Pathogenesis of Ischemic Stroke: Role of Epigenetic Mechanisms


Abstract
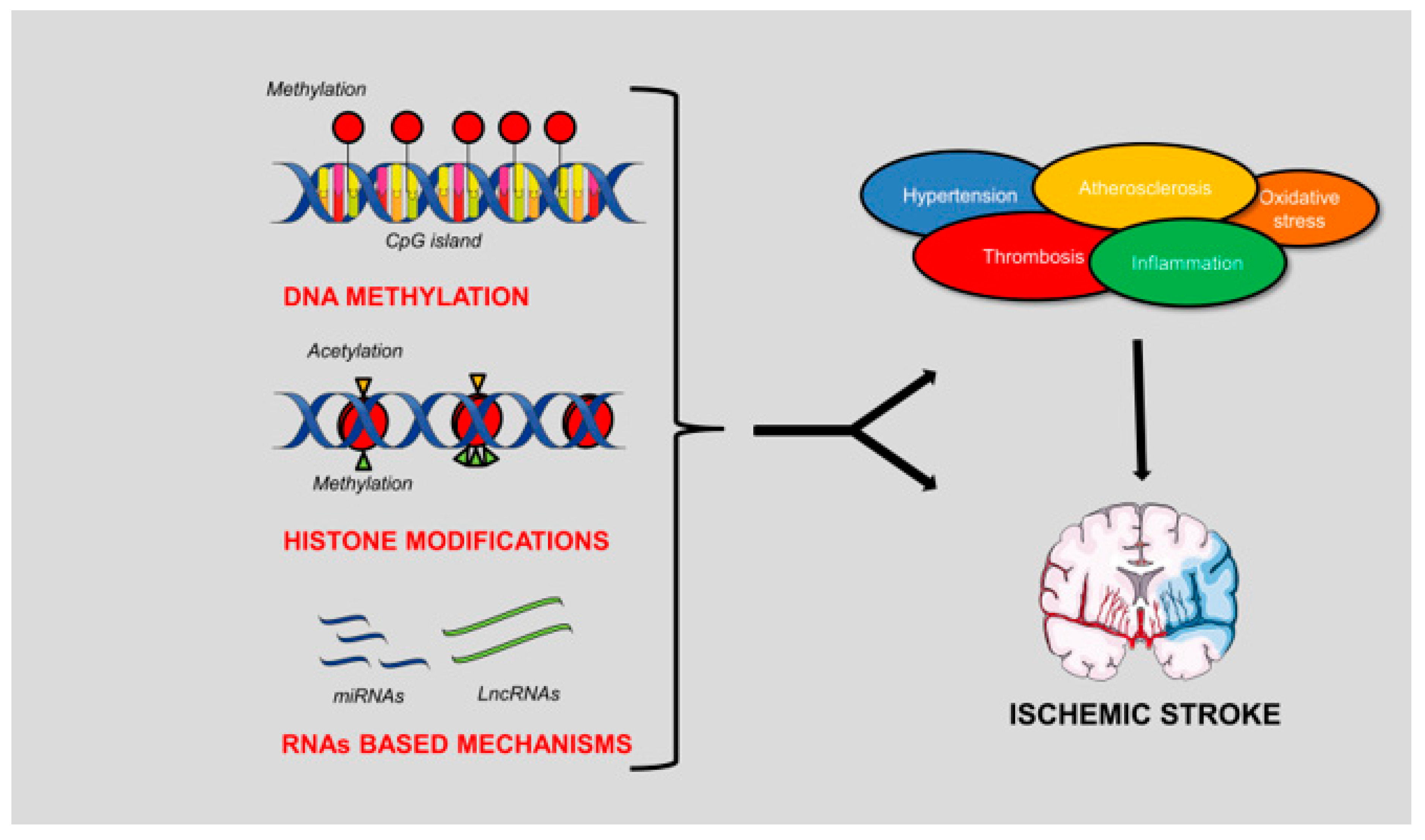
1. Introduction
2. Overview of Epigenetic Mechanisms of Gene Regulation
3. DNA Methylation in IS
4. Histone Modifications in IS
5. RNA-Based Mechanisms in IS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hajat, C.; Heuschmann, P.U.; Coshall, C.; Padayachee, S.; Chambers, J.; Rudd, A.G.; Wolfe, C.D. Incidence of aetiological subtypes of stroke in a multi-ethnic population based study: The South London Stroke Register. J. Neurol. Neurosurg. Psychiatry 2011, 82, 527–533. [Google Scholar] [CrossRef]
- Favate, A.S.; Younger, D.S. Epidemiology of Ischemic Stroke. Neurol. Clin. 2016, 34, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Raichle, M.E. The pathophysiology of brain ischemia. Ann. Neurol. 1983, 13, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Chehaibi, K.; Trabelsi, I.; Mahdouani, K.; Slimane, M.N. Correlation of Oxidative Stress Parameters and Inflammatory Markers in Ischemic Stroke Patients. J. Stroke Cerebrovasc. Dis. 2016, 25, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Giliberti, R.; Volpe, M. Etiology and pathophysiology of stroke as a complex trait. Am. J. Hypertens. 2000, 13, 1139–1148. [Google Scholar] [CrossRef]
- Hassan, A.; Markus, H.S. Genetics and ischaemic stroke. Brain 2000, 123 Pt 9, 1784–1812. [Google Scholar] [CrossRef]
- Rubattu, S.; Di Angelantonio, E.; Stanzione, R.; Zanda, B.; Evangelista, A.; Pirisi, A.; De Paolis, P.; Cota, L.; Brunetti, E.; Volpe, M. Gene polymorphisms of the renin-angiotensin-aldosterone system and the risk of ischemic stroke: A role of the A1166C/AT1 gene variant. J. Hypertens. 2004, 22, 2129–2134. [Google Scholar] [CrossRef]
- Rubattu, S.; Stanzione, R.; Di Angelantonio, E.; Zanda, B.; Evangelista, A.; Tarasi, D.; Gigante, B.; Pirisi, A.; Brunetti, E.; Volpe, M. Atrial natriuretic peptide gene polymorphisms and risk of ischemic stroke in humans. Stroke 2004, 35, 814–818. [Google Scholar] [CrossRef]
- Catto, A.J. Genetic aspects of the hemostatic system in cerebrovascular disease. Neurology 2001, 57 (Suppl. 2), S24–S30. [Google Scholar] [CrossRef]
- Rubattu, S.; Di Angelantonio, E.; Nitsch, D.; Gigante, B.; Zanda, B.; Stanzione, R.; Evangelista, A.; Pirisi, A.; Rosati, G.; Volpe, M. Polymorphisms in prothrombotic genes and their impact on ischemic stroke in a Sardinian population. Thromb. Haemost. 2005, 93, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Ballabio, E.; Bresolin, N.; Candelise, L. Genetic polymorphisms for the study of multifactorial stroke. Hum. Mutat. 2008, 29, 776–795. [Google Scholar] [CrossRef] [PubMed]
- Picascia, A.; Grimaldi, V.; Iannone, C.; Soricelli, A.; Napoli, C. Innate and adaptive immune response in stroke: Focus on epigenetic regulation. J. Neuroimmunol. 2015, 289, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Flex, A.; Gaetani, E.; Papaleo, P.; Straface, G.; Proia, A.S.; Pecorini, G.; Tondi, P.; Pola, P.; Pola, R. Proinflammatory genetic profiles in subjects with history of ischemic stroke. Stroke 2004, 35, 2270–2275. [Google Scholar] [CrossRef]
