Functionally Annotating Regulatory Elements in the Equine Genome Using Histone Mark ChIP-Seq

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chromatin Extraction
2.2. Immunoprecipitation
2.3. Sequencing
2.4. Data Processing
2.5. Data Analysis
3. Results
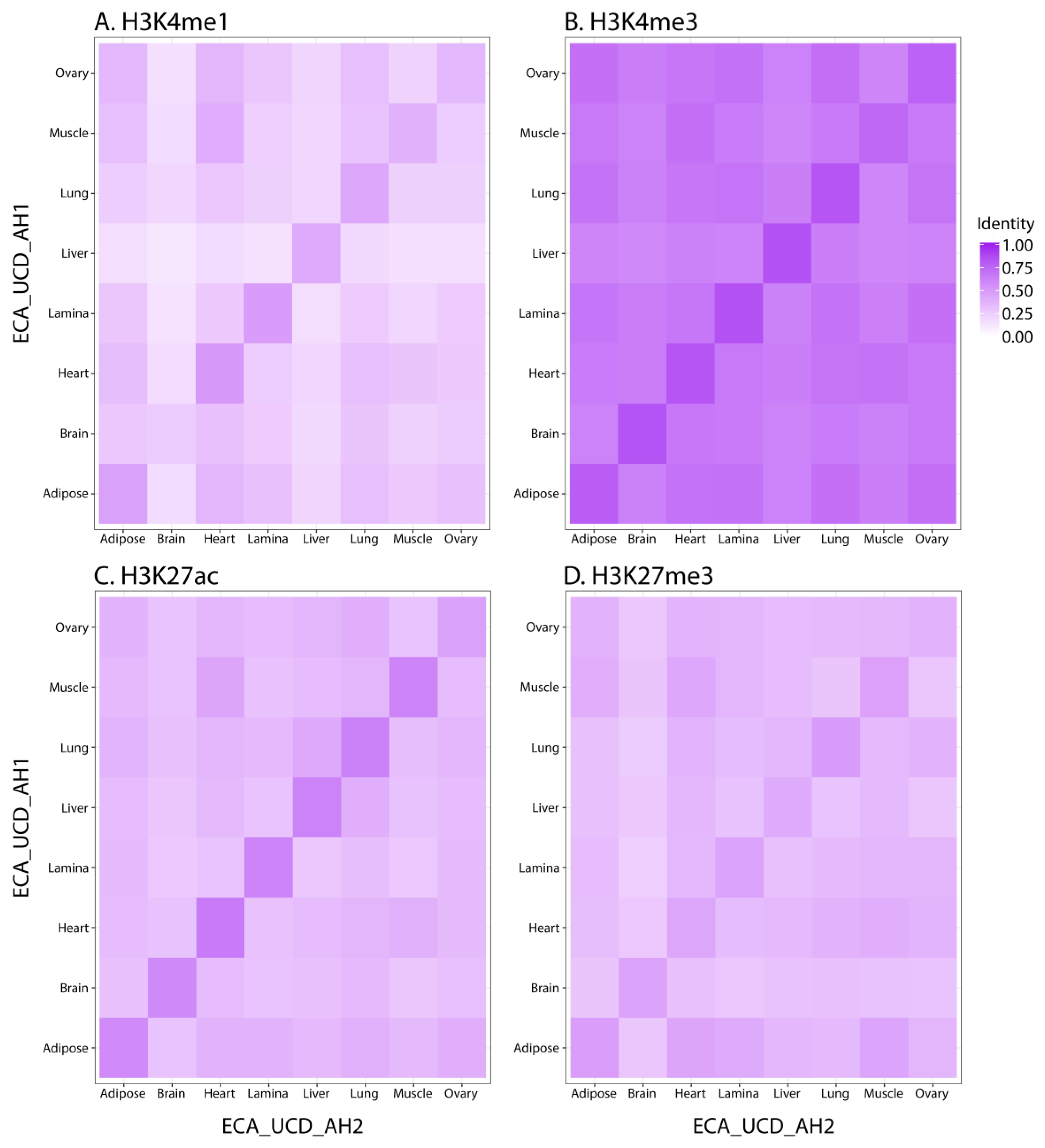
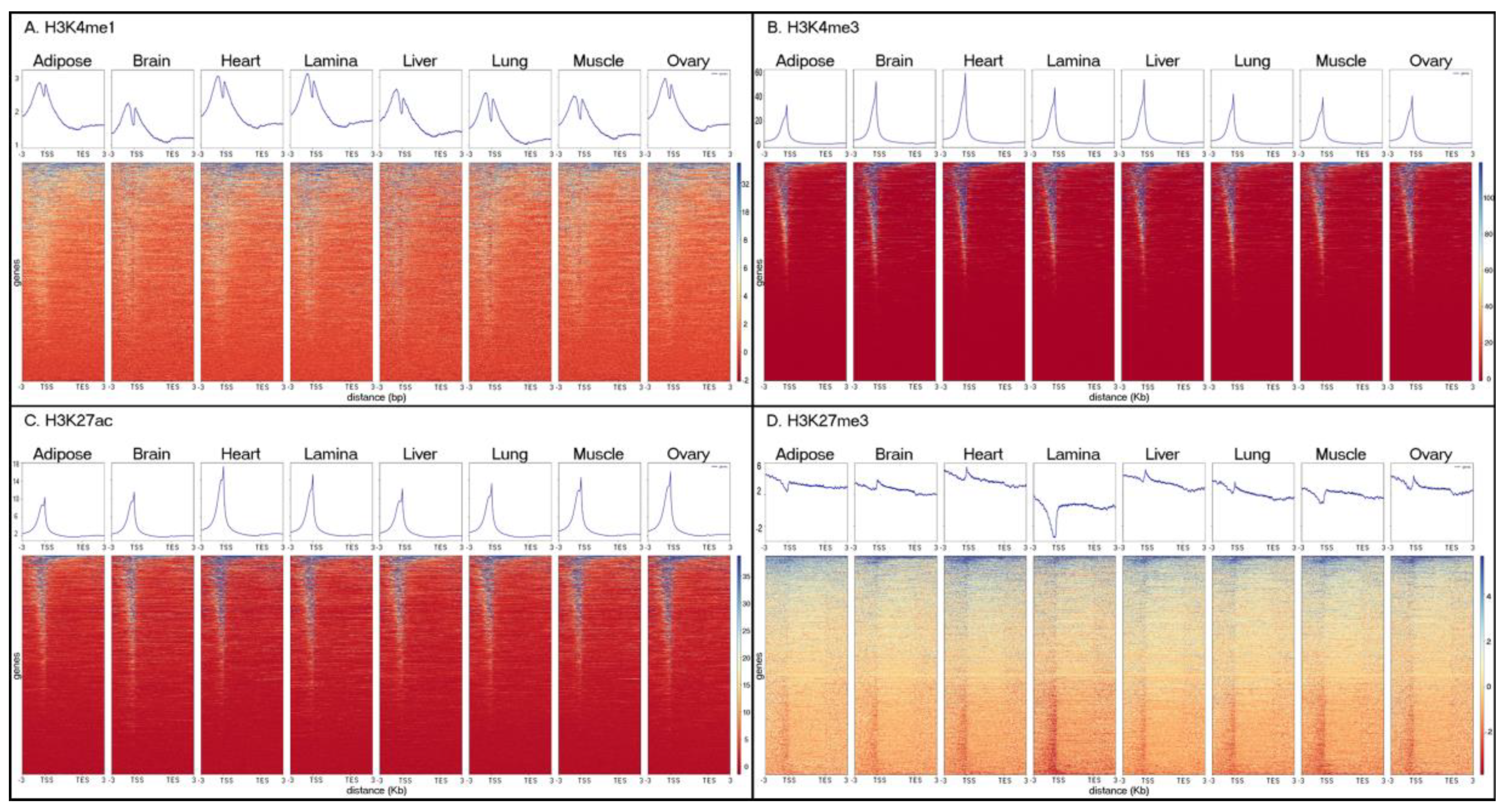
3.1. Assessing Data Quality
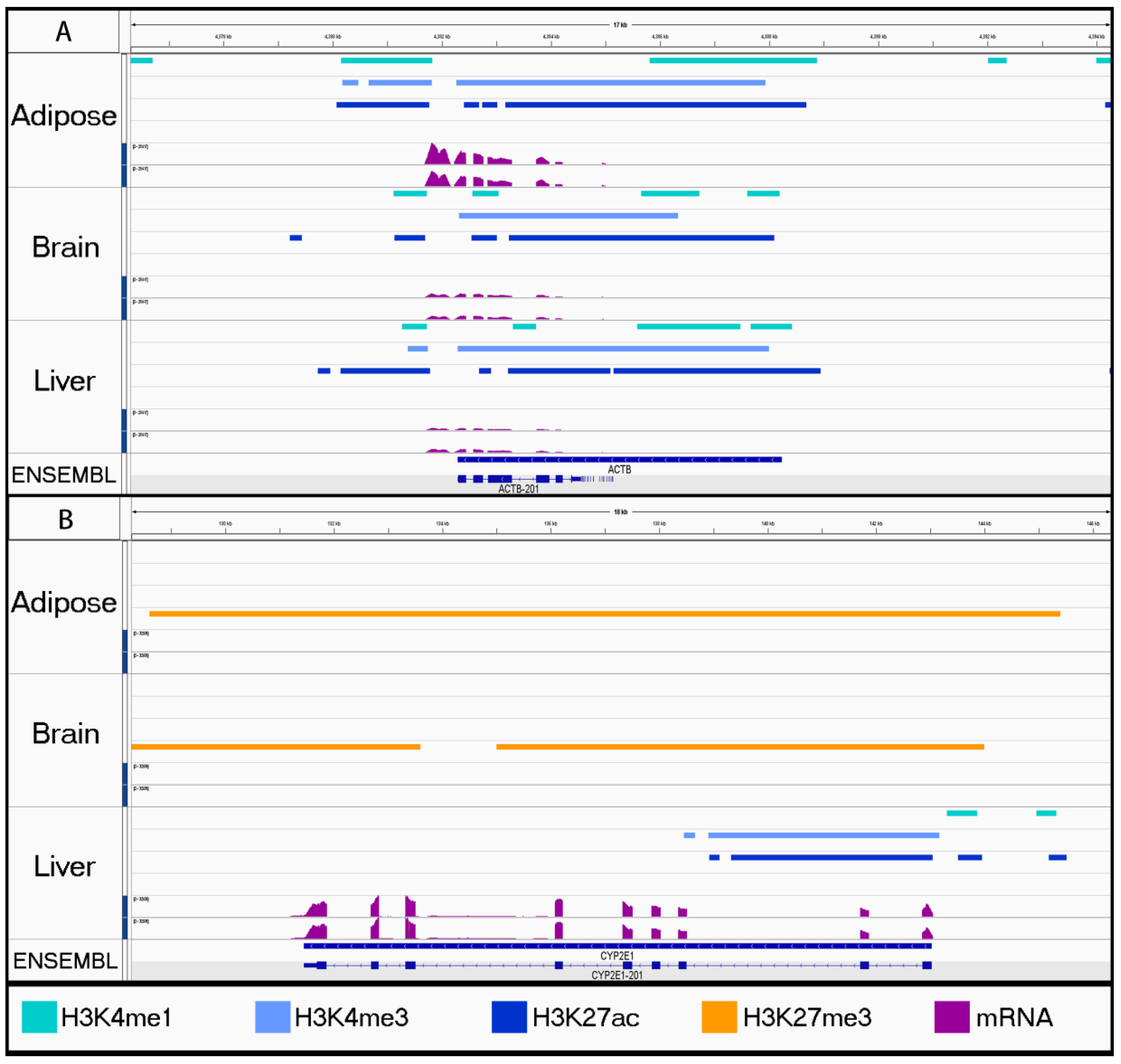
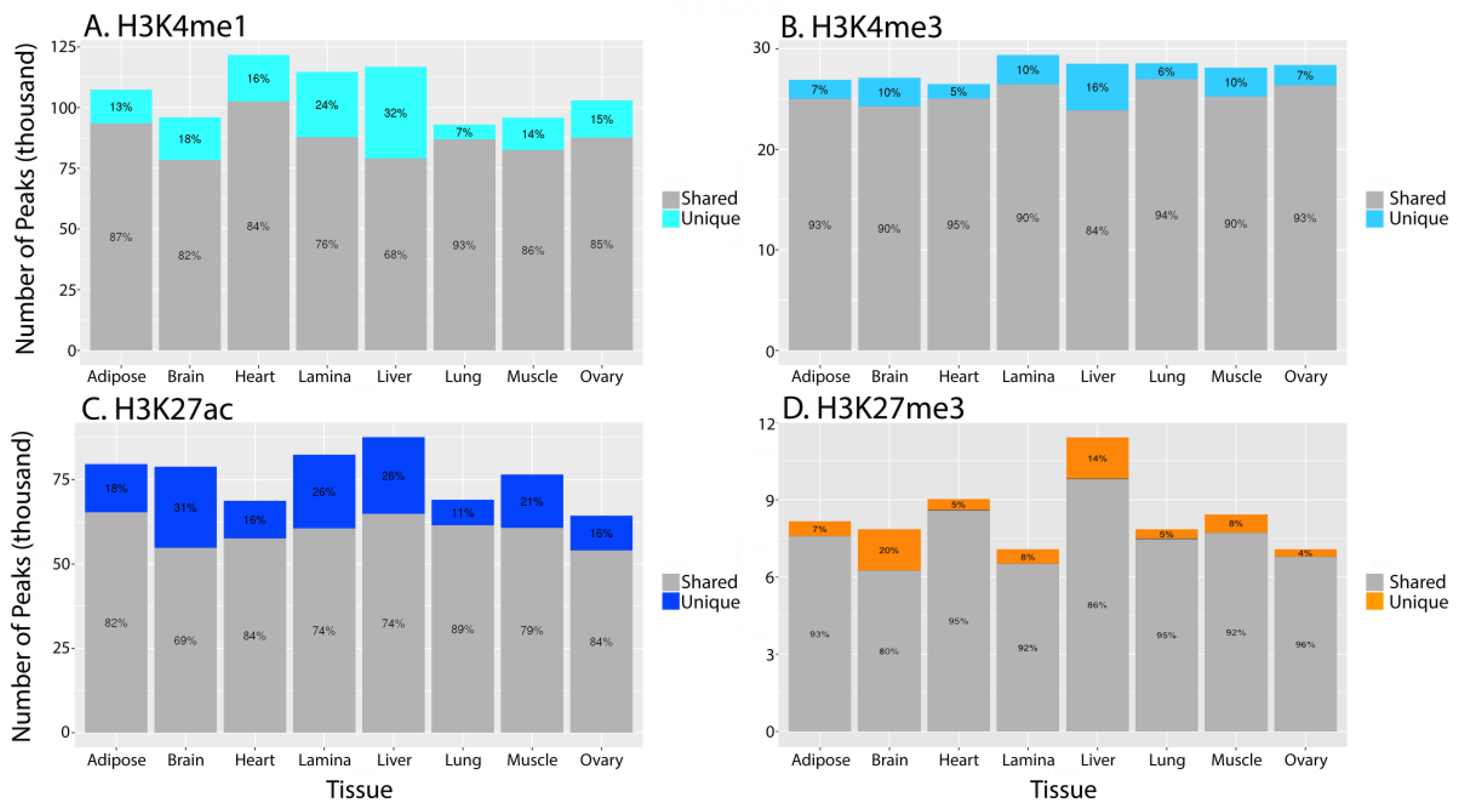
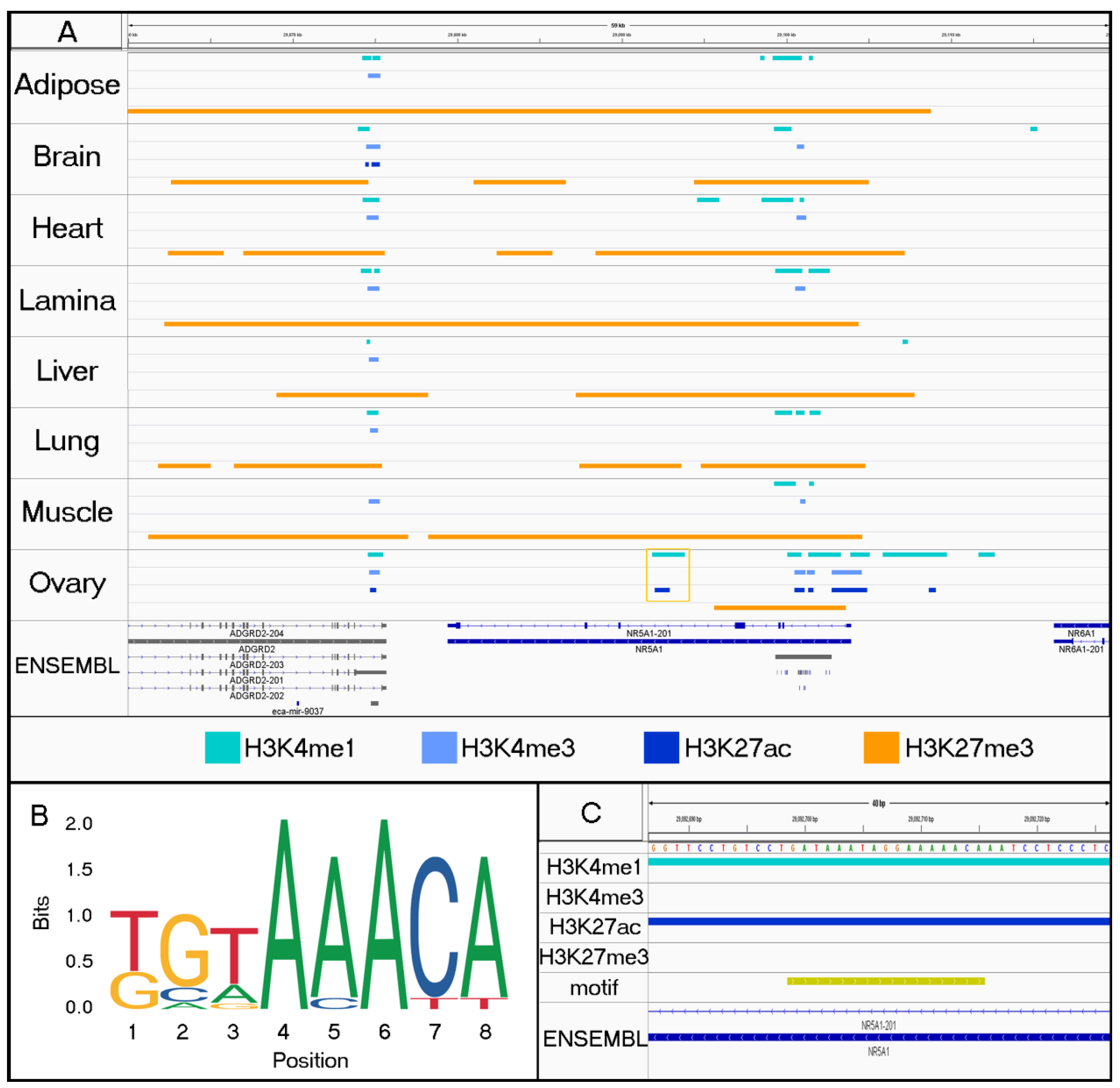
3.2. Characterizing Tissue-Specific Features
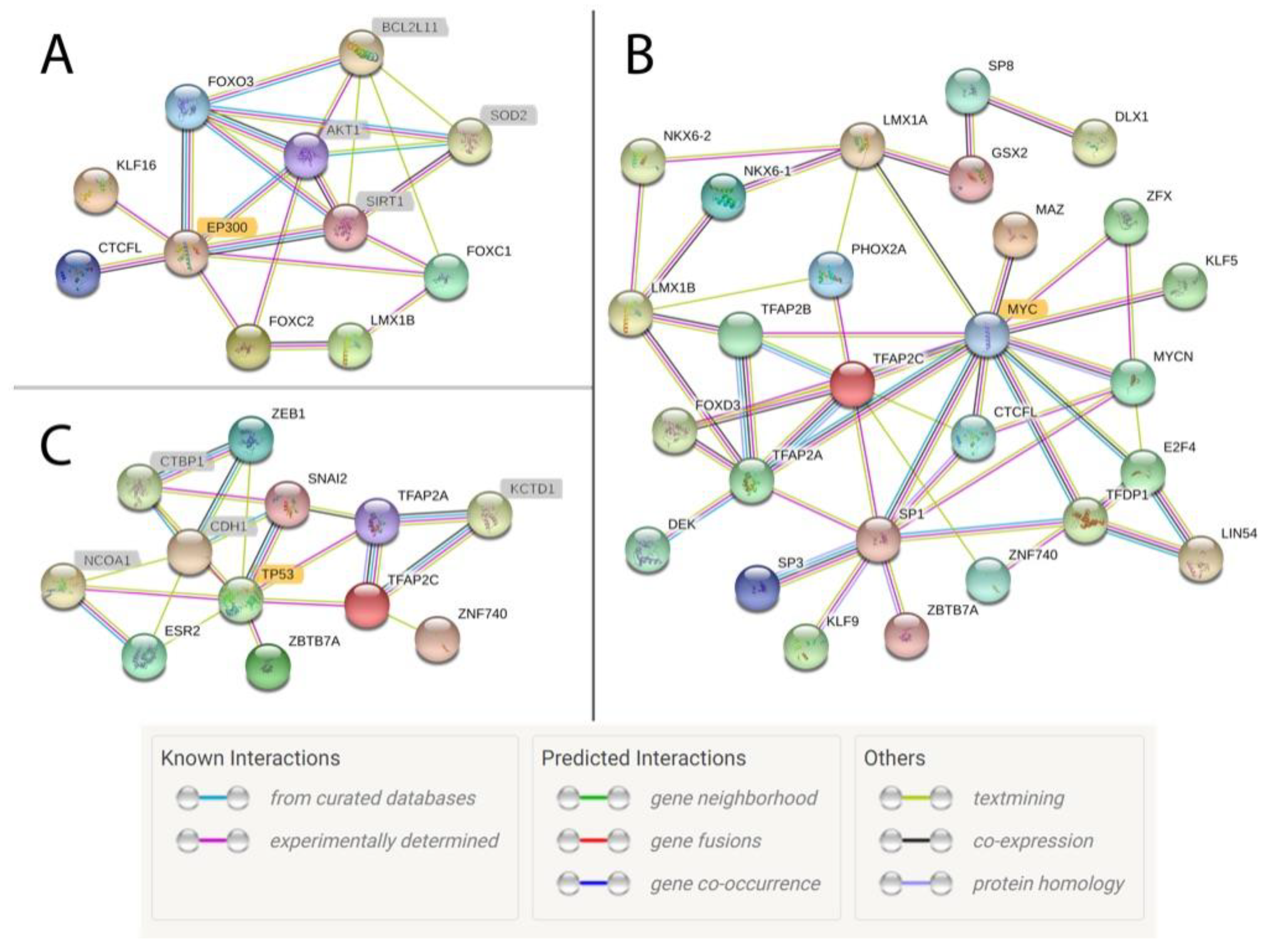
3.3. Identifying Motifs and Biological Process GO Terms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rudolph, J.A.; Spier, S.J.; Byrans, G.; Hoffman, E.P. Linkage of hyperkalaemic periodic paralysis in Quarter horses to the horse adult skeletal muscle sodium channel gene. Anim. Genet. 1992, 23, 241–250. [Google Scholar] [CrossRef]
- Finno, C.J.; Bannasch, D.L. Applied equine genetics. Equine Vet. J. 2014, 46, 538–544. [Google Scholar] [CrossRef]
- Raudsepp, T.; Finno, C.J.; Bellone, R.R.; Petersen, J.J. Ten years of the horse reference genome: Insights into equine biology, domestication, and population dynamics in the post-genome era. Anim. Genet. 2019, 50, 569–597. [Google Scholar] [CrossRef]
