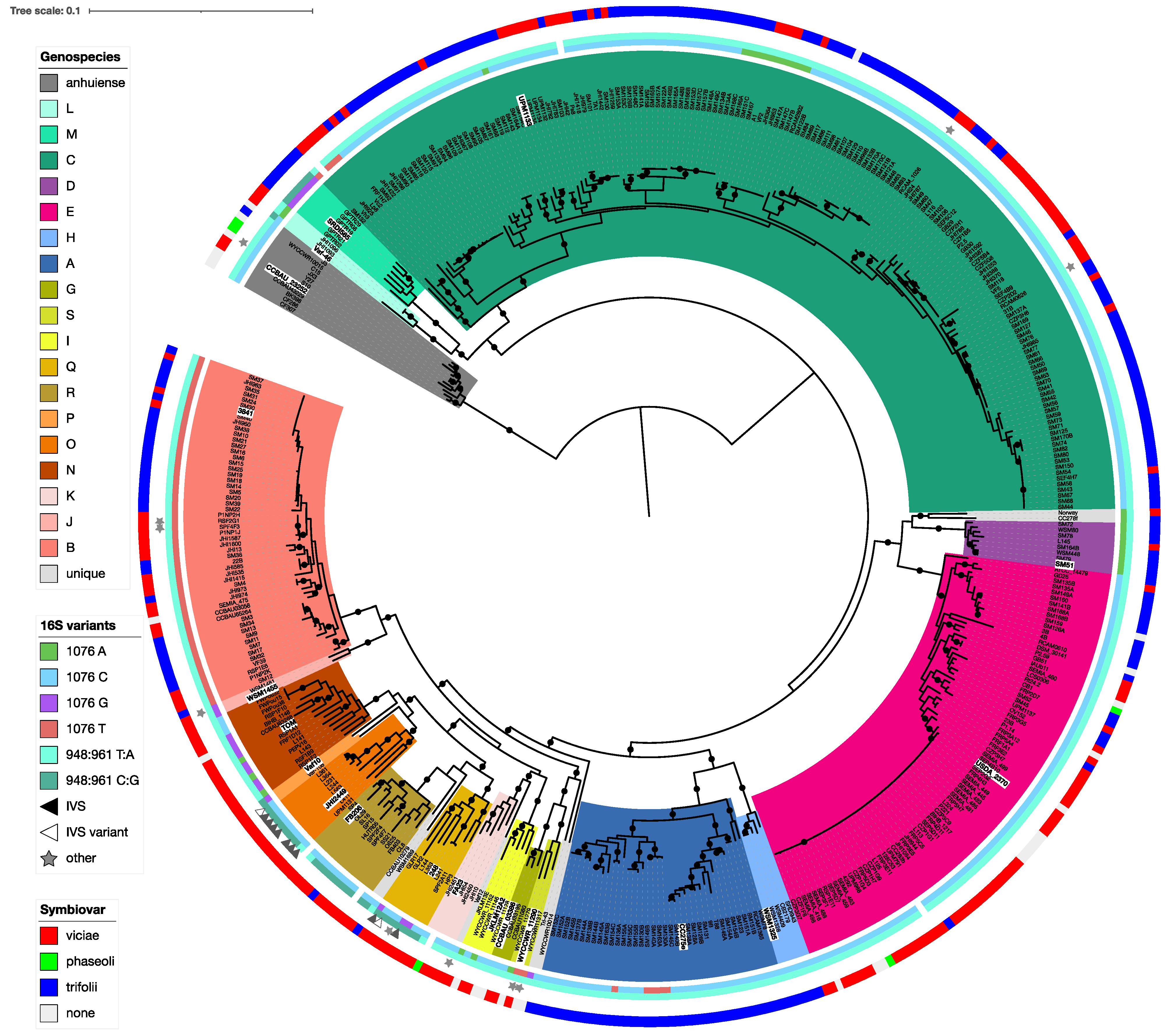
3.3. Genospecies Can Be Defined within the Rlc
3.4. The Genospecies Are Consistent with Genomic Taxonomy Databases
3.5. The Genospecies Could Be the Basis for New Formal Taxonomic Names
3.6. Housekeeping Gene Amplicons Can Identify Isolates to Genospecies
3.7. 16S rRNA Sequence Is Not Indicative of Genospecies
3.8. Nodulation Specificity Is Not a Useful Taxonomic Character
3.9. Genospecies Have Distinct Plasmid-Borne Sequences
3.10. Genospecies Have Distinct Gene Complements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vandamme, P.; Pot, B.; Gillis, M.; Vos, P.D.; Kersters, K.; Swings, J. Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol. Rev. 1996, 60, 407–438. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, P.; Peeters, C. Time to revisit polyphasic taxonomy. Antonie van Leeuwenhoek 2014, 106, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Fanning, G.R.; Rake, A.V.; Johnson, K.E. Batch procedure for thermal elution of DNA from hydroxyapatite. Anal. Biochem. 1969, 28, 447–459. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.G.E.; Stackebrandt, E.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bact. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Bact. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Mukherjee, S.; Seshadri, R.; Varghese, N.J.; Eloe-Fadrosh, E.A.; Meier-Kolthoff, J.P.; Göker, M.; Coates, R.C.; Hadjithomas, M.; Pavlopoulos, G.A.; Paez-Espino, D.; et al. 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat. Biotechnol. 2017, 35, 676–683. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; Meyer, S.D.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Wu, L.; Ma, J. The Global Catalogue of Microorganisms (GCM) 10K type strain sequencing project: Providing services to taxonomists for standard genome sequencing and annotation. Int. J. Syst. Evol. Microbiol. 2019, 69, 895–898. [Google Scholar] [CrossRef]
- Chun, J.; Rainey, F.A. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 2014, 64, 316–324. [Google Scholar] [CrossRef]
- Whitman, W.B. Genome sequences as the type material for taxonomic descriptions of prokaryotes. Syst. Appl. Microbiol. 2015, 38, 217–222. [Google Scholar] [CrossRef]
- Thompson, C.C.; Amaral, G.R.; Campeão, M.; Edwards, R.A.; Polz, M.F.; Dutilh, B.E.; Ussery, D.W.; Sawabe, T.; Swings, J.; Thompson, F.L. Microbial taxonomy in the post-genomic era: Rebuilding from scratch? Arch. Microbiol. 2015, 197, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Garrity, G.M. A New Genomics-Driven Taxonomy of Bacteria and Archaea: Are We There Yet? J. Clin. Microbiol. 2016, 54, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.E.; Freudenstein, J.; Gribaldo, S.; Hatzenpichler, R.; Hugenholtz, P.; Kämpfer, P.; Konstantinidis, K.T.; Lane, C.E.; Papke, R.T.; Parks, D.H.; et al. Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol. 2020, 5, 987–994. [Google Scholar] [CrossRef]