- Cotlarciuc, I.; Malik, R.; Holliday, E.G.; Ahmadi, K.R.; Pare, G.; Psaty, B.M.; Fornage, M.; Hasan, N.; Rinne, P.E.; Ikram, M.A.; et al. Effect of genetic variants associated with plasma homocysteine levels on stroke risk. Stroke 2014, 45, 1920–1924. [Google Scholar] [CrossRef]
- Di Castro, S.; Scarpino, S.; Marchitti, S.; Bianchi, F.; Stanzione, R.; Cotugno, M.; Sironi, L.; Gelosa, P.; Duranti, E.; Ruco, L.; et al. Differential modulation of uncoupling protein 2 in kidneys of stroke-prone spontaneously hypertensive rats under high-salt/low-potassium diet. Hypertension 2013, 61, 534–541. [Google Scholar] [CrossRef]
- Rubattu, S.; Stanzione, R.; Bianchi, F.; Cotugno, M.; Forte, M.; Della Ragione, F.; Fioriniello, S.; D’Esposito, M.; Marchitti, S.; Madonna, M.; et al. Reduced brain UCP2 expression mediated by microRNA-503 contributes to increased stroke susceptibility in the high-salt fed stroke-prone spontaneously hypertensive rat. Cell Death Dis. 2017, 8, e2891. [Google Scholar] [CrossRef]
- Rubattu, S.; Di Castro, S.; Schulz, H.; Geurts, A.M.; Cotugno, M.; Bianchi, F.; Maatz, H.; Hummel, O.; Falak, S.; Stanzione, R.; et al. Ndufc2 Gene Inhibition Is Associated With Mitochondrial Dysfunction and Increased Stroke Susceptibility in an Animal Model of Complex Human Disease. J. Am. Heart Assoc. 2016, 5, e002701. [Google Scholar] [CrossRef]
- Forte, M.; Bianchi, F.; Cotugno, M.; Marchitti, S.; De Falco, E.; Raffa, S.; Stanzione, R.; Di Nonno, F.; Chimenti, I.; Palmerio, S.; et al. Pharmacological restoration of autophagy reduces hypertension-related stroke occurrence. Autophagy 2019, 1–14. [Google Scholar] [CrossRef]
- Forte, M.; Palmerio, S.; Bianchi, F.; Volpe, M.; Rubattu, S. Mitochondrial complex I deficiency and cardiovascular diseases: Current evidence and future directions. J. Mol. Med. (Berl.) 2019, 97, 579–591. [Google Scholar] [CrossRef]
- Gretarsdottir, S.; Thorleifsson, G.; Manolescu, A.; Styrkarsdottir, U.; Helgadottir, A.; Gschwendtner, A.; Kostulas, K.; Kuhlenbaumer, G.; Bevan, S.; Jonsdottir, T.; et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann. Neurol. 2008, 64, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Traylor, M.; Farrall, M.; Holliday, E.G.; Sudlow, C.; Hopewell, J.C.; Cheng, Y.C.; Fornage, M.; Ikram, M.A.; Malik, R.; Bevan, S.; et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): A meta-analysis of genome-wide association studies. Lancet Neurol. 2012, 11, 951–962. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediators Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [PubMed]
- Udali, S.; Guarini, P.; Moruzzi, S.; Choi, S.W.; Friso, S. Cardiovascular epigenetics: From DNA methylation to microRNAs. Mol. Asp. Med. 2013, 34, 883–901. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Ann. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15 (Suppl. 1), R17–R29. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kaminska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Cai, R.; Wang, G.; Shu, X.; Pang, W. Long noncoding RNA: Multiple players in gene expression. BMB Rep. 2018, 51, 280–289. [Google Scholar] [CrossRef]
- Maass, P.G.; Luft, F.C.; Bahring, S. Long non-coding RNA in health and disease. J. Mol. Med. (Berl.) 2014, 92, 337–346. [Google Scholar] [CrossRef]