- OMIA—Online Mendelian Inheritance in Animals. Available online: https://omia.org/home/ (accessed on 4 January 2019).
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome Sequence, Comparative Analysis, and Population Genetics of the Domestic Horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef]
- Mathelier, A.; Shi, W.; Wasserman, W.W. Identification of altered cis-regulatory elements in human disease. Trends Genet. 2015, 31, 67–76. [Google Scholar] [CrossRef]
- Koufariotis, L.; Chen, Y.P.; Bolormaa, S.; Hayes, B.J. Regulatory and coding genome regions are enriched for trait associated variants in dairy and beef cattle. BMC Genom. 2014, 15, 436. [Google Scholar] [CrossRef]
- Wang, M.; Hancock, T.P.; MacLeod, I.M.; Pryce, J.E.; Cocks, B.G.; Hayes, B.J. Putative enhancer sites in the bovine genome are enriched with variants affecting complex traits. Genet. Sel. Evol. 2017, 49, 56. [Google Scholar] [CrossRef]
- Danko, C.G.; Choate, L.A.; Marks, B.A.; Rice, E.J.; Wang, Z.; Chu, T.; Martins, A.L.; Dukler, N.; Coonrod, S.A.; Tait Wojno, E.D.; et al. Dynamic evolution of regulatory element ensembles in primate CD4+ T cells. Nat. Ecol. Evol. 2018, 2, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, L.; Gaulton, K.J.; Rodriguez-Segui, S.A.; Mularoni, L.; Miguel-Escalada, I.; Akerman, I.; Tena, J.J.; Moran, I.; Gomez-Marin, C.; van de Bunt, M.; et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 2014, 46, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Poschmann, J.; del Rosario, R.C.H.