Genomic Divergence in Swedish Warmblood Horses Selected for Equestrian Disciplines

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Definition of SWB Subpopulations
2.2. DNA Isolation, Genotyping and Quality Control
2.3. Genomic Structure of the SWB Population
2.4. Divergent Selection Between Subpopulations at Genotype Level
2.5. Divergent Selection Between Subpopulations at Haplotype Level
2.6. Candidate Genes
2.7. Ethical Approval
3. Results
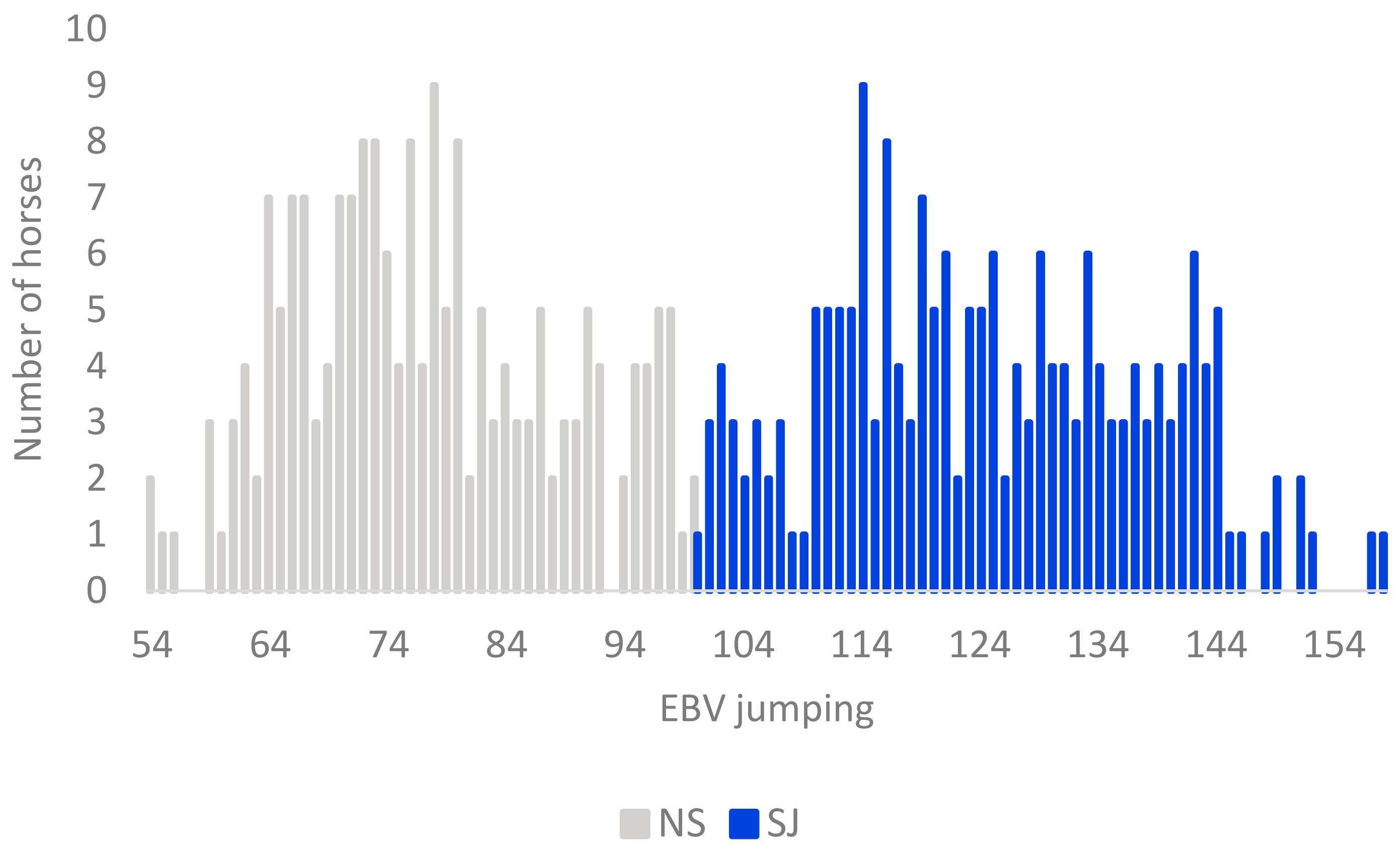