- Parker, C.T.; Tindall, B.J.; Garrity, G.M. International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2015, 69, S1–S111. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Palmer, M.; Steenkamp, E.T.; Blom, J.; Hedlund, B.P.; Venter, S.N. All ANIs are not created equal: Implications for prokaryotic species boundaries and integration of ANIs into polyphasic taxonomy. Int. J. Syst. Evol. Microbiol. 2020, 70, 2937–2948. [Google Scholar] [CrossRef]
- Young, J.P.W. Bacteria Are Smartphones and Mobile Genes Are Apps. Trends Microbiol. 2016, 24, 931–932. [Google Scholar] [CrossRef][Green Version]
- Klenk, H.-P.; Göker, M. En route to a genome-based classification of Archaea and Bacteria? Syst. Appl. Microbiol. 2010, 33, 175–182. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Cavassim, M.I.A.; Moeskjaer, S.; Moslemi, C.; Fields, B.; Bachmann, A.; Vilhjálmsson, B.J.; Schierup, M.H.; Young, J.P.W.; Andersen, S.U. Symbiosis genes show a unique pattern of introgression and selection within a Rhizobium leguminosarum species complex. Microbial. Genom. 2020, 89. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M.; et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Ravin, A.W. Experimental Approaches to the Study of Bacterial Phylogeny. Am. Nat. 1963, 97, 307–318. [Google Scholar] [CrossRef]
- Mayr, E. Systematics and the Origin of Species from the Viewpoint of a Zoologist; Harvard University Press: Cambridge, MA, USA, 1942. [Google Scholar]
- Hanage, W.P.; Spratt, B.G.; Turner, K.M.E.; Fraser, C. Modelling bacterial speciation. Phil. Trans. R. Soc. B 2006, 361, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.; Hanage, W.P.; Spratt, B.G. Recombination and the Nature of Bacterial Speciation. Science 2007, 315, 476–480. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Nat. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Varghese, N.J.; Mukherjee, S.; Ivanova, N.; Konstantinidis, K.T.; Mavrommatis, K.; Kyrpides, N.C.; Pati, A. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015, 43, 6761–6771. [Google Scholar] [CrossRef]
- Kumar, N.; Lad, G.; Giuntini, E.; Kaye, M.E.; Udomwong, P.; Shamsani, N.J.; Young, J.P.W.; Bailly, X. Bacterial genospecies that are not ecologically coherent: Population genomics of Rhizobium leguminosarum. Open Biol. 2015, 5, 140133. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Lindström, K.; Mousavi, S.A. Effectiveness of nitrogen fixation in rhizobia. Microb. Biotechnol. 2020, 13, 1314–1335. [Google Scholar] [CrossRef]
- Mahmud, K.; Makaju, S.; Ibrahim, R.; Missaoui, A. Current Progress in Nitrogen Fixing Plants and Microbiome Research. Plants 2020, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.B. Über die Pilzsymbiose der Leguminosen. Berichte der Deutschen Botanischen Gesellschaft 1889, 7, 332–346. [Google Scholar]
- Buchanan, R.E. What Names Should Be Used for the Organisms Producing Nodules on the Roots of Leguminous Plants? Proc. Iowa Acad. Sci. 1926, 33, 81–90. [Google Scholar]
- Dangeard, P.A. Recherches sur les Tubercles Radicaux des Légumineuses; Ed. du Botaniste: Feucherolles, France, 1926. [Google Scholar]
- Jarvis, B.D.W.; Pankhurst, C.E.; Patel, J.J. Rhizobium loti, a New Species of Legume Root Nodule Bacteria. Int. J. Syst. Bact. 1982, 32, 378–380. [Google Scholar] [CrossRef]
