Suprabasin—A Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
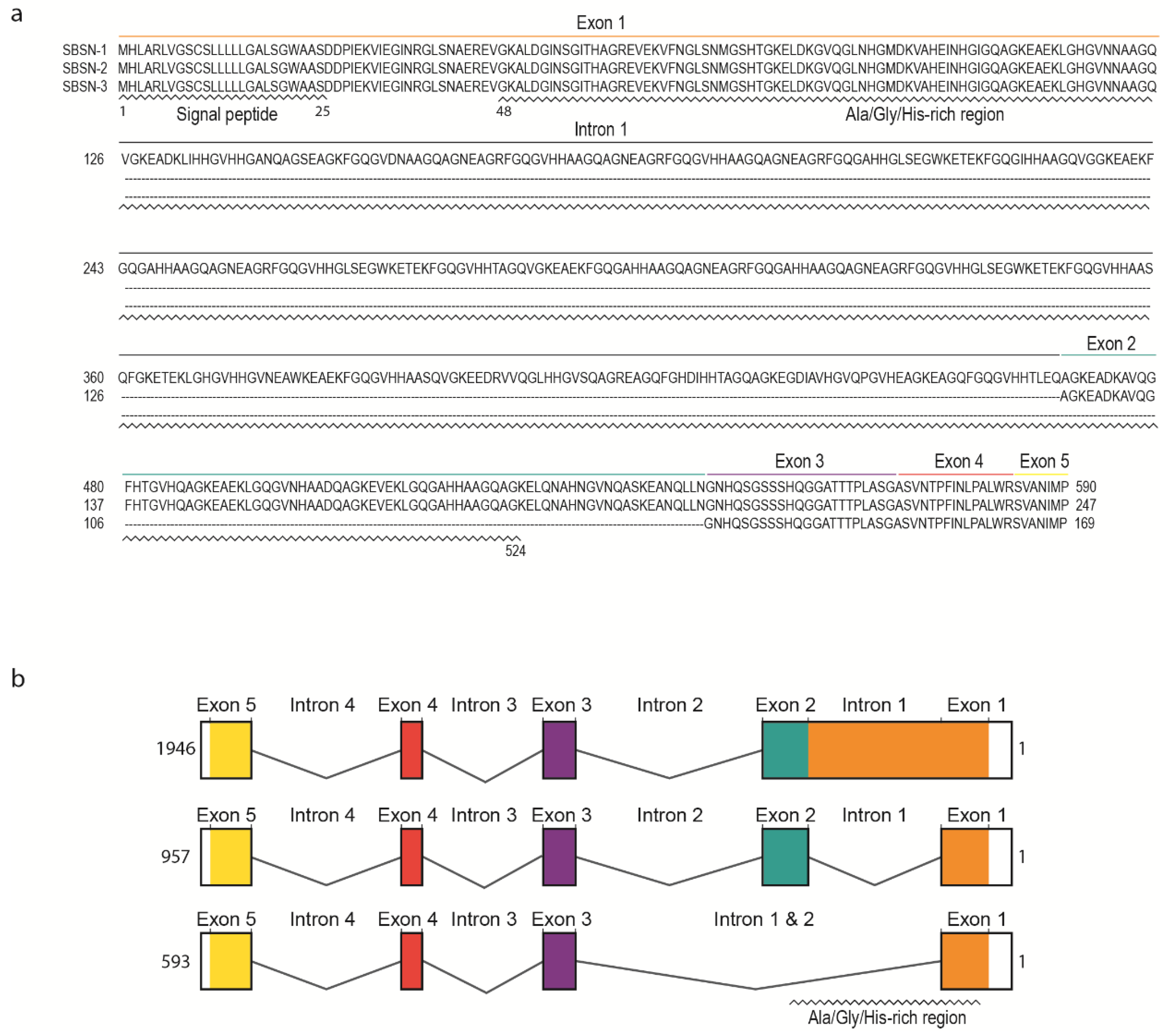
2. SBSN Gene Organization
3. SBSN Protein Structure
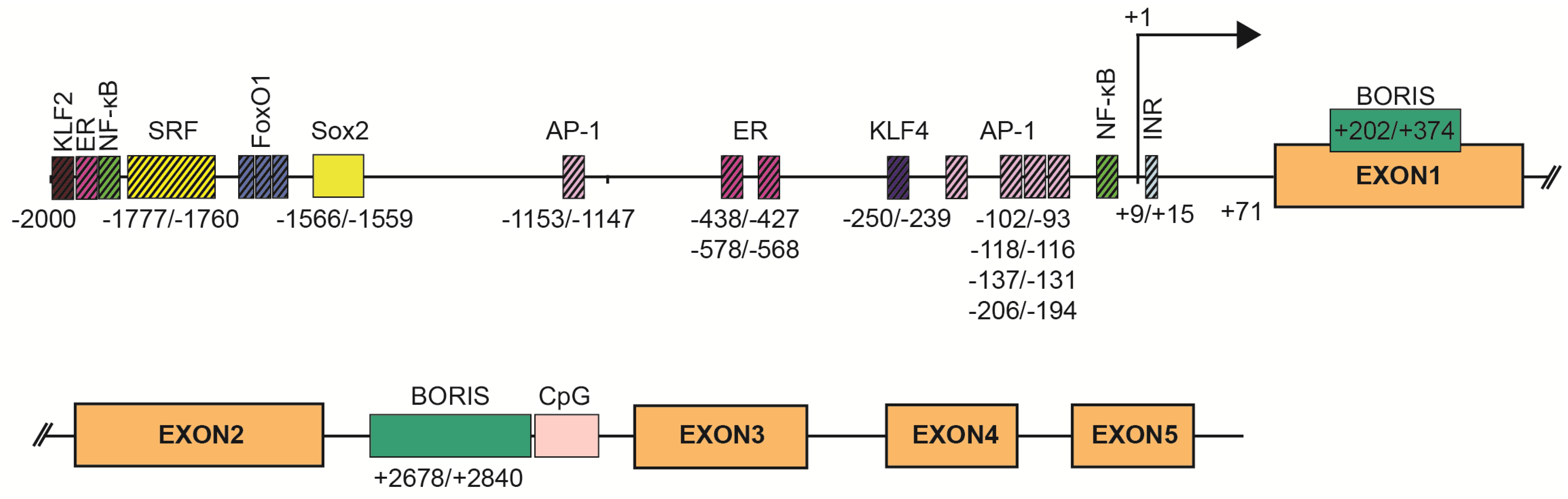
4. Regulation of SBSN Expression
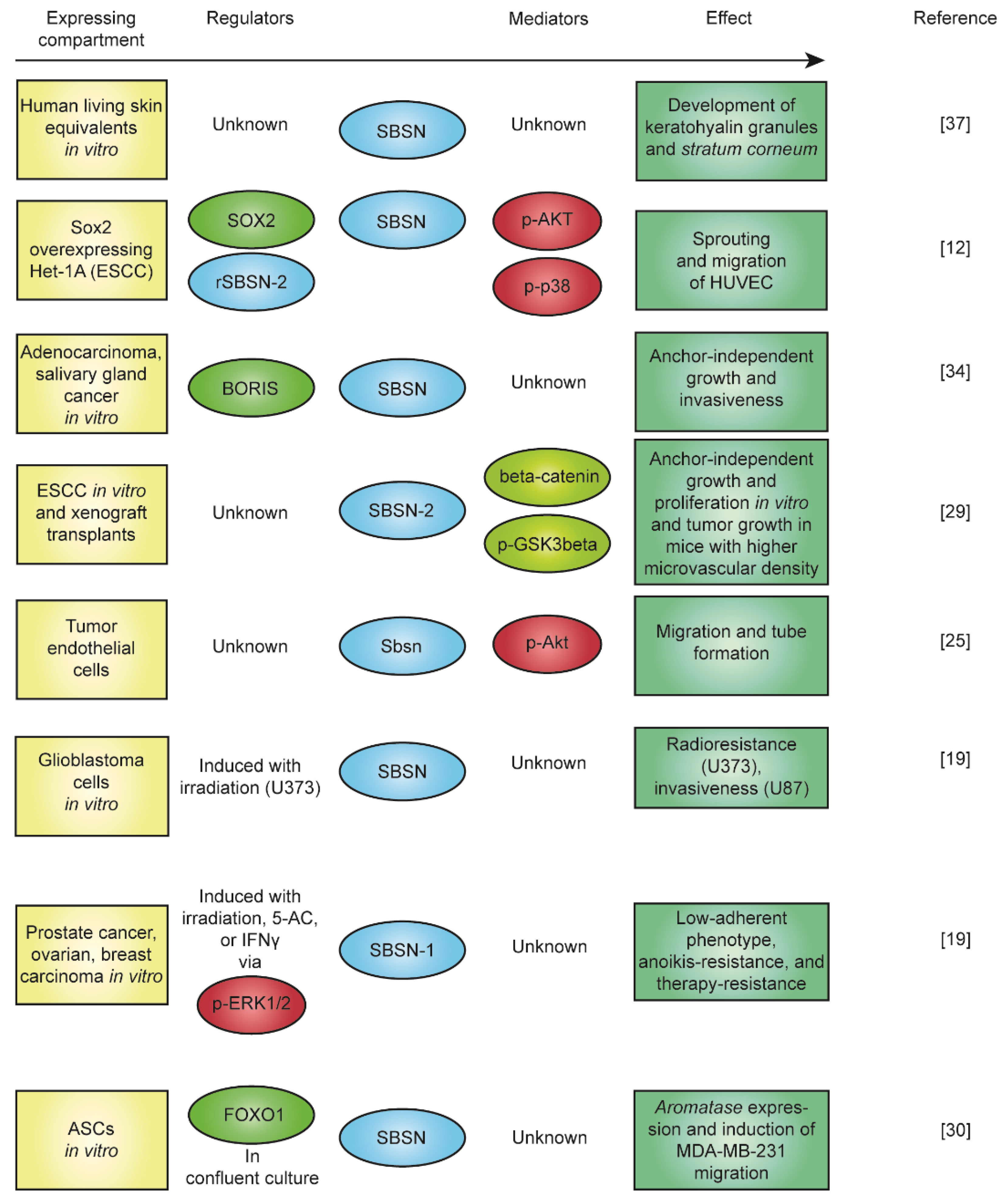
5. The Function of SBSN in Context of Physiology and Pathology
6. SBSN in Cancer
7. SBSN in Other Human Pathologies
8. SBSN, Bacterial Infection, Immune Response, and Obesity
9. Conclusions
10. Future Perspectives
- (1)
- Is SBSN a ligand and an inducer of signalling pathway?