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.; et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 2016, 167, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- The FAANG Consortium; Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Tuggle, C.K.; Guiffra, E.; White, S.N.; Clarke, L.; Zhou, H.; Ross, P.J.; Acloque, H.; Reecy, J.M.; Archibald, A.; Bellone, R.R.; et al. GO-FAANG meeting: A Gathering On Functional Annotation of Animal Genomes. Anim. Genet. 2016, 47, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Guiffra, E.; Tuggle, C.K.; FAANG Consortium. Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2019, 7, 65–88. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Howe, F.S.; Fischl, H.; Murray, S.C.; Mellor, J. Is H3K4me3 instructive for transcription activation? Bioessays 2016, 39, 1–12. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef]
- Entrevan, M.; Schuettengruber, B.; Cavalli, G. Regulation of Genome Architecture and Function by Polycomb Proteins. Trends Cell Biol. 2016, 26, 511–525. [Google Scholar] [CrossRef]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signaling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental states. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kousarides, T.; Schreiber, S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8695–8700. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wang, Z. Histone Modification Patterns and Their Responses to Environment. Curr. Environ. Health Rep. 2014, 1, 11–21. [Google Scholar] [CrossRef]
- Burns, E.N.; Bordbari, M.H.; Mienaltowski, M.J.; Affolter, V.K.; Barro, M.V.; Gianino, F.; Gianino, G.; Giulotto, E.; Kalbfleisch, T.S.; Katzman, S.A.; et al. Generation of an equine biobank to be used for Functional Annotation of Animal Genomes project. Anim. Genet. 2018, 49, 564–570. [Google Scholar] [CrossRef]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Berstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Rickter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Trim Galore. Version 0.4.0. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- Martin, M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O’Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Saremi, N.F.; et al. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 2018, 1, 197. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Picard Toolkit, GitHub Repository. Version 2.7.1. Available online: http://broadinstitute.github.io/picard/.