- Jordan, D.C. Transfer of Rhizobium japonicum Buchanan 1980 to Bradyrhizobium gen. nov., a Genus of Slow-Growing, Root Nodule Bacteria from Leguminous Plants. Int. J. Syst. Bact. 1982, 32, 136–139. [Google Scholar] [CrossRef]
- de Lajudie, P.; Willems, A.; Pot, B.; Dewettinck, D.; Maestrojuan, G.; Neyra, M.; Collins, M.D.; Dreyfus, B.; Kersters, K.; Gillis, M. Polyphasic Taxonomy of Rhizobia: Emendation of the Genus Sinorhizobium and Description of Sinorhizobium meliloti comb. nov., Sinorhizobium saheli sp. nov., and Sinorhizobium teranga sp. nov. Int. J. Syst. Evol. Microbiol. 1994, 44, 715–733. [Google Scholar] [CrossRef]
- Jarvis, B.D.W.; Berkum, P.V.; Chen, W.X.; Nour, S.M.; Fernandez, M.P.; Cleyet-Marel, J.C.; Gillis, M. Transfer of Rhizobium loti, Rhizobium huakuii, Rhizobium ciceri, Rhizobium mediterraneum, and Rhizobium tianshanense to Mesorhizobium gen. nov. Int. J. Syst. Evol. Microb. 1997, 47, 895–898. [Google Scholar] [CrossRef]
- Jordan, D.C. Genus I. Rhizobium Frank 1889, 338AL. In Bergey’s Manual of Systematic Bacteriology; Krieg, N.R., Holt, J.G., Eds.; Williams & Wilkins: Baltimore, MD, USA, 1984; Volume 1, pp. 235–242. [Google Scholar]
- Johnston, A.W.B.; Beynon, J.L.; Buchanan-Wollaston, A.V.; Setchell, S.M.; Hirsch, P.R.; Beringer, J.E. High frequency transfer of nodulating ability between strains and species of Rhizobium. Nature 1978, 276, 634–636. [Google Scholar] [CrossRef]
- Young, J.P.W. Rhizobium population genetics: Enzyme polymorphism in isolates from peas, clover, beans and lucerne grown at the same site. J. Gen. Microbiol. 1985, 131, 2399–2408. [Google Scholar] [CrossRef][Green Version]
- Young, J.P.W.; Wexler, M. Sym Plasmid and Chromosomal Genotypes are Correlated in Field Populations of Rhizobium leguminosarum. J. Gen. Microbiol. 1988, 134, 2731–2739. [Google Scholar] [CrossRef][Green Version]
- de Lajudie, P.M.; Andrews, M.; Ardley, J.; Eardly, B.; Jumas-Bilak, E.; Kuzmanović, N.; Lassalle, F.; Lindström, K.; Mhamdi, R.; Martínez-Romero, E.; et al. Minimal standards for the description of new genera and species of rhizobia and agrobacteria. Int. J. Syst. Evol. Microbiol. 2019, 67, 2485. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.A.; Ormeño-Orrillo, E.; Martínez-Romero, E. Symbiovars in rhizobia reflect bacterial adaptation to legumes. Syst. Appl. Microbiol. 2011, 34, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Remigi, P.; Zhu, J.; Young, J.P.W.; Masson-Boivin, C. Symbiosis within symbiosis: Evolving nitrogen-fixing legume symbionts. Trends Microbiol. 2016, 24, 63–75. [Google Scholar] [CrossRef]
- Ramirez-Bahena, M.H.; Garcia-Fraile, P.; Peix, A.; Valverde, A.; Rivas, R.; Igual, J.M.; Mateos, P.F.; Martinez-Molina, E.; Velazquez, E. Revision of the taxonomic status of the species Rhizobium leguminosarum (Frank 1879) Frank 1889AL, Rhizobium phaseoli Dangeard 1926AL and Rhizobium trifolii Dangeard 1926AL. R. trifolii is a later synonym of R. leguminosarum. Reclassification of the strain R. leguminosarum DSM 30132 (=NCIMB 11478) as Rhizobium pisi sp. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 2484–2490. [Google Scholar] [CrossRef] [PubMed]