- (2)
- What stimulates aberrant expression of SBSN under malignant conditions?
- (3)
- What is the functional role of individual SBSN isoforms?
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aa | Amino acids |
| AGT | Angiotensinogen |
| AKT | Thymomas of AKR (AKV retrovirus) mice |
| AP-1 | Activator protein-1 |
| AXIN2 | Axis inhibition protein 2 |
| bFGF | Basic fibroblast growth factor |
| BORIS | Brother of the regulator of imprinted sites |
| CCL2 | C-C motif chemokine ligand 2 |
| CDK | Cyclin-dependent kinase |
| CEBP | CCAAT enhancer-binding protein |
| CpGs | Cytosine phosphate guanine sites |
| CTCFL | CCCTC-binding factor like |
| Da | Daltons |
| DMBA | 7,12-Dimethylbenz[a]anthracene |
| EGF | Epidermal growth factor |
| ERK | Extracellular signal-regulated kinase |
| EST | Expressed sequence tag |
| FOS | Finkel-Biskis-Jinkins murine osteosarcoma |
| FOXO | Forkhead box |
| FRA | Fos-related antigen |
| GSK3beta | Glycogen synthase kinase 3 β |
| HUVEC | Human umbilical vein endothelial cells |
| IFN | Interferon |
| IgE | Immunoglobulin E |
| IL | Interleukin |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| kbp | kilobase pair |
| kDa | kilodaltons |
| Kdap | Keratinocyte differentiation-associated protein |
| KLF | Kruppel like factor |
| LYVE1 | Lymphatic vessel endothelial hyaluronan receptor 1 |
| MAL/SRF | Megakaryoblastic leukaemia 1/ serum response factor |
| MAPK | Mitogen-activated protein kinase |
| MMP7 | Matrix metallopeptidase 7 |
| NF-kappaB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLR | Nod-like receptors |
| PKC | Protein kinase C |
| PLG | Plasminogen |
| PMA | Phorbol 12-myristate 13-acetate |
| SERPINA1 | Serpin family a member 1 |
| SILAC | Stable isotope labelling with amino acids in cell culture |
| SOX2 | SRY (sex determining region Y)-box 2 |
| SP1 | Specificity protein 1 |
| SPRR | Small proline rich protein |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TLR | Toll-like receptors |
| VEGF | Vascular endothelial growth factor |
| WNT | Wingless-type |
References
- Park, G.T.; Lim, S.E.; Jang, S.I.; Morasso, M.I. Suprabasin, a novel epidermal differentiation marker and potential cornified envelope precursor. J. Biol. Chem. 2002, 277, 45195–45202. [Google Scholar] [CrossRef]
- Matsui, T.; Hayashi-Kisumi, F.; Kinoshita, Y.; Katahira, S.; Morita, K.; Miyachi, Y.; Ono, Y.; Imai, T.; Tanigawa, Y.; Komiya, T.; et al. Identification of novel keratinocyte-secreted peptides dermokine-α/- β and a new stratified epithelium-secreted protein gene complex on human chromosome 19q13.1. Genomics 2004, 84, 384–397. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Clark, H.F. The Secreted Protein Discovery Initiative (SPDI), a Large-Scale Effort to Identify Novel Human Secreted and Transmembrane Proteins: A Bioinformatics Assessment. Genome Res. 2003, 13, 2265–2270. [Google Scholar] [CrossRef]
- Consortium, T.U. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar] [CrossRef]
- Moffatt, P.; Salois, P.; St-Amant, N.; Gaumond, M.H.; Lanctôt, C. Identification of a conserved cluster of skin-specific genes encoding secreted proteins. Gene 2004, 334, 123–131. [Google Scholar] [CrossRef]
- Sancandi, M.; Uysal-Onganer, P.; Kraev, I.; Mercer, A.; Lange, S. Protein deimination signatures in plasma and plasma-evs and protein deimination in the brain vasculature in a rat model of pre-motor parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 2743. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.-B.G.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Michael, J.E.S. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2016, 10, 845–858. [Google Scholar] [CrossRef]
- Mathelier, A.; Zhao, X.; Zhang, A.W.; Parcy, F.; Worsley-Hunt, R.; Arenillas, D.J.; Buchman, S.; Chen, C.Y.; Chou, A.; Ienasescu, H.; et al. JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014, 42, 142–147. [Google Scholar] [CrossRef]