- Sallam, T.; Sandhu, J.; Tontonoz, P. Long Noncoding RNA Discovery in Cardiovascular Disease: Decoding Form to Function. Circ. Res. 2018, 122, 155–166. [Google Scholar] [CrossRef]
- Deng, G.X.; Xu, N.; Huang, Q.; Tan, J.Y.; Zhang, Z.; Li, X.F.; Wei, J.R. Association between promoter DNA methylation and gene expression in the pathogenesis of ischemic stroke. Aging (Albany NY) 2019, 11, 7663–7677. [Google Scholar] [CrossRef]
- Tang, J.; Zhuang, S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin. Sci. (Lond.) 2019, 133, 597–609. [Google Scholar] [CrossRef]
- Endres, M.; Meisel, A.; Biniszkiewicz, D.; Namura, S.; Prass, K.; Ruscher, K.; Lipski, A.; Jaenisch, R.; Moskowitz, M.A.; Dirnagl, U. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 2000, 20, 3175–3181. [Google Scholar] [CrossRef]
- Dock, H.; Theodorsson, A.; Theodorsson, E. DNA Methylation Inhibitor Zebularine Confers Stroke Protection in Ischemic Rats. Transl. Stroke Res. 2015, 6, 296–300. [Google Scholar] [CrossRef]
- Faraco, G.; Pancani, T.; Formentini, L.; Mascagni, P.; Fossati, G.; Leoni, F.; Moroni, F.; Chiarugi, A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharmacol. 2006, 70, 1876–1884. [Google Scholar] [CrossRef]
- Lee, H.A.; Hong, S.H.; Kim, J.W.; Jang, I.S. Possible involvement of DNA methylation in NKCC1 gene expression during postnatal development and in response to ischemia. J. Neurochem. 2010, 114, 520–529. [Google Scholar] [CrossRef]
- Hu, C.J.; Chen, S.D.; Yang, D.I.; Lin, T.N.; Chen, C.M.; Huang, T.H.; Hsu, C.Y. Promoter region methylation and reduced expression of thrombospondin-1 after oxygen-glucose deprivation in murine cerebral endothelial cells. J. Cereb. Blood Flow Metab. 2006, 26, 1519–1526. [Google Scholar] [CrossRef]
- van Meurs, J.B.; Pare, G.; Schwartz, S.M.; Hazra, A.; Tanaka, T.; Vermeulen, S.H.; Cotlarciuc, I.; Yuan, X.; Malarstig, A.; Bandinelli, S.; et al. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am. J. Clin. Nutr. 2013, 98, 668–676. [Google Scholar] [CrossRef]
- Wang, C.; Xu, G.; Wen, Q.; Peng, X.; Chen, H.; Zhang, J.; Xu, S.; Zhang, C.; Zhang, M.; Ma, J.; et al. CBS promoter hypermethylation increases the risk of hypertension and stroke. Clinics (Sao Paulo) 2019, 74, e630. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, L.; Zhang, W.; Wang, X.; Zhou, S. Plasma homocysteine involved in methylation and expression of thrombomodulin in cerebral infarction. Biochem. Biophys. Res. Commun. 2016, 473, 1218–1222. [Google Scholar] [CrossRef]
- Tziomalos, K.; Athyros, V.G.; Karagiannis, A.; Mikhailidis, D.P. Dyslipidemia as a risk factor for ischemic stroke. Curr. Top. Med. Chem. 2009, 9, 1291–1297. [Google Scholar] [CrossRef]
- Imamura, T.; Doi, Y.; Arima, H.; Yonemoto, K.; Hata, J.; Kubo, M.; Tanizaki, Y.; Ibayashi, S.; Iida, M.; Kiyohara, Y. LDL cholesterol and the development of stroke subtypes and coronary heart disease in a general Japanese population: The Hisayama study. Stroke 2009, 40, 382–388. [Google Scholar] [CrossRef]
- Tirschwell, D.L.; Smith, N.L.; Heckbert, S.R.; Lemaitre, R.N.; Longstreth, W.T., Jr.; Psaty, B.M. Association of cholesterol with stroke risk varies in stroke subtypes and patient subgroups. Neurology 2004, 63, 1868–1875. [Google Scholar] [CrossRef]