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Berntein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Wei, L.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- SICERpy, GitHub Repository. Available online: https://github.com/dariober/SICERpy (accessed on 10 October 2019).
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Favorov, A.; Mularoni, L.; Cope, L.M.; Medvedeva, Y.; Mironov, A.A.; Makeev, V.J.; Wheelan, S.J. Exploring Massive, Genome Scale Datasets with the GenometriCorr Package. PLoS Comput. Biol. 2012, 8, e1002529. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic featues. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; Version 3.4.3; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org/.
- McLeary, R.; Bailey, T.L. Motif Enrichment Analysis: A unified framework and method evaluation. BMC Bioinform. 2010, 11, 165. [Google Scholar]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nature Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrel, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; Garcia Giron, C.; Hourlier, T.; et al. The Ensembl gene annotation system. Database 2016, 2016, baw093. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1141–R1155. [Google Scholar] [CrossRef] [PubMed]
- Drogemuller, M.; Jagannathan, V.; Welle, M.M.; Graubner, C.; Straub, R.; Gerber, V.; Burger, D.; Signer-Hasler, H.; Poncet, P.A.; Klopfenstein, S. Congenital hepatic fibrosis in the Franches-Montagnes horse is associated with the polycystic kidney and hepatic disease 1 (PKHD1) gene. PLoS ONE 2014, 9, e110125. [Google Scholar] [CrossRef] [PubMed]
- Velie, B.D.; Lillie, M.; Fegraeus, K.J.; Rosengren, M.K.; Sole, M.; Wiklund, M.; Ihler, C.F.; Strand, E.; Lindgren, G. Exploring the genetics of trotting racing ability in horses using a unique Nordic horse model. BMC Genom. 2019, 20, 104. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Ferraz-de-Souza, B.; Lin, L.; Achermann, J.C. Steroidogenic factor-1 (SF1, NR5A1) and human disease. Mol. Cell. Endocrinol. 2011, 336, 198–205. [Google Scholar] [CrossRef]
- Szalkowski, A.M.; Schmid, C.D. Rapid innovation in ChIP-seq peak-calling algorithms is outdistancing benchmarking efforts. Brief. Bioinform. 2010, 12, 626–633. [Google Scholar] [CrossRef]
- Steinhauser, S.; Kurzawa, N.; Eils, R.; Herrmann, C. A comprehensive comparison of tools for differential ChIP-seq analysis. Brief. Bioinform. 2016, 17, 953–966. [Google Scholar] [CrossRef]
- Xu, S.; Grullon, S.; Ge, K.; Peng, W. Spatial Clustering for Identification of ChIP-Enriched Regions (SICER) to Map Regions of Histone Methylation Patterns in Embryonic Stem Cells. In Stem Cell Transcriptional Networks; Kidder, B., Ed.; Humana Press: New York, NY, USA, 2014; Volume 1150, pp. 97–111. [Google Scholar]
- Masland, R.H. Neuronal cell types. Curr. Biol. 2004, 14, R497–R500. [Google Scholar] [CrossRef]
- Belknap, J.K. Genomics of laminitis. In Equine Genomics, 1st ed.; Chowdhary, B.P., Ed.; John Wiley & Sons, Inc.: Ames, IA, USA, 2013; pp. 255–264. [Google Scholar]
- Al-Agele, R.; Paul, E.; Kubale Dvojmoc, V.; Sturrock, C.J.; Rauch, C.; Rutland, C.S. The Anatomy, Histology and Physiology of the Healthy and Lame Equine Hoof. In Veterinary Anatomy and Physiology; Rutland, C., Kubale, V., Eds.; IntechOpen: London, UK, 2019; pp. 1–18. [Google Scholar]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The Transcriptional Coactivators p300 and CBP Are Histone Acetyletransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Song, C.; Keller, K.; Chen, Y.; Chen, Y.; Murata, K.; Stamatoyannopoulos, G. Transcription coactivator CBP has direct DNA binding activity and stimulates transcription factor DNA binding through small domains. Biochem. Biophys. Res. Commun. 2002, 296, 118–124. [Google Scholar] [CrossRef]
- Oster, S.K.; Ho, C.S.; Soucie, E.L.; Penn, L.Z. The myc oncogene: MarvelouslY Complex. Adv. Cancer Res. 2002, 84, 81–154. [Google Scholar] [PubMed]
- Mogi, A.; Kuwano, H. TP53 Mutations in Nonsmall Cell Lung Cancer. J. Biomed. Biotechnol. 2010, 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Widmann, M.; Nieß, A.M.; Munz, B. Physical Exercise and Epigenetic Modifications in Skeletal Muscle. Sports Med. 2019, 49, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Fages, A.; Hanghoj, K.; Khan, N.; Gaunitz, C.; Seguin-Orlando, A.; Leonardi, M.; McCrory Constantz, C.; Gamba, C.; Al-Rasheid, K.A.S.; Albizuri, S.; et al. Tracking Five Millennia of Horse Management with Extensive Ancient Genome Time Series. Cell 2019, 177, 1419–1435. [Google Scholar] [CrossRef] [PubMed]