- Takahashi, K.; Asano, N.; Imatani, A.; Kondo, Y.; Saito, M.; Takeuchi, A.; Jin, X.; Saito, M.; Hatta, W.; Asanuma, K.; et al. Sox2 induces tumorigenesis and angiogenesis of early stage esophagealsquamous cell carcinoma through secretion of Suprabasin. Carcinogenesis 2020, 26, 1–15. [Google Scholar] [CrossRef]
- Gaykalova, D.; Vatapalli, R.; Glazer, C.A.; Bhan, S.; Shao, C.; Sidransky, D.; Ha, P.K.; Califano, J.A. Dose-Dependent Activation of Putative Oncogene SBSN by BORIS. PLoS ONE 2012, 7, e40389. [Google Scholar] [CrossRef]
- Li, J.; Zheng, L.; Uchiyama, A.; Bin, L.; Mauro, T.M.; Elias, P.M.; Pawelczyk, T.; Sakowicz-Burkiewicz, M.; Trzeciak, M.; Leung, D.Y.M.; et al. A data mining paradigm for identifying key factors in biological processes using gene expression data. Sci. Rep. 2018, 8, 9083. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Bazzi, H.; Fantauzzo, K.A.; Richardson, G.D.; Jahoda, C.A.B.; Christiano, A.M. Transcriptional profiling of developing mouse epidermis reveals novel patterns of coordinated gene expression. Dev. Dyn. 2007, 236, 961–970. [Google Scholar] [CrossRef]
- Brass, E.P.; Peters, M.A.; Hinchcliff, K.W.; He, Y.D.; Ulrich, R.G. Temporal pattern of skeletal muscle gene expression following endurance exercise in Alaskan sled dogs. J. Appl. Physiol. 2009, 107, 605–612. [Google Scholar] [CrossRef]
- Ichinose, K.; Ohyama, K.; Furukawa, K.; Higuchi, O.; Mukaino, A.; Satoh, K.; Nakane, S.; Shimizu, T.; Umeda, M.; Fukui, S.; et al. Novel anti-suprabasin antibodies may contribute to the pathogenesis of neuropsychiatric systemic lupus erythematosus. Clin. Immunol. 2018, 193, 123–130. [Google Scholar] [CrossRef]
- Hubackova, S.; Pribyl, M.; Kyjacova, L.; Moudra, A.; Dzijak, R.; Salovska, B.; Strnad, H.; Tambor, V.; Imrichova, T.; Svec, J.; et al. Interferon-regulated suprabasin is essential for stress-induced stem-like cell conversion and therapy resistance of human malignancies. Mol. Oncol. 2019, 13, 1467–1489. [Google Scholar] [CrossRef]
- Mehic, D.; Bakiri, L.; Ghannadan, M.; Wagner, E.F.; Tschachler, E. Fos and Jun Proteins Are Specifically Expressed During Differentiation of Human Keratinocytes. J. Investig. Dermatol. 2005, 124, 212–220. [Google Scholar] [CrossRef]
- Connelly, J.T.; Gautrot, J.E.; Trappmann, B.; Tan, D.W.-M.; Donati, G.; Huck, W.T.S.; Watt, F.M. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat. Cell Biol. 2010, 12, 711–718. [Google Scholar] [CrossRef]
- Zhu, A.J.; Watt, F.M. beta-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development 1999, 126, 2285–2298. [Google Scholar]
- Dubash, A.D.; Koetsier, J.L.; Amargo, E.V.; Najor, N.A.; Harmon, R.M.; Green, K.J. The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein-1. J. Cell Biol. 2013, 202, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Luxenburg, C.; Pasolli, H.A.; Williams, S.E.; Fuchs, E. Developmental roles for Srf, cortical cytoskeleton and cell shape in epidermal spindle orientation. Nat. Cell Biol. 2011, 13, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Nagao-Kitamoto, H.; Ohga, N.; Akiyama, K.; Maishi, N.; Kawamoto, T.; Shinohara, N.; Taketomi, A.; Shindoh, M.; Hida, Y.; et al. Suprabasin as a novel tumor endothelial cell marker. Cancer Sci. 2014, 105, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Novak, N.; Baumann, M.; Koehn, J.; Borth, N. Bioinformatic Identification of Chinese Hamster Ovary (CHO) Cold-Shock Genes and Biological Evidence of their Cold-Inducible Promoters. Biotechnol. J. 2019, e1900359. [Google Scholar] [CrossRef] [PubMed]
- Thaisuchat, H.; Baumann, M.; Pontiller, J.; Hesse, F.; Ernst, W. Identification of a novel temperature sensitive promoter in cho cells. BMC Biotechnol. 2011, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Longo, L.D. Acclimatization to long-term hypoxia: Gene expression in ovine carotid arteries. Physiol. Genom. 2014, 46, 725–734. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, G.; Li, Q.; Gong, H.; Song, J.; Cao, L.; Wu, S.; Song, L.; Jiang, L. Overexpression of Suprabasin is Associated with Proliferation and Tumorigenicity of Esophageal Squamous Cell Carcinoma. Sci. Rep. 2016, 6, 21549. [Google Scholar] [CrossRef]