- Tanne, D.; Koren-Morag, N.; Graff, E.; Goldbourt, U. Blood lipids and first-ever ischemic stroke/transient ischemic attack in the Bezafibrate Infarction Prevention (BIP) Registry: High triglycerides constitute an independent risk factor. Circulation 2001, 104, 2892–2897. [Google Scholar] [CrossRef]
- Wannamethee, S.G.; Shaper, A.G.; Ebrahim, S. HDL-Cholesterol, total cholesterol, and the risk of stroke in middle-aged British men. Stroke 2000, 31, 1882–1888. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- McCarron, M.O.; Delong, D.; Alberts, M.J. APOE genotype as a risk factor for ischemic cerebrovascular disease: A meta-analysis. Neurology 1999, 53, 1308–1311. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, X.; Wang, C.; Du, R.; Wang, X.; Fu, J.; Sun, Q. A Preliminary Study of the Association between Apolipoprotein E Promoter Methylation and Atherosclerotic Cerebral Infarction. J. Stroke Cerebrovasc. Dis. 2019, 28, 1056–1061. [Google Scholar] [CrossRef]
- Zhou, S.; Cai, B.; Zhang, Z.; Zhang, Y.; Wang, L.; Liu, K.; Zhang, H.; Sun, L.; Cai, H.; Lu, G.; et al. CDKN2B Methylation and Aortic Arch Calcification in Patients with Ischemic Stroke. J. Atheroscler. Thromb. 2017, 24, 609–620. [Google Scholar] [CrossRef]
- Ridker, P.M. Role of inflammatory biomarkers in prediction of coronary heart disease. Lancet 2001, 358, 946–948. [Google Scholar] [CrossRef]
- Malik, I.; Danesh, J.; Whincup, P.; Bhatia, V.; Papacosta, O.; Walker, M.; Lennon, L.; Thomson, A.; Haskard, D. Soluble adhesion molecules and prediction of coronary heart disease: A prospective study and meta-analysis. Lancet 2001, 358, 971–976. [Google Scholar] [CrossRef]
- Baccarelli, A.; Tarantini, L.; Wright, R.O.; Bollati, V.; Litonjua, A.A.; Zanobetti, A.; Sparrow, D.; Vokonas, P.S.; Schwartz, J. Repetitive element DNA methylation and circulating endothelial and inflammation markers in the VA normative aging study. Epigenetics 2010, 5, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Fabrega, C.; Carrera, C.; Reny, J.L.; Fontana, P.; Slowik, A.; Pera, J.; Pezzini, A.; Serrano-Heras, G.; Segura, T.; Marti-Fabregas, J.; et al. TRAF3 Epigenetic Regulation Is Associated With Vascular Recurrence in Patients With Ischemic Stroke. Stroke 2016, 47, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Fabrega, C.; Carrera, C.; Reny, J.L.; Fontana, P.; Slowik, A.; Pera, J.; Pezzini, A.; Serrano-Heras, G.; Segura, T.; Bin Dukhyil, A.A.; et al. PPM1A Methylation Is Associated With Vascular Recurrence in Aspirin-Treated Patients. Stroke 2016, 47, 1926–1929. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.S.; Chen, P.S.; Chuang, D.M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Hong, L.; Zhang, M.; Zhang, W. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012, 90, 463–468. [Google Scholar] [CrossRef]
- Ren, M.; Leng, Y.; Jeong, M.; Leeds, P.R.; Chuang, D.M. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: Potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 2004, 89, 1358–1367. [Google Scholar] [CrossRef]