- Pujar, S.; Meyers-Wallen, V.N. Sequence variations in equine candidate genes for XX and XY inherited disorders of sexual development. Reprod. Domest. Anim. 2012, 47, 827–834. [Google Scholar] [CrossRef]
- Achermann, J.C.; Ozisik, G.; Ito, M.; Orun, U.A.; Harmanci, K.; Gurakan, B.; Jameson, J.L. Gonadal Determination and Adrenal Development Are Regulated by the Orphan Nuclear Receptor Steroidogenic Factor-1, in a Dose-Dependent Manner. J. Clin. Endocrinol. Metab. 2002, 87, 1829–1833. [Google Scholar] [CrossRef]
- Splinter, E.; Heath, H.; Kooren, J.; Palstra, R.; Klous, P.; Grosveld, F.; Galiart, N.; de Laat, W.L. CTCF mediates long-range chromatin looping and local histone modification in the β-globin locus. Genes Dev. 2006, 20, 2349–2354. [Google Scholar] [CrossRef]
- Hou, C.; Dale, R.; Dean, A. Cell type specificity of chromatin organization mediated by CTCF and cohesion. Proc. Natl. Acad. Sci. USA 2010, 107, 3651–3656. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Adipose | Brain | Heart | Lamina | Liver | Lung | Muscle | Ovary |
|---|---|---|---|---|---|---|---|---|
| Starting Tissue (mg) | 220 | 90 | 105 | 100 | 40 | 40 | 100 | 85 |
| Homogenization Time (min) | 8 | 5 | 5 | 9 | n/a | 5 | 5 | 5 |
| Duration Fixed (min) | 10 | 9 | 9 | 9 | 9 | 9 | 9 | 9 |
| Fixation Temp. (°C) | 37 | 23 | 23 | 23 | 23 | 23 | 23 | 23 |
| Shearing Volume (uL) | 400 | 1500 | 1800 | 1800 | 1500 | 1500 | 1800 | 1800 |
| Shearing Cycles | 5 × 8 1 | 10 | 13 | 10 | 12 | 10 | 12 | 10 |
| Chromatin per IP (ng) | 700 | 300 | 500 | 700 | 450 | 800 | 260 | 1200 |
| Software | Parameter | H3K4me1 | H3K4me3 | H3K27ac | H3K27me3 |
|---|---|---|---|---|---|
| MACS2 | Filtering | strict | strict | strict | strict |
| Size | narrow 1 | narrow | narrow | broad | |
| Size Flag | none | none | none | --broad | |
| Model | --fix-bimodal | --fix-bimodal | --fix-bimodal | --fix-bimodal | |
| Genome Size | 2,409,143,234 | 2,409,143,234 | 2,409,143,234 | 2,409,143,234 | |
| both | Fragment Size | 200 | 200 | 200 | 200 |
| FDR | 0.05 | 0.01 | 0.01 | 0.1 | |
| SICERpy 2 | Gap Size | n/a | n/a | n/a | 4 |
| Window Size | n/a | n/a | n/a | 200 | |
| Genome Fraction | n/a | n/a | n/a | 0.63 |
| Mark | Tissue | Software | Combined Peak Number | Percent Genome Covered | AH1 Peak Number | AH2 Peak Number |
|---|---|---|---|---|---|---|
| H3K4me1 | Adipose | MACS2 | 107,318 | 5.1 | 130,242 | 157,497 |
| Brain | MACS2 | 95,918 | 3.1 | 143,328 | 65,327 | |
| Heart | MACS2 | 121,663 | 4.9 | 137,385 | 155,881 | |
| Lamina | MACS2 | 114,708 | 4.2 | 137,575 | 124,150 | |
| Liver | MACS2 | 116,760 | 3.6 | 97,863 | 135,122 | |
| Lung | MACS2 | 92,972 | 2.9 | 90,687 | 109,001 | |
| Muscle | MACS2 | 95,816 | 3.7 | 137,322 | 100,999 | |
| Ovary | MACS2 | 102,986 | 4.3 | 166,303 | 133,209 | |
| H3K4me3 | Adipose | MACS2 | 26,905 | 1.7 | 26,286 | 29,121 |
| Brain | MACS2 | 27,101 | 1.6 | 25,473 | 28,028 | |
| Heart | MACS2 | 26,475 | 1.4 | 24,101 | 27,985 | |
| Lamina | MACS2 | 29,380 | 1.6 | 29,023 | 19,742 | |
| Liver | MACS2 | 28,498 | 1.5 | 28,204 | 28,222 | |
| Lung | MACS2 | 28,546 | 1.6 | 30,048 | 27,779 | |
| Muscle | MACS2 | 28,110 | 1.6 | 30,428 | 25,123 | |
| Ovary | MACS2 | 28,378 | 1.7 | 30,522 | 29,192 | |
| H3K27ac | Adipose | MACS2 | 79,620 | 3.3 | 75,823 | 99,249 |
| Brain | MACS2 | 78,823 | 3.2 | 89,445 | 73,795 | |
| Heart | MACS2 | 68,728 | 2.9 | 71,462 | 7192 | |
| Lamina | MACS2 | 82,394 | 2.9 | 91,345 | 78,953 | |
| Liver | MACS2 | 87,589 | 3.1 | 84,814 | 96,238 | |
| Lung | MACS2 | 69,054 | 2.9 | 69,621 | 75,299 | |
| Muscle | MACS2 | 76,495 | 2.9 | 78,047 | 86,524 | |
| Ovary | MACS2 | 64,318 | 3.3 | 94,817 | 82,318 | |
| H3K27me3 | Adipose | MACS2 | 25,183 | 0.6 | 8948 | 29,906 |
| Brain | MACS2 | 24,243 | 0.6 | 16,055 | 23,411 | |
| Heart | MACS2 | 68,113 | 1.8 | 31,455 | 88,818 | |
| Lamina | MACS2 | 37,366 | 0.8 | 31,839 | 28,508 | |
| Liver | MACS2 | 63,874 | 1.3 | 93,423 | 23,888 | |
| Lung | MACS2 | 30,191 | 0.7 | 32,385 | 18,124 | |