- Ghosh, S.; Dean, A.; Walter, M.; Bao, Y.; Hu, Y.; Ruan, J.; Li, R. Cell density-dependent transcriptional activation of endocrine-related genes in human adipose tissue-derived stem cells. Exp. Cell Res. 2010, 316, 2087–2098. [Google Scholar] [CrossRef]
- Stanton, A.; Mowbray, C.; Lanz, M.; Brown, K.; Hilton, P.; Tyson-Capper, A.; Pickard, R.S.; Ali, A.S.M.; Hall, J. Topical Estrogen Treatment Augments the Vaginal Response to Escherichia coli Flagellin. Sci. Rep. 2020, 10, 8473. [Google Scholar] [CrossRef]
- Pribyl, M.; Hubackova, S.; Moudra, A.; Vancurova, M.; Polackova, H.; Stopka, T.; Jonasova, A.; Bokorova, R.; Fuchs, O.; Stritesky, J.; et al. Aberrantly elevated suprabasin in the bone marrow as a candidate biomarker of advanced disease state in myelodysplastic syndromes. Mol. Oncol. 2020, 14, 2403–2419. [Google Scholar] [CrossRef]
- Glazer, C.A.; Smith, I.M.; Ochs, M.F.; Begum, S.; Westra, W.; Chang, S.S.; Sun, W.; Bhan, S.; Khan, Z.; Ahrendt, S.; et al. Integrative Discovery of Epigenetically Derepressed Cancer Testis Antigens in NSCLC. PLoS ONE 2009, 4, e8189. [Google Scholar] [CrossRef]
- Shao, C.; Tan, M.; Bishop, J.A.; Liu, J.; Bai, W.; Gaykalova, D.A.; Ogawa, T.; Vikani, A.R.; Agrawal, Y.; Li, R.J.; et al. Suprabasin Is Hypomethylated and Associated with Metastasis in Salivary Adenoid Cystic Carcinoma. PLoS ONE 2012, 7, 1–7. [Google Scholar] [CrossRef]
- Sangwung, P.; Zhou, G.; Nayak, L.; Chan, E.R.; Kumar, S.; Kang, D.-W.; Zhang, R.; Liao, X.; Lu, Y.; Sugi, K.; et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017, 2, e91700. [Google Scholar] [CrossRef]
- Nakazawa, S.; Shimauchi, T.; Funakoshi, A.; Aoshima, M.; Phadungsaksawasdi, P.; Sakabe, J.; Asakawa, S.; Hirasawa, N.; Ito, T.; Tokura, Y. Suprabasin-null mice retain skin barrier function and show high contact hypersensitivity to nickel upon oral nickel loading. Sci. Rep. 2020, 10, 14559. [Google Scholar] [CrossRef]
- Aoshima, M.; Phadungsaksawasdi, P.; Nakazawa, S.; Iwasaki, M.; Sakabe, J.; Umayahara, T.; Yatagai, T.; Ikeya, S.; Shimauchi, T.; Tokura, Y. Decreased expression of suprabasin induces aberrant differentiation and apoptosis of epidermal keratinocytes: Possible role for atopic dermatitis. J. Dermatol. Sci. 2019, 95, 107–112. [Google Scholar] [CrossRef]
- Formolo, C.A.; Williams, R.; Gordish-Dressman, H.; MacDonald, T.J.; Lee, N.H.; Hathout, Y. Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 2011, 10, 3149–3159. [Google Scholar] [CrossRef]
- Sheng, S.H.; Zhu, H.L. Proteomic analysis of pleural effusion from lung adenocarcinoma patients by shotgun strategy. Clin. Transl. Oncol. 2014, 16, 153–157. [Google Scholar] [CrossRef]
- Ambekar, A.S.; Kelkar, D.S.; Pinto, S.M.; Sharma, R.; Hinduja, I.; Zaveri, K.; Pandey, A.; Prasad, T.S.K.; Gowda, H.; Mukherjee, S. Proteomics of follicular fluid from women with polycystic ovary syndrome suggests molecular defects in follicular development. J. Clin. Endocrinol. Metab. 2015, 100, 744–753. [Google Scholar] [CrossRef]
- Kuuselo, R.; Simon, R.; Karhu, R.; Tennstedt, P.; Marx, A.H.; Izbicki, J.R.; Yekebas, E.; Sauter, G.; Kallioniemi, A. 19q13 amplification is associated with high grade and stage in pancreatic cancer. Genes. Chromosom. Cancer 2010, 49, 569–575. [Google Scholar] [CrossRef]
- Thompson, F.H.; Nelson, M.A.; Trent, J.M.; Guan, X.-Y.; Liu, Y.; Yang, J.-M.; Emerson, J.; Adair, L.; Wymer, J.; Balfour, C.; et al. Amplification of 19q13.1–q13.2 sequences in ovarian cancer. Cancer Genet. Cytogenet. 1996, 87, 55–62. [Google Scholar] [CrossRef]
- Muleris, M.; Almeida, A.; Gerbault-Seureau, M.; Malfoy, B.; Dutrillaux, B. Identification of amplified DNA sequences in breast cancer and their organization within homogeneously staining regions. Genes Chromosom. Cancer 1995, 14, 155–163. [Google Scholar] [CrossRef]