- Langley, B.; Brochier, C.; Rivieccio, M.A. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke 2009, 40, 2899–2905. [Google Scholar] [CrossRef]
- Kong, Q.; Hao, Y.; Li, X.; Wang, X.; Ji, B.; Wu, Y. HDAC4 in ischemic stroke: Mechanisms and therapeutic potential. Clin. Epigenet. 2018, 10, 117. [Google Scholar] [CrossRef]
- Majdzadeh, N.; Wang, L.; Morrison, B.E.; Bassel-Duby, R.; Olson, E.N.; D’Mello, S.R. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev. Neurobiol. 2008, 68, 1076–1092. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Wang, Z.J.; Sun, D.M.; Wang, Y.; Xu, P.; Wu, W.J.; Liu, X.H.; Zhu, Y.Z. Novel Therapeutic Effects of Leonurine On Ischemic Stroke: New Mechanisms of BBB Integrity. Oxid. Med. Cell. Longev. 2017, 2017, 7150376. [Google Scholar] [CrossRef]
- Liu, X.S.; Chopp, M.; Kassis, H.; Jia, L.F.; Hozeska-Solgot, A.; Zhang, R.L.; Chen, C.; Cui, Y.S.; Zhang, Z.G. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 2012, 220, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Patnala, R.; Arumugam, T.V.; Gupta, N.; Dheen, S.T. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia During Ischemic Stroke. Mol. Neurobiol. 2017, 54, 6391–6411. [Google Scholar] [CrossRef]
- Kao, M.H.; Lin, T.N. Histone deacetylases in stroke. Chin. J. Physiol. 2019, 62, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Brookes, R.L.; Crichton, S.; Wolfe, C.D.A.; Yi, Q.; Li, L.; Hankey, G.J.; Rothwell, P.M.; Markus, H.S. Sodium Valproate, a Histone Deacetylase Inhibitor, Is Associated With Reduced Stroke Risk After Previous Ischemic Stroke or Transient Ischemic Attack. Stroke 2018, 49, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Rink, C.; Khanna, S. MicroRNA in ischemic stroke etiology and pathology. Physiol. Genom. 2011, 43, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.W.; Wang, Y.L.; Lin, J.X.; Li, N.; Zhao, X.Q.; Liu, G.F.; Liu, L.P.; Jiao, Y.; Gu, W.K.; Wang, D.Z.; et al. Circulating MicroRNAs as potential risk biomarkers for hematoma enlargement after intracerebral hemorrhage. CNS Neurosci. Ther. 2012, 18, 1003–1011. [Google Scholar] [CrossRef]
- Mirzaei, H.; Momeni, F.; Saadatpour, L.; Sahebkar, A.; Goodarzi, M.; Masoudifar, A.; Kouhpayeh, S.; Salehi, H.; Mirzaei, H.R.; Jaafari, M.R. MicroRNA: Relevance to stroke diagnosis, prognosis, and therapy. J. Cell. Physiol. 2018, 233, 856–865. [Google Scholar] [CrossRef]
- Eyileten, C.; Wicik, Z.; De Rosa, S.; Mirowska-Guzel, D.; Soplinska, A.; Indolfi, C.; Jastrzebska-Kurkowska, I.; Czlonkowska, A.; Postula, M. MicroRNAs as Diagnostic and Prognostic Biomarkers in Ischemic Stroke-A Comprehensive Review and Bioinformatic Analysis. Cells 2018, 7, 249. [Google Scholar] [CrossRef]
- Laterza, O.F.; Lim, L.; Garrett-Engele, P.W.; Vlasakova, K.; Muniappa, N.; Tanaka, W.K.; Johnson, J.M.; Sina, J.F.; Fare, T.L.; Sistare, F.D.; et al. Plasma MicroRNAs as sensitive and specific biomarkers of tissue injury. Clin. Chem. 2009, 55, 1977–1983. [Google Scholar] [CrossRef]
- Dharap, A.; Bowen, K.; Place, R.; Li, L.C.; Vemuganti, R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J. Cereb. Blood Flow Metab. 2009, 29, 675–687. [Google Scholar] [CrossRef]