- Bailly, X.; Giuntini, E.; Sexton, M.C.; Lower, R.P.; Harrison, P.W.; Kumar, N.; Young, J.P.W. Population genomics of Sinorhizobium medicae based on low-coverage sequencing of sympatric isolates. ISME J. 2011, 5, 1722–1734. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Boivin, S.; Lahmidi, N.A.; Sherlock, D.; Bonhomme, M.; Dijon, D.; Heulin-Gotty, K.; Le-Queré, A.; Pervent, M.; Tauzin, M.; Carlsson, G.; et al. Host-specific competitiveness to form nodules in Rhizobium leguminosarum symbiovar viciae. New Phytol. 2020, 226, 555–568. [Google Scholar] [CrossRef]
- Saïdi, S.; Ramírez-Bahena, M.-H.; Santillana, N.; Zúñiga, D.; Álvarez-Martínez, E.; Peix, A.; Mhamdi, R.; Velázquez, E. Rhizobium laguerreae sp. nov. nodulates Vicia faba on several continents. Int. J. Syst. Evol. Microbiol. 2014, 64, 242–247. [Google Scholar] [CrossRef]
- Seshadri, R.; Reeve, W.G.; Ardley, J.K.; Tennessen, K.; Woyke, T.; Kyrpides, N.C.; Ivanova, N.N. Discovery of Novel Plant Interaction Determinants from the Genomes of 163 Root Nodule Bacteria. Sci. Rep. 2015, 5, 16825. [Google Scholar] [CrossRef]
- Boivin, S.; Mahé, F.; Pervent, M.; Tancelin, M.; Tauzin, M.; Wielbo, J.; Mazurier, S.; Young, J.P.W.; Lepetit, M. Genetic variation in host-specific competitiveness of the symbiont Rhizobium leguminosarum symbiovar viciae. Authorea 2020. [Google Scholar] [CrossRef]
- Afonin, A.; Sulima, A.; Zhernakov, A.; Zhukov, V. Draft genome of the strain RCAM1026 Rhizobium leguminosarum bv. viciae. Genomics Data 2017, 11, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cañizares, C.; Jorrin, B.; Durán, D.; Nadendla, S.; Albareda, M.; Rubio-Sanz, L.; Lanza, M.; González-Guerrero, M.; Prieto, R.; Brito, B.; et al. Genomic Diversity in the Endosymbiotic Bacterium Rhizobium leguminosarum. Genes 2018, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Hoffrichter, A.; Brachmann, A.; Marín, M. Complete genome of Rhizobium leguminosarum Norway, an ineffective Lotus micro-symbiont. Stand. Genom. Sci. 2018, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chirak, E.R.; Kimeklis, A.K.; Karasev, E.S.; Kopat, V.V.; Safronova, V.I.; Belimov, A.A.; Aksenova, T.S.; Kabilov, M.R.; Provorov, N.A.; Andronov, E.E. Search for Ancestral Features in Genomes of Rhizobium leguminosarum bv. viciae Strains Isolated from the Relict Legume Vavilovia formosa. Genes 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Jorrin, B.; Palacios, J.M.; Peix, Á.; Imperial, J. Rhizobium ruizarguesonis sp. nov., isolated from nodules of Pisum sativum L. Syst. Appl. Microbiol. 2020, 43, 126090. [Google Scholar] [CrossRef]
- Rahi, P.; Giram, P.; Chaudhari, D.; diCenzo, G.C.; Kiran, S.; Khullar, A.; Chandel, M.; Gawari, S.; Mohan, A.; Chavan, S.; et al. Rhizobium indicum sp. nov., isolated from root nodules of pea (Pisum sativum) cultivated in the Indian trans-Himalayas. Syst. Appl. Microbiol. 2020, 43, 126127. [Google Scholar] [CrossRef]
- Ayuso-Calles, M.; García-Estévez, I.; Jiménez-Gómez, A.; Flores-Félix, J.D.; Escribano-Bailón, M.T.; Rivas, R. Rhizobium laguerreae Improves Productivity and Phenolic Compound Content of Lettuce (Lactuca sativa L.) under Saline Stress Conditions. Foods 2020, 9, 1166. [Google Scholar] [CrossRef]