- Rao, P.H.; Arias-Pulido, H.; Lu, X.-Y.; Harris, C.P.; Vargas, H.; Zhang, F.F.; Narayan, G.; Schneider, A.; Terry, M.B.; Murty, V.V. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer 2004, 4, 5. [Google Scholar] [CrossRef]
- Hong, J.A.; Kang, Y.; Abdullaev, Z.; Flanagan, P.T.; Pack, S.D.; Fischette, M.R.; Adnani, M.T.; Loukinov, D.I.; Vatolin, S.; Risinger, J.I.; et al. Reciprocal Binding of CTCF and BORIS to the NY-ESO-1 Promoter Coincides with Derepression of this Cancer-Testis Gene in Lung Cancer Cells. Cancer Res. 2005, 65, 7763–7774. [Google Scholar] [CrossRef]
- Vatolin, S.; Abdullaev, Z.; Pack, S.D.; Flanagan, P.T.; Custer, M.; Loukinov, D.I.; Pugacheva, E.; Hong, J.A.; Morse, H.; Schrump, D.S.; et al. Conditional Expression of the CTCF-Paralogous Transcriptional Factor BORIS in Normal Cells Results in Demethylation and Derepression of MAGE-A1 and Reactivation of Other Cancer-Testis Genes. Cancer Res. 2005, 65, 7751–7762. [Google Scholar] [CrossRef]
- Bhan, S.; Negi, S.S.; Shao, C.; Glazer, C.A.; Chuang, A.; Gaykalova, D.A.; Sun, W.; Sidransky, D.; Ha, P.K.; Califano, J.A. BORIS Binding to the Promoters of Cancer Testis Antigens, MAGEA2, MAGEA3, and MAGEA4, Is Associated with Their Transcriptional Activation in Lung Cancer. Clin. Cancer Res. 2011, 17, 4267–4276. [Google Scholar] [CrossRef]
- Soltanian, S.; Dehghani, H. BORIS: A key regulator of cancer stemness. Cancer Cell Int. 2018, 18, 154. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, Q.; Su, Y.; Pan, L. Network-Based Differential Analysis to Identify Molecular Features of Tumorigenesis for Esophageal Squamous Carcinoma. Molecules 2018, 23, 88. [Google Scholar] [CrossRef]
- Kenagy, R.D.; Civelek, M.; Kikuchi, S.; Chen, L.; Grieff, A.; Sobel, M.; Lusis, A.J.; Clowes, A.W. Scavenger receptor class A member 5 (SCARA5) and suprabasin (SBSN) are hub genes of coexpression network modules associated with peripheral vein graft patency. J. Vasc. Surg. 2016, 64, 202–209.e6. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Liu, B.; Huang, G.; Zhu, H.; Ma, Z.; Tian, X.; Yin, L.; Gao, X.; He, X. Analysis of gene co-expression network reveals prognostic significance of CNFN in patients with head and neck cancer. Oncol. Rep. 2019, 41, 2168–2180. [Google Scholar] [CrossRef]
- Winter, S.; Shoaie, S.; Kordasti, S.; Platzbecker, U. Integrating the “Immunome” in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef]
- Chrastinová, L.; Pastva, O.; Bocková, M.; Lynn, N.S.; Šácha, P.; Hubálek, M.; Suttnar, J.; Kotlín, R.; Štikarová, J.; Hlaváčková, A.; et al. A New Approach for the Diagnosis of Myelodysplastic Syndrome Subtypes Based on Protein Interaction Analysis. Sci. Rep. 2019, 9, 12647. [Google Scholar] [CrossRef]
- Druhan, L.J.; Lance, A.; Li, S.; Price, A.E.; Emerson, J.T.; Baxter, S.A.; Gerber, J.M.; Avalos, B.R. Leucine Rich α-2 Glycoprotein: A Novel Neutrophil Granule Protein and Modulator of Myelopoiesis. PLoS ONE 2017, 12, e0170261. [Google Scholar] [CrossRef]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef]
- Campos, D.; Freitas, D.; Gomes, J.; Magalhães, A.; Steentoft, C.; Gomes, C.; Vester-Christensen, M.B.; Ferreira, J.A.; Afonso, L.P.; Santos, L.L.; et al. Probing the O-Glycoproteome of Gastric Cancer Cell Lines for Biomarker Discovery. Mol. Cell. Proteom. 2015, 14, 1616–1629. [Google Scholar] [CrossRef]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef]
- Ku, T.K.S.; Crowe, D.L. Impaired T lymphocyte function increases tumorigenicity and decreases tumor latency in a mouse model of head and neck cancer. Int. J. Oncol. 2009, 35, 1211–1221. [Google Scholar] [CrossRef][Green Version]
- Fugmann, T.; Sofron, A.; Ritz, D.; Bootz, F.; Neri, D. The MHC Class II Immunopeptidome of Lymph Nodes in Health and in Chemically Induced Colitis. J. Immunol. 2017, 198, 1357–1364. [Google Scholar] [CrossRef]
- Richardson, M.R.; Segu, Z.M.; Price, M.O.; Lai, X.; Witzmann, F.A.; Mechref, Y.; Yoder, M.C.; Price, F.W. Alterations in the aqueous humor proteome in patients with Fuchs endothelial corneal dystrophy. Mol. Vis. 2010, 16, 2376–2383. [Google Scholar]