- Rubattu, S.; Volpe, M.; Kreutz, R.; Ganten, U.; Ganten, D.; Lindpaintner, K. Chromosomal mapping of quantitative trait loci contributing to stroke in a rat model of complex human disease. Nat. Genet. 1996, 13, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Stanzione, R.; Bianchi, F.; Cotugno, M.; Marchitti, S.; Forte, M.; Busceti, C.; Ryskalin, L.; Fornai, F.; Volpe, M.; Rubattu, S. A Decrease of Brain MicroRNA-122 Level Is an Early Marker of Cerebrovascular Disease in the Stroke-Prone Spontaneously Hypertensive Rat. Oxid. Med. Cell Longev. 2017, 2017, 1206420. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem. 2002, 277, 47129–47135. [Google Scholar] [CrossRef] [PubMed]
- Sekar, D.; Venugopal, B.; Sekar, P.; Ramalingam, K. Role of microRNA 21 in diabetes and associated/related diseases. Gene 2016, 582, 14–18. [Google Scholar] [CrossRef]
- Sekar, D.; Shilpa, B.R.; Das, A.J. Relevance of microRNA 21 in Different Types of Hypertension. Curr. Hypertens Rep. 2017, 19, 57. [Google Scholar] [CrossRef]
- Jazbutyte, V.; Thum, T. MicroRNA-21: From cancer to cardiovascular disease. Curr. Drug Targets 2010, 11, 926–935. [Google Scholar] [CrossRef]
- Xu, L.F.; Wu, Z.P.; Chen, Y.; Zhu, Q.S.; Hamidi, S.; Navab, R. MicroRNA-21 (miR-21) regulates cellular proliferation, invasion, migration, and apoptosis by targeting PTEN, RECK and Bcl-2 in lung squamous carcinoma, Gejiu City, China. PLoS ONE 2014, 9, e103698. [Google Scholar] [CrossRef]
- Buller, B.; Liu, X.; Wang, X.; Zhang, R.L.; Zhang, L.; Hozeska-Solgot, A.; Chopp, M.; Zhang, Z.G. MicroRNA-21 protects neurons from ischemic death. FEBS J. 2010, 277, 4299–4307. [Google Scholar] [CrossRef]
- Wu, J.; Fan, C.L.; Ma, L.J.; Liu, T.; Wang, C.; Song, J.X.; Lv, Q.S.; Pan, H.; Zhang, C.N.; Wang, J.J. Distinctive expression signatures of serum microRNAs in ischaemic stroke and transient ischaemic attack patients. Thromb. Haemost. 2017, 117, 992–1001. [Google Scholar] [CrossRef]
- Tan, K.S.; Armugam, A.; Sepramaniam, S.; Lim, K.Y.; Setyowati, K.D.; Wang, C.W.; Jeyaseelan, K. Expression profile of MicroRNAs in young stroke patients. PLoS ONE 2009, 4, e7689. [Google Scholar] [CrossRef]
- Zhao, B.; Zhu, Z.; Hao, J.; Wan, Z.; Guo, X. Decreased plasma miR-335 expression in patients with acute ischemic stroke and its association with calmodulin expression. J. Int. Med. Res. 2016, 44, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yuan, H.; Denton, K.; Li, X.J.; McCullough, L.; Li, J. Calcium/calmodulin-dependent protein kinase kinase beta is neuroprotective in stroke in aged mice. Eur. J. Neurosci. 2016, 44, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, T.; Yu, H.; Sun, K.; Shi, Y.; Song, W.; Bai, Y.; Wang, X.; Lou, K.; Song, Y.; et al. A functional variant in the 3’-UTR of angiopoietin-1 might reduce stroke risk by interfering with the binding efficiency of microRNA 211. Hum. Mol. Genet. 2010, 19, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Garcia-Cardena, G.; Dengler, T.J.; Maisonpierre, P.C.; Yancopoulos, G.D.; Sessa, W.C. Direct actions of angiopoietin-1 on human endothelium: Evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab. Investig. 1999, 79, 213–223. [Google Scholar] [PubMed]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef]