- Afonin, A.M.; Gribchenko, E.S.; Sulima, A.S.; Zhukov, V.A.; Newton, I.L.G. Complete Genome Sequence of an Efficient Rhizobium leguminosarum bv. viciae Strain, A1. Microbiol. Resour. Announc. 2020, 9, 143. [Google Scholar] [CrossRef]
- Perry, B.J.; Ferguson, S.; Laugraud, A.; Wakelin, S.A.; Reeve, W.; Ronson, C.W. Complete Genome Sequences of Trifolium spp. Inoculant Strains Rhizobium leguminosarum sv. trifolii TA1 and CC275e: Resources for Genomic Study of the Rhizobium—Trifolium Symbiosis. Mol. Plant-Microbe Interact. 2020. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Reeve, W.; O’Hara, G.; Chain, P.; Ardley, J.; Bräu, L.; Nandesena, K.; Tiwari, R.; Copeland, A.; Nolan, M.; Han, C.; et al. Complete genome sequence of Rhizobium leguminosarum bv. trifolii strain WSM1325, an effective microsymbiont of annual Mediterranean clovers. Stand Genom. Sci. 2010, 2, 347–356. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Young, J.P.W.; Crossman, L.C.; Johnston, A.W.; Thomson, N.R.; Ghazoui, Z.F.; Hull, K.H.; Wexler, M.; Curson, A.R.; Todd, J.D.; Poole, P.S.; et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7, R34. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Shen, X.-X.; Hittinger, C.T.; Rokas, A. Evaluating Fast Maximum Likelihood-Based Phylogenetic Programs Using Empirical Phylogenomic Data Sets. Mol. Biol. Evol. 2017, 35, 486–503. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2019, 37, 291–294. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Gaunt, M.W.; Turner, S.L.; Rigottier-Gois, L.; Lloyd-Macgilp, S.A.; Young, J.P.W. Phylogenies of atpD and recA support the small subunit rRNA-based classification of rhizobia. Int. J. Syst. Evol. Microbiol. 2001, 51, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Dawyndt, P.; Coopman, R.; Gillis, M.; Vos, P.D.; Willems, A. Advantages of multilocus sequence analysis for taxonomic studies: A case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int. J. Syst. Evol. Microbiol. 2008, 58, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Marek-Kozaczuk, M.; Leszcz, A.; Wielbo, J.; Wdowiak-Wróbel, S.; Skorupska, A. Rhizobium pisi sv. trifolii K3.22 harboring nod genes of the Rhizobium leguminosarum sv. trifolii cluster. Syst. Appl. Microbiol. 2013, 36, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Efstathiadou, E.; Savvas, D.; Tampakaki, A.P. Genetic diversity and phylogeny of indigenous rhizobia nodulating faba bean (Vicia faba L.) in Greece. Syst. Appl. Microbiol. 2020, 43, 126149. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Fields, B.; Moeskjær, S.; Friman, V.; Andersen, S.U.; Young, J.P.W. MAUI-seq: Metabarcoding using amplicons with unique molecular identifiers to improve error correction. Mol. Ecol. Resour. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- de Lajudie, P.; Laurent-Fulele, E.; Willems, A.; Torek, U.; Coopman, R.; Collins, M.D.; Kersters, K.; Dreyfus, B.; Gillis, M. Allorhizobium undicola gen. nov., sp. nov., nitrogen-fixing bacteria that efficiently nodulate Neptunia natans in Senegal. Int. J. Syst. Evol. Microbiol. 1998, 48, 1277–1290. [Google Scholar] [CrossRef]