- Nanda, G.G.; Alone, D.P. REVIEW: Current understanding of the pathogenesis of Fuchs’ endothelial corneal dystrophy. Mol. Vis. 2019, 25, 295–310. [Google Scholar]
- Liu, L.; Watanabe, N.; Akatsu, H.; Nishimura, M. Neuronal expression of ILEI/FAM3C and its reduction in Alzheimer’s disease. Neuroscience 2016, 330, 236–246. [Google Scholar] [CrossRef]
- Theriot, C.A.; Zanello, S.B. Molecular Effects of Spaceflight in the Mouse Eye after Space Shuttle Mission. Gravit. Space Res. 2014, 2, 3–24. [Google Scholar]
- Mao, X.; Byrum, S.; Nishiyama, N.; Pecaut, M.; Sridharan, V.; Boerma, M.; Tackett, A.; Shiba, D.; Shirakawa, M.; Takahashi, S.; et al. Impact of Spaceflight and Artificial Gravity on the Mouse Retina: Biochemical and Proteomic Analysis. Int. J. Mol. Sci. 2018, 19, 2546. [Google Scholar] [CrossRef]
- Mao, X.W.; Nishiyama, N.C.; Byrum, S.D.; Stanbouly, S.; Jones, T.; Drew, A.; Sridharan, V.; Boerma, M.; Tackett, A.J.; Zawieja, D.; et al. Characterization of mouse ocular response to a 35-day spaceflight mission: Evidence of blood-retinal barrier disruption and ocular adaptations. Sci. Rep. 2019, 9, 8215. [Google Scholar] [CrossRef]
- Chidambaram, J.D.; Kannambath, S.; Srikanthi, P.; Shah, M.; Lalitha, P.; Elakkiya, S.; Bauer, J.; Prajna, N.V.; Holland, M.J.; Burton, M.J. Persistence of Innate Immune Pathways in Late Stage Human Bacterial and Fungal Keratitis: Results from a Comparative Transcriptome Analysis. Front. Cell. Infect. Microbiol. 2017, 7, 193. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Alexandraki, K.; Piperi, C.; Protogerou, A.; Katsikis, I.; Paterakis, T.; Lekakis, J.; Panidis, D. Inflammatory and endothelial markers in women with polycystic ovary syndrome. Eur. J. Clin. Investig. 2006, 36, 691–697. [Google Scholar] [CrossRef]
- Orio, F.; Palomba, S.; Spinelli, L.; Cascella, T.; Tauchmanovà, L.; Zullo, F.; Lombardi, G.; Colao, A. The Cardiovascular Risk of Young Women with Polycystic Ovary Syndrome: An Observational, Analytical, Prospective Case-Control Study. J. Clin. Endocrinol. Metab. 2004, 89, 3696–3701. [Google Scholar] [CrossRef]
- Kelly, C.C.J.; Lyall, H.; Petrie, J.R.; Gould, G.W.; Connell, J.M.C.; Sattar, N. Low Grade Chronic Inflammation in Women with Polycystic Ovarian Syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 2453–2455. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Maruyama, N. Importance of research on peptidylarginine deiminase and citrullinated proteins in age-related disease. Geriatr. Gerontol. Int. 2010, 10, S53–S58. [Google Scholar] [CrossRef] [PubMed]
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolino, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, X.; Peng, X.; Li, M.; Ren, B.; Cheng, G.; Cheng, L. Porphyromonas gingivalis Promotes Immunoevasion of Oral Cancer by Protecting Cancer from Macrophage Attack. J. Immunol. 2020, 205, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, J.; Mahendra, L.; Kurian, V.M.; Jaishankar, K.; Mythilli, R. Prevalence of periodontal pathogens in coronary atherosclerotic plaque of patients undergoing coronary artery bypass graft surgery. J. Maxillofac. Oral Surg. 2009, 8, 108–113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mougeot, J.-L.C.; Stevens, C.B.; Paster, B.J.; Brennan, M.T.; Lockhart, P.B.; Mougeot, F.K.B. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J. Oral Microbiol. 2017, 9, 1281562. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, 1–22. [Google Scholar] [CrossRef]
- Haditsch, U.; Roth, T.; Rodriguez, L.; Hancock, S.; Cecere, T.; Nguyen, M.; Arastu-Kapur, S.; Broce, S.; Raha, D.; Lynch, C.C.; et al. Alzheimer’s Disease-Like Neurodegeneration in Porphyromonas gingivalis Infected Neurons with Persistent Expression of Active Gingipains. J. Alzheimer’s Dis. 2020, 75, 1361–1376. [Google Scholar] [CrossRef]