- Stanzione, R.; Sciarretta, S.; Marchitti, S.; Bianchi, F.; Di Castro, S.; Scarpino, S.; Cotugno, M.; Frati, G.; Volpe, M.; Rubattu, S. C2238/alphaANP modulates apolipoprotein E through Egr-1/miR199a in vascular smooth muscle cells in vitro. Cell Death Dis. 2015, 6, e2033. [Google Scholar] [CrossRef]
- Dykstra-Aiello, C.; Jickling, G.C.; Ander, B.P.; Shroff, N.; Zhan, X.; Liu, D.; Hull, H.; Orantia, M.; Stamova, B.S.; Sharp, F.R. Altered Expression of Long Noncoding RNAs in Blood After Ischemic Stroke and Proximity to Putative Stroke Risk Loci. Stroke 2016, 47, 2896–2903. [Google Scholar] [CrossRef]
- Zhao, F.; Qu, Y.; Liu, J.; Liu, H.; Zhang, L.; Feng, Y.; Wang, H.; Gan, J.; Lu, R.; Mu, D. Microarray Profiling and Co-Expression Network Analysis of LncRNAs and mRNAs in Neonatal Rats Following Hypoxic-ischemic Brain Damage. Sci. Rep. 2015, 5, 13850. [Google Scholar] [CrossRef]
- Bao, M.H.; Szeto, V.; Yang, B.B.; Zhu, S.Z.; Sun, H.S.; Feng, Z.P. Long non-coding RNAs in ischemic stroke. Cell Death Dis. 2018, 9, 281. [Google Scholar] [CrossRef]
- Chen, R.; Xu, X.; Huang, L.; Zhong, W.; Cui, L. The Regulatory Role of Long Noncoding RNAs in Different Brain Cell Types Involved in Ischemic Stroke. Front. Mol. Neurosci. 2019, 12, 61. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, D.; Ji, T.F.; Shi, L.; Yu, J.L. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-kappaB signaling pathway in a rat model. Oncotarget 2017, 8, 17347–17359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, X.; Liu, K.; Hamblin, M.H.; Yin, K.J. Long Noncoding RNA Malat1 Regulates Cerebrovascular Pathologies in Ischemic Stroke. J. Neurosci. 2017, 37, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yuan, J.; Gao, L.; Rao, J.; Hu, J. Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience 2016, 337, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Karatas, H.; Liu, Y.; Yalcin, A.; Montaner, J.; Lo, E.H.; van Leyen, K. STAT-dependent upregulation of 12/15-lipoxygenase contributes to neuronal injury after stroke. J. Cereb. Blood Flow Metab. 2015, 35, 2043–2051. [Google Scholar] [CrossRef]
- Liu, X.; Hou, L.; Huang, W.; Gao, Y.; Lv, X.; Tang, J. The Mechanism of Long Non-coding RNA MEG3 for Neurons Apoptosis Caused by Hypoxia: Mediated by miR-181b-12/15-LOX Signaling Pathway. Front. Cell. Neurosci. 2016, 10, 201. [Google Scholar] [CrossRef]
- Chen, C.; Cheng, G.; Yang, X.; Li, C.; Shi, R.; Zhao, N. Tanshinol suppresses endothelial cells apoptosis in mice with atherosclerosis via lncRNA TUG1 up-regulating the expression of miR-26a. Am. J. Transl. Res. 2016, 8, 2981–2991. [Google Scholar]
- Li, J.; Zhang, M.; An, G.; Ma, Q. LncRNA TUG1 acts as a tumor suppressor in human glioma by promoting cell apoptosis. Exp. Biol. Med. (Maywood) 2016, 241, 644–649. [Google Scholar] [CrossRef]
- Chen, S.; Wang, M.; Yang, H.; Mao, L.; He, Q.; Jin, H.; Ye, Z.M.; Luo, X.Y.; Xia, Y.P.; Hu, B. LncRNA TUG1 sponges microRNA-9 to promote neurons apoptosis by up-regulated Bcl2l11 under ischemia. Biochem. Biophys. Res. Commun. 2017, 485, 167–173. [Google Scholar] [CrossRef]