| Muscle | MACS2 | 42,610 | 0.9 | 39,076 | 29,579 | |
| Ovary | MACS2 | 40,825 | 1.2 | 43,036 | 33,220 | |
| H3K27me3 | Adipose | SICER | 8167 | 4.9 | 13,540 | 14,571 |
| Brain | SICER | 7860 | 3.4 | 11,386 | 13,603 | |
| Heart | SICER | 9032 | 3.3 | 12,192 | 18,903 | |
| Lamina | SICER | 7072 | 3.8 | 11,933 | 11,694 | |
| Liver | SICER | 11,430 | 3.7 | 22,270 | 16,099 | |
| Lung | SICER | 7863 | 2.6 | 12,668 | 11,715 | |
| Muscle | SICER | 8437 | 4.6 | 17,073 | 10,987 | |
| Ovary | SICER | 7083 | 3.0 | 14,731 | 11,124 |
| Mark | Tissue | Rep | NRF | PBC1 | PBC2 | NSC | RSC | JSD |
|---|---|---|---|---|---|---|---|---|
| Thresholds | (>0.5) | (>0.5) | (>1) | (>1.05) | (>0.8) | (>0.05) | ||
| H3K4me1 | Adipose | AH2 | 0.677 | 0.673 | 3.017 | 1.068 | 1.249 | 0.281 |
| Adipose | AH1 | 0.621 | 0.617 | 2.595 | 1.067 | 1.147 | 0.239 | |
| Brain | AH2 | 0.435 | 0.443 | 1.908 | 1.055 | 1.243 | 0.186 | |
| Brain | AH1 | 0.754 | 0.756 | 4.128 | 1.074 | 1.275 | 0.228 | |
| Heart | AH2 | 0.708 | 0.708 | 3.444 | 1.086 | 1.790 | 0.321 | |
| Heart | AH1 | 0.497 | 0.496 | 2.023 | 1.071 | 1.657 | 0.259 | |
| Lamina | AH2 | 0.606 | 0.606 | 2.551 | 1.093 | 1.628 | 0.281 | |
| Lamina | AH1 | 0.561 | 0.562 | 2.311 | 1.088 | 1.809 | 0.283 | |
| Liver | AH2 | 0.760 | 0.762 | 4.226 | 1.097 | 1.240 | 0.226 | |
| Liver | AH1 | 0.838 | 0.842 | 6.462 | 1.117 | 1.289 | 0.252 | |
| Lung | AH2 | 0.736 | 0.736 | 3.796 | 1.079 | 1.123 | 0.199 | |
| Lung | AH1 | 0.667 | 0.665 | 2.980 | 1.069 | 1.063 | 0.178 | |
| Muscle | AH2 | 0.706 | 0.706 | 3.418 | 1.077 | 1.030 | 0.210 | |
| Muscle | AH1 | 0.576 | 0.573 | 2.338 | 1.084 | 1.200 | 0.259 | |
| Ovary | AH2 | 0.712 | 0.712 | 3.488 | 1.077 | 1.265 | 0.245 | |
| Ovary | AH1 | 0.692 | 0.691 | 3.240 | 1.085 | 2.117 | 0.313 | |
| H3K4me3 | Adipose | AH2 | 0.595 | 0.604 | 2.581 | 1.322 | 1.391 | 0.382 |
| Adipose | AH1 | 0.559 | 0.571 | 2.389 | 1.313 | 1.501 | 0.354 | |
| Brain | AH2 | 0.497 | 0.515 | 2.167 | 1.366 | 1.198 | 0.516 | |
| Brain | AH1 | 0.333 | 0.362 | 1.813 | 1.360 | 1.249 | 0.528 | |
| Heart | AH2 | 0.410 | 0.435 | 1.905 | 1.467 | 1.364 | 0.540 | |
| Heart | AH1 | 0.337 | 0.374 | 1.857 | 1.399 | 1.639 | 0.548 | |
| Lamina | AH2 | 0.529 | 0.551 | 2.345 | 1.384 | 1.188 | 0.467 | |
| Lamina | AH1 | 0.571 | 0.594 | 2.606 | 1.380 | 1.289 | 0.465 | |
| Liver | AH2 | 0.452 | 0.471 | 1.996 | 1.407 | 1.196 | 0.517 | |
| Liver | AH1 | 0.421 | 0.444 | 1.926 | 1.385 | 1.282 | 0.537 | |
| Lung | AH2 | 0.610 | 0.628 | 2.813 | 1.354 | 1.154 | 0.387 | |
| Lung | AH1 | 0.580 | 0.600 | 2.634 | 1.344 | 1.117 | 0.452 | |
| Muscle | AH2 | 0.240 | 0.277 | 1.818 | 1.340 | 1.354 | 0.441 | |
| Muscle | AH1 | 0.559 | 0.567 | 2.350 | 1.350 | 1.164 | 0.448 | |
| Ovary | AH2 | 0.633 | 0.646 | 2.926 | 1.315 | 1.191 | 0.428 | |
| Ovary | AH1 | 0.603 | 0.622 | 2.779 | 1.335 | 1.220 | 0.439 | |
| H3K27ac | Adipose | AH2 | 0.678 | 0.677 | 3.087 | 1.223 | 1.605 | 0.313 |
| Adipose | AH1 | 0.537 | 0.532 | 2.129 | 1.250 | 1.800 | 0.333 | |
| Brain | AH2 | 0.495 | 0.493 | 2.001 | 1.202 | 1.320 | 0.310 | |
| Brain | AH1 | 0.655 | 0.657 | 2.939 | 1.200 | 1.341 | 0.326 | |
| Heart | AH2 | 0.493 | 0.489 | 1.970 | 1.316 | 2.193 | 0.402 | |
| Heart | AH1 | 0.573 | 0.573 | 2.361 | 1.331 | 1.856 | 0.376 | |
| Lamina | AH2 | 0.597 | 0.596 | 2.486 | 1.296 | 1.655 | 0.351 | |
| Lamina | AH1 | 0.657 | 0.662 | 3.006 | 1.304 | 1.711 | 0.345 | |
| Liver | AH2 | 0.719 | 0.722 | 3.651 | 1.258 | 1.225 | 0.347 | |
| Liver | AH1 | 0.721 | 0.724 | 3.674 | 1.242 | 1.237 | 0.298 | |
| Lung | AH2 | 0.500 | 0.499 | 2.008 | 1.241 | 1.290 | 0.327 | |
| Lung | AH1 | 0.654 | 0.658 | 2.956 | 1.208 | 1.281 | 0.299 | |
| Muscle | AH2 | 0.605 | 0.604 | 2.524 | 1.291 | 1.306 | 0.335 | |
| Muscle | AH1 | 0.510 | 0.511 | 2.072 | 1.285 | 1.335 | 0.381 | |
| Ovary | AH2 | 0.733 | 0.736 | 3.816 | 1.254 | 1.309 | 0.374 | |
| Ovary | AH1 | 0.678 | 0.678 | 3.112 | 1.224 | 1.461 | 0.391 | |
| H3K27me3 | Adipose | AH2 | 0.646 | 0.641 | 2.751 | 1.057 | 0.659 | 0.101 |
| Adipose | AH1 | 0.650 | 0.647 | 2.809 | 1.053 | 0.592 | 0.067 | |
| Brain | AH2 | 0.511 | 0.510 | 2.077 | 1.060 | 0.400 | 0.101 | |
| Brain | AH1 | 0.616 | 0.614 | 2.587 | 1.067 | 0.477 | 0.088 | |
| Heart | AH2 | 0.407 | 0.414 | 1.834 | 1.070 | 0.595 | 0.106 | |
| Heart | AH1 | 0.287 | 0.315 | 1.778 | 1.090 | 0.649 | 0.102 | |
| Lamina | AH2 | 0.459 | 0.460 | 1.919 | 1.069 | 0.656 | 0.071 | |
| Lamina | AH1 | 0.429 | 0.436 | 1.885 | 1.076 | 0.732 | 0.065 | |
| Liver | AH2 | 0.545 | 0.537 | 2.140 | 1.076 | 0.648 | 0.093 | |
| Liver | AH1 | 0.454 | 0.451 | 1.871 | 1.084 | 0.661 | 0.123 | |
| Lung | AH2 | 0.619 | 0.615 | 2.575 | 1.072 | 0.617 | 0.072 | |
| Lung | AH1 | 0.550 | 0.545 | 2.199 | 1.084 | 0.671 | 0.088 | |
| Muscle | AH2 | 0.534 | 0.526 | 2.098 | 1.070 | 0.597 | 0.070 | |
| Muscle | AH1 | 0.476 | 0.472 | 1.914 | 1.079 | 0.689 | 0.103 | |
| Ovary | AH2 | 0.524 | 0.520 | 2.103 | 1.071 | 0.587 | 0.066 | |
| Ovary | AH1 | 0.495 | 0.489 | 1.970 | 1.077 | 0.688 | 0.101 | |
| Rank | Motif ID | Consensus | Adjusted p-Value | UniProt Entry |
|---|---|---|---|---|
| Adipose | ||||
| 1 | SP3 | VCCACGCCCMC | 1.49 × 10−10 | Q02447 |
| 2 | TFDP1 | VSGCGGGAAVN | 1.74 × 10−10 | Q14186 |
| 3 | TFAP2A | HGCCYSAGGCD | 3.27 × 10−10 | P05549 |
| 4 | TFAP2C | YGCCYBVRGGCA | 4.56 × 10−10 | Q92754 |
| 6 | KLF16 | GMCACGCCCCC | 5.81 × 10−9 | Q9BXK1 |
| Brain | ||||
| 1 | TFAP2A(var.2) | YGCCCBVRGGCR | 1.82 × 10−16 | P05549 |
| 2 | TFAP2B | YGCCCBVRGGCA | 1.29 × 10−13 | Q92481 |
| 3 | SP3 | VCCACGCCCMC | 2.69 × 10−13 | Q02447 |
| 4 | TFAP2C | YGCCYBVRGGCA | 4.99 × 10−13 | Q92754 |
| 5 | KLF16 | GMCACGCCCCC | 1.06 × 10−12 | Q9BXK1 |
| Heart | ||||
| 1 | MZF1 | BGGGGA | 2.23 × 10−5 | P28698 |
| 2 | Ascl2 | ARCAGCTGCY | 7.06 × 10−4 | Q99929 |
| 3 | ASCL1 | VSAGCAGCTGSNN | 9.41 × 10−4 | P50553 |
| 4 | SP3 | VCCACGCCCMC | 1.42 × 10−3 | Q02447 |
| 5 | NEUROD1 | NRACAGATGGYNN | 1.60 × 10−3 | Q13562 |
| Lamina | ||||
| 1 | SP2 | GYCCCGCCYCYBSSS | 8.51 × 10−15 | Q02086 |
| 2 | SP1 | GCCCCKCCCCC | 5.98 × 10−14 | P08047 |
| 3 | SP3 | VCCACGCCCMC | 2.84 × 10−13 | Q02447 |
| 4 | KLF16 | GMCACGCCCCC | 3.22 × 10−13 | Q9BXK1 |
| 7 | Zfx | SSSGCCBVGGCCTS | 1.06 × 10−11 | P17010 |
| Liver | ||||
| 1 | SP1 | GCCCCKCCCCC | 7.81 × 10−13 | P08047 |
| 2 | TFAP2B | YGCCCBVRGGCA | 4.23 × 10−12 | Q92481 |
| 3 | TFAP2C | YGCCYBVRGGCA | 1.87 × 10−11 | Q92754 |
| 4 | TFAP2A | HGCCYSAGGCD | 4.97 × 10−11 | P05549 |
| 5 | ZNF740 | MCCCCCCCAC | 8.99 × 10−11 | Q8NDX6 |
| Lung | ||||
| 1 | THAP1 | YTGCCCDBA | 5.09 × 10−9 | Q9NVV9 |
| 3 | ESR2 | AGGTCASVNTGMCCY | 1.08 × 10−8 | Q92731 |
| 4 | Zfx | SSSGCCBVGGCCTS | 1.44 × 10−8 | P17010 |
| 5 | ZBTB7A | NVCCGGAAGTGSV | 1.46 × 10−8 | O95365 |
| 6 | TFAP2A(var.2) | YGCCCBVRGGCR | 6.51 × 10−8 | P05549 |
| Muscle | ||||
| 1 | SP1 | GCCCCKCCCCC | 1.45 × 10−9 | P08047 |
| 2 | SP2 | GYCCCGCCYCYBSSS | 4.21 × 10−9 | Q02086 |
| 3 | SP8 | RCCACGCCCMCY | 1.15 × 10−8 | Q8IXZ3 |
| 4 | CTCFL | CRSCAGGGGGCRSB | 3.44 × 10−8 | Q8NI51 |
| 5 | KLF16 | GMCACGCCCCC | 4.36 × 10−7 | Q9BXK1 |
| Ovary | ||||
| 1 | FOXO3 1 | DAAAYA | 7.23 × 10−7 | O43524 |
| 3 | KLF16 | GMCACGCCCCC | 1.93 × 10−4 | Q9BXK1 |
| 4 | FOXC1 1 | WAWGTAAAYAW | 2.39 × 10−4 | Q12948 |
| 6 | CTCFL | CRSCAGGGGGCRSB | 4.38 × 10−4 | Q8NI51 |
| 7 | Arid5a | SYAATATTGVDANH | 4.99 × 10−4 | Q03989 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kingsley, N.B.; Kern, C.; Creppe, C.; Hales, E.N.; Zhou, H.; Kalbfleisch, T.S.; MacLeod, J.N.; Petersen, J.L.; Finno, C.J.; Bellone, R.R. Functionally Annotating Regulatory Elements in the Equine Genome Using Histone Mark ChIP-Seq. Genes 2020, 11, 3. https://doi.org/10.3390/genes11010003
Kingsley NB, Kern C, Creppe C, Hales EN, Zhou H, Kalbfleisch TS, MacLeod JN, Petersen JL, Finno CJ, Bellone RR. Functionally Annotating Regulatory Elements in the Equine Genome Using Histone Mark ChIP-Seq. Genes. 2020; 11(1):3. https://doi.org/10.3390/genes11010003
Chicago/Turabian StyleKingsley, N. B., Colin Kern, Catherine Creppe, Erin N. Hales, Huaijun Zhou, T. S. Kalbfleisch, James N. MacLeod, Jessica L. Petersen, Carrie J. Finno, and Rebecca R. Bellone. 2020. "Functionally Annotating Regulatory Elements in the Equine Genome Using Histone Mark ChIP-Seq" Genes 11, no. 1: 3. https://doi.org/10.3390/genes11010003
APA StyleKingsley, N. B., Kern, C., Creppe, C., Hales, E. N., Zhou, H., Kalbfleisch, T. S., MacLeod, J. N., Petersen, J. L., Finno, C. J., & Bellone, R. R. (2020). Functionally Annotating Regulatory Elements in the Equine Genome Using Histone Mark ChIP-Seq. Genes, 11(1), 3. https://doi.org/10.3390/genes11010003