- Yoshio, T.; Okamoto, H.; Kurasawa, K.; Dei, Y.; Hirohata, S.; Minota, S. IL-6, IL-8, IP-10, MCP-1 and G-CSF are significantly increased in cerebrospinal fluid but not in sera of patients with central neuropsychiatric lupus erythematosus. Lupus 2016, 25, 997–1003. [Google Scholar] [CrossRef]
- Wang, J.B.; Li, H.; Wang, L.L.; Liang, H.D.; Zhao, L.; Dong, J. Role of IL-1β, IL-6, IL-8 and IFN-γ in pathogenesis of central nervous system neuropsychiatric systemic lupus erythematous. Int. J. Clin. Exp. Med. 2015, 8, 16658–16663. [Google Scholar] [PubMed]
- Castillo, C.; Carrillo, I.; Libisch, G.; Juiz, N.; Schijman, A.; Robello, C.; Kemmerling, U. Host-parasite interaction: Changes in human placental gene expression induced by Trypanosoma cruzi. Parasit. Vectors 2018, 11, 479. [Google Scholar] [CrossRef]
- Lorey, M.B.; Rossi, K.; Eklund, K.K.; Nyman, T.A.; Matikainen, S. Global characterization of protein secretion from human macrophages following non-canonical caspase-4/5 inflammasome activation. Mol. Cell. Proteom. 2017, 16, S187–S199. [Google Scholar] [CrossRef]
- Keegan, C. Mycobacterium Tuberculosis tRNA Triggers TLR8 to Induce a Pathway for Th1 Cell Instruction. Ph.D. Thesis, University of California, Oakland, CA, USA, 2016. [Google Scholar]
- Hummelen, R.; Macklaim, J.M.; Bisanz, J.E.; Hammond, J.A.; McMillan, A.; Vongsa, R.; Koenig, D.; Gloor, G.B.; Reid, G. Vaginal microbiome and epithelial gene array in post-menopausal women with moderate to severe dryness. PLoS ONE 2011, 6, e26602. [Google Scholar] [CrossRef]
- Orikasa, S.; Hinman, F. Reaction of the vesical wall to bacterial penetration: Resistance to attachment, desquamation, and leukocytic activity. Investig. Urol. 1977, 15, 185–193. [Google Scholar]
- Mulvey, M.A. Induction and Evasion of Host Defenses by Type 1-Piliated Uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Hultgren, S.J.; Gordon, J.I. Functional genomic studies of uropathogenic Escherichia coli and host urothelial cells when intracellular bacterial communities are assembled. J. Biol. Chem. 2007, 282, 21259–21267. [Google Scholar] [CrossRef]
- Carregaro, F.; Stefanini, A.C.B.; Henrique, T.; Tajara, E.H. Study of small proline-rich proteins (SPRRs) in health and disease: A review of the literature. Arch. Dermatol. Res. 2013, 305, 857–866. [Google Scholar] [CrossRef]
- Li, R.W.; Meyer, M.J.; Van Tassell, C.P.; Sonstegard, T.S.; Connor, E.E.; Van Amburgh, M.E.; Boisclair, Y.R.; Capuco, A.V. Identification of estrogen-responsive genes in the parenchyma and fat pad of the bovine mammary gland by microarray analysis. Physiol. Genom. 2006, 27, 42–53. [Google Scholar] [CrossRef]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y.M. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef]
- Hamilton, J.D.; Suárez-Fariñas, M.; Dhingra, N.; Cardinale, I.; Li, X.; Kostic, A.; Ming, J.E.; Radin, A.R.; Krueger, J.G.; Graham, N.; et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 1293–1300. [Google Scholar] [CrossRef]
- Sehra, S.; Yao, Y.; Howell, M.D.; Nguyen, E.T.; Kansas, G.S.; Leung, D.Y.M.; Travers, J.B.; Kaplan, M.H. IL-4 Regulates Skin Homeostasis and the Predisposition toward Allergic Skin Inflammation. J. Immunol. 2010, 184, 3186–3190. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2009, 40, 22–35. [Google Scholar] [CrossRef]
- Gerada, C.; Ryan, K.M. Autophagy, the innate immune response and cancer. Mol. Oncol. 2020, 14, 1913–1929. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pribyl, M.; Hodny, Z.; Kubikova, I. Suprabasin—A Review. Genes 2021, 12, 108. https://doi.org/10.3390/genes12010108
Pribyl M, Hodny Z, Kubikova I. Suprabasin—A Review. Genes. 2021; 12(1):108. https://doi.org/10.3390/genes12010108
Chicago/Turabian StylePribyl, Miroslav, Zdenek Hodny, and Iva Kubikova. 2021. "Suprabasin—A Review" Genes 12, no. 1: 108. https://doi.org/10.3390/genes12010108
APA StylePribyl, M., Hodny, Z., & Kubikova, I. (2021). Suprabasin—A Review. Genes, 12(1), 108. https://doi.org/10.3390/genes12010108