- Zhu, W.; Tian, L.; Yue, X.; Liu, J.; Fu, Y.; Yan, Y. LncRNA Expression Profiling of Ischemic Stroke During the Transition From the Acute to Subacute Stage. Front. Neurol. 2019, 10, 36. [Google Scholar] [CrossRef]
- Tang, Y.; Lin, Y.H.; Ni, H.Y.; Dong, J.; Yuan, H.J.; Zhang, Y.; Liang, H.Y.; Yao, M.C.; Zhou, Q.G.; Wu, H.Y.; et al. Inhibiting Histone Deacetylase 2 (HDAC2) Promotes Functional Recovery From Stroke. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Santini, V.; Melnick, A.; Maciejewski, J.P.; Duprez, E.; Nervi, C.; Cocco, L.; Ford, K.G.; Mufti, G. Epigenetics in focus: Pathogenesis of myelodysplastic syndromes and the role of hypomethylating agents. Crit. Rev. Oncol. Hematol. 2013, 88, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tang, X.; Sun, P.; Shi, Y.; Liu, K.; Hassan, S.H.; Stetler, R.A.; Chen, J.; Yin, K.J. MicroRNA-15a/16-1 Antagomir Ameliorates Ischemic Brain Injury in Experimental Stroke. Stroke 2017, 48, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Mallick, B. AGO-Driven Non-Coding RNAs: Codes to Decode the Therapeutics of Diseases, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128156698. [Google Scholar]


{kind=link}
| Study Model | Condition/Effects | Outcomes | References |
|---|---|---|---|
| MCAO rat | Pharmacological inhibition of DNMT |  Infarct sizeCerebral ischemic damage Infarct sizeCerebral ischemic damage | [41,42,43] |
| MCAO rat | Increased demethylation of NKCC1 |  Cerebral ischemic injury Cerebral ischemic injury | [44] |
| Rat endothelial cells under oxygen-glucose deprivation (OGD) | Hypermethylation of THBS1 | Angiogenesis | [45] |
| Hypertensive and stroke patients | Hypermethylation of CBS | Homocysteinemiastroke occurrence | [47] |
| Stroke patients | Hypermethylation of TM | Vascular endothelial damage Risk of IS | [48] |
| Meta-analysis study of stroke patients Stroke patients | Hypermethylation of ApoE | Lipoprotein accumulationAtherosclerotic cerebral infarction | [56,57] |
| Stroke patients | Hypermethylation of CDKN2B | Aortic arch calcification | [58] |
| Elderly patients | Hypomethylation of LINE-1 | VCAM-1 level | [61] |
| Stroke patients receiving antiplatelet drugs | Hypomethylation of TRAF3 and PPM1A | Stroke occurrence | [62,63] |
indicates decrease whereas indicates increase.© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanzione, R.; Cotugno, M.; Bianchi, F.; Marchitti, S.; Forte, M.; Volpe, M.; Rubattu, S. Pathogenesis of Ischemic Stroke: Role of Epigenetic Mechanisms. Genes 2020, 11, 89. https://doi.org/10.3390/genes11010089
Stanzione R, Cotugno M, Bianchi F, Marchitti S, Forte M, Volpe M, Rubattu S. Pathogenesis of Ischemic Stroke: Role of Epigenetic Mechanisms. Genes. 2020; 11(1):89. https://doi.org/10.3390/genes11010089
Chicago/Turabian StyleStanzione, Rosita, Maria Cotugno, Franca Bianchi, Simona Marchitti, Maurizio Forte, Massimo Volpe, and Speranza Rubattu. 2020. "Pathogenesis of Ischemic Stroke: Role of Epigenetic Mechanisms" Genes 11, no. 1: 89. https://doi.org/10.3390/genes11010089
APA StyleStanzione, R., Cotugno, M., Bianchi, F., Marchitti, S., Forte, M., Volpe, M., & Rubattu, S. (2020). Pathogenesis of Ischemic Stroke: Role of Epigenetic Mechanisms. Genes, 11(1), 89. https://doi.org/10.3390/genes11010089