- Mousavi, S.A.; Willems, A.; Nesme, X.; de Lajudie, P.; Lindström, K. Revised phylogeny of Rhizobiaceae: Proposal of the delineation of Pararhizobium gen. nov., and 13 new species combinations. Syst. Appl. Microbiol. 2015, 38, 84–90. [Google Scholar] [CrossRef]
- Mousavi, S.A.; Österman, J.; Wahlberg, N.; Nesme, X.; Lavire, C.; Vial, L.; Paulin, L.; de Lajudie, P.; Lindström, K. Phylogeny of the Rhizobium–Allorhizobium–Agrobacterium clade supports the delineation of Neorhizobium gen. nov. Syst. Appl. Microbiol. 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kimes, N.E.; López-Pérez, M.; Flores-Félix, J.D.; Ramírez-Bahena, M.-H.; Igual, J.M.; Peix, A.; Rodriguez-Valera, F.; Velázquez, E. Pseudorhizobium pelagicum gen. nov., sp. nov. isolated from a pelagic Mediterranean zone. Syst. Appl. Microbiol. 2015, 38, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, F.; Dastgheib, S.M.M.; Zhao, F.-J.; Zhang, J.; Verbarg, S.; Frühling, A.; Brinkmann, H.; Osborne, T.H.; Sikorski, J.; Balloux, F.; et al. Phylogenomic analysis reveals the basis of adaptation of Pseudorhizobium species to extreme environments. bioRxiv 2019, 54, 690347. [Google Scholar] [CrossRef]
- Ramírez-Bahena, M.H.; Vial, L.; Lassalle, F.; Diel, B.; Chapulliot, D.; Daubin, V.; Nesme, X.; Muller, D. Single acquisition of protelomerase gave rise to speciation of a large and diverse clade within the Agrobacterium/Rhizobium supercluster characterized by the presence of a linear chromid. Mol. Phylogenet. Evol. 2014, 73, 202–207. [Google Scholar] [CrossRef]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.-M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1,000+ Type-Strain Genomes Substantially Improves Taxonomic Classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 3156. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Hang, P.; Zhang, L.; Zhou, X.-Y.; Hu, Q.; Jiang, J.-D. Rhizobium album sp. nov., isolated from a propanil-contaminated soil. Antonie Van Leeuwenhoek 2019, 112, 319–327. [Google Scholar] [CrossRef]
- Jiao, Y.S.; Yan, H.; Ji, Z.J.; Liu, Y.H.; Sui, X.H.; Wang, E.T.; Guo, B.L.; Chen, W.X.; Chen, W.F. Rhizobium sophorae sp. nov. and Rhizobium sophoriradicis sp. nov., nitrogen-fixing rhizobial symbionts of the medicinal legume Sophora flavescens. Int. J. Syst. Evol. Microbiol. 2015, 65, 497–503. [Google Scholar] [CrossRef]
- Youseif, S.H.; El-Megeed, F.H.A.; Mohamed, A.H.; Ageez, A.; Veliz, E.; Martínez-Romero, E. Diverse Rhizobium strains isolated from root nodules of Trifolium alexandrinum in Egypt and symbiovars. Syst. Appl. Microbiol. 2020, 126156. [Google Scholar] [CrossRef]
- Flores-Félix, J.D.; Sánchez-Juanes, F.; García-Fraile, P.; Valverde, A.; Mateos, P.F.; Gónzalez-Buitrago, J.M.; Velázquez, E.; Rivas, R. Phaseolus vulgaris is nodulated by the symbiovar viciae of several genospecies of Rhizobium laguerreae complex in a Spanish region where Lens culinaris is the traditionally cultivated legume. Syst. Appl. Microbiol. 2019, 42, 240–247. [Google Scholar] [CrossRef]
- Mutch, L.A.; Young, J.P.W. Diversity and specificity of Rhizobium leguminosarum biovar viciae on wild and cultivated legumes. Mol. Ecol. 2004, 13, 2435–2444. [Google Scholar] [CrossRef]
- Harrison, P.W.; Lower, R.P.J.; Kim, N.K.D.; Young, J.P.W. Introducing the bacterial ‘chromid’: Not a chromosome, not a plasmid. Trends Microbiol. 2010, 18, 141–148. [Google Scholar] [CrossRef]
- Lawrence, J.G. Gene transfer, speciation, and the evolution of bacterial genomes. Curr. Opin. Microbiol. 1999, 2, 519–523. [Google Scholar] [CrossRef]
- Gevers, D.; Cohan, F.M.; Lawrence, J.G.; Spratt, B.G.; Coenye, T.; Feil, E.J.; Stackebrandt, E.; de Peer, Y.V.; Vandamme, P.; Thompson, F.L.; et al. Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 2005, 3, 733–739. [Google Scholar] [CrossRef]
- Barraclough, T.G.; Balbi, K.J.; Ellis, R.J. Evolving Concepts of Bacterial Species. Evol. Biol. 2012, 39, 148–157. [Google Scholar] [CrossRef]
- Shapiro, B.J.; Polz, M.F. Microbial Speciation. CSH Perspect Biol. 2015, 7, a018143. [Google Scholar] [CrossRef]
- Arevalo, P.; VanInsberghe, D.; Elsherbini, J.; Gore, J.; Polz, M.F. A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations. Cell 2019, 178, 820–834.e14. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genospecies or Strain | GTDB Species |
|---|---|
| R. anhuiense | s__Rhizobium anhuiense |
| L | s__Rhizobium leguminosarum_D |
| M | s__Rhizobium leguminosarum_I |
| C | s__Rhizobium leguminosarum_C |
| D + CC278f + Norway | s__Rhizobium leguminosarum_K |
| E | s__Rhizobium leguminosarum |
| H | s__Rhizobium leguminosarum_J |
| A | s__Rhizobium leguminosarum_E |
| WYCCWR10014 | s__Rhizobium sp001657485 |
| Tri-43 | s__Rhizobium leguminosarum_M |
| G | not represented |
| S | not represented |
| I | not represented |
| Q, WSM1689, CCBAU10279, R, P, O, N | s__Rhizobium laguerreae |
| Vaf12 | s__Rhizobium sp005860925 |
| K, J, B | s__Rhizobium leguminosarum_L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, J.P.W.; Moeskjær, S.; Afonin, A.; Rahi, P.; Maluk, M.; James, E.K.; Cavassim, M.I.A.; Rashid, M.H.-o.; Aserse, A.A.; Perry, B.J.; et al. Defining the Rhizobium leguminosarum Species Complex. Genes 2021, 12, 111. https://doi.org/10.3390/genes12010111
Young JPW, Moeskjær S, Afonin A, Rahi P, Maluk M, James EK, Cavassim MIA, Rashid MH-o, Aserse AA, Perry BJ, et al. Defining the Rhizobium leguminosarum Species Complex. Genes. 2021; 12(1):111. https://doi.org/10.3390/genes12010111
Chicago/Turabian StyleYoung, J. Peter W., Sara Moeskjær, Alexey Afonin, Praveen Rahi, Marta Maluk, Euan K. James, Maria Izabel A. Cavassim, M. Harun-or Rashid, Aregu Amsalu Aserse, Benjamin J. Perry, and et al. 2021. "Defining the Rhizobium leguminosarum Species Complex" Genes 12, no. 1: 111. https://doi.org/10.3390/genes12010111
APA StyleYoung, J. P. W., Moeskjær, S., Afonin, A., Rahi, P., Maluk, M., James, E. K., Cavassim, M. I. A., Rashid, M. H.-o., Aserse, A. A., Perry, B. J., Wang, E. T., Velázquez, E., Andronov, E. E., Tampakaki, A., Flores Félix, J. D., Rivas González, R., Youseif, S. H., Lepetit, M., Boivin, S., ... Tian, C.-F. (2021). Defining the Rhizobium leguminosarum Species Complex. Genes, 12(1), 111. https://doi.org/10.3390/genes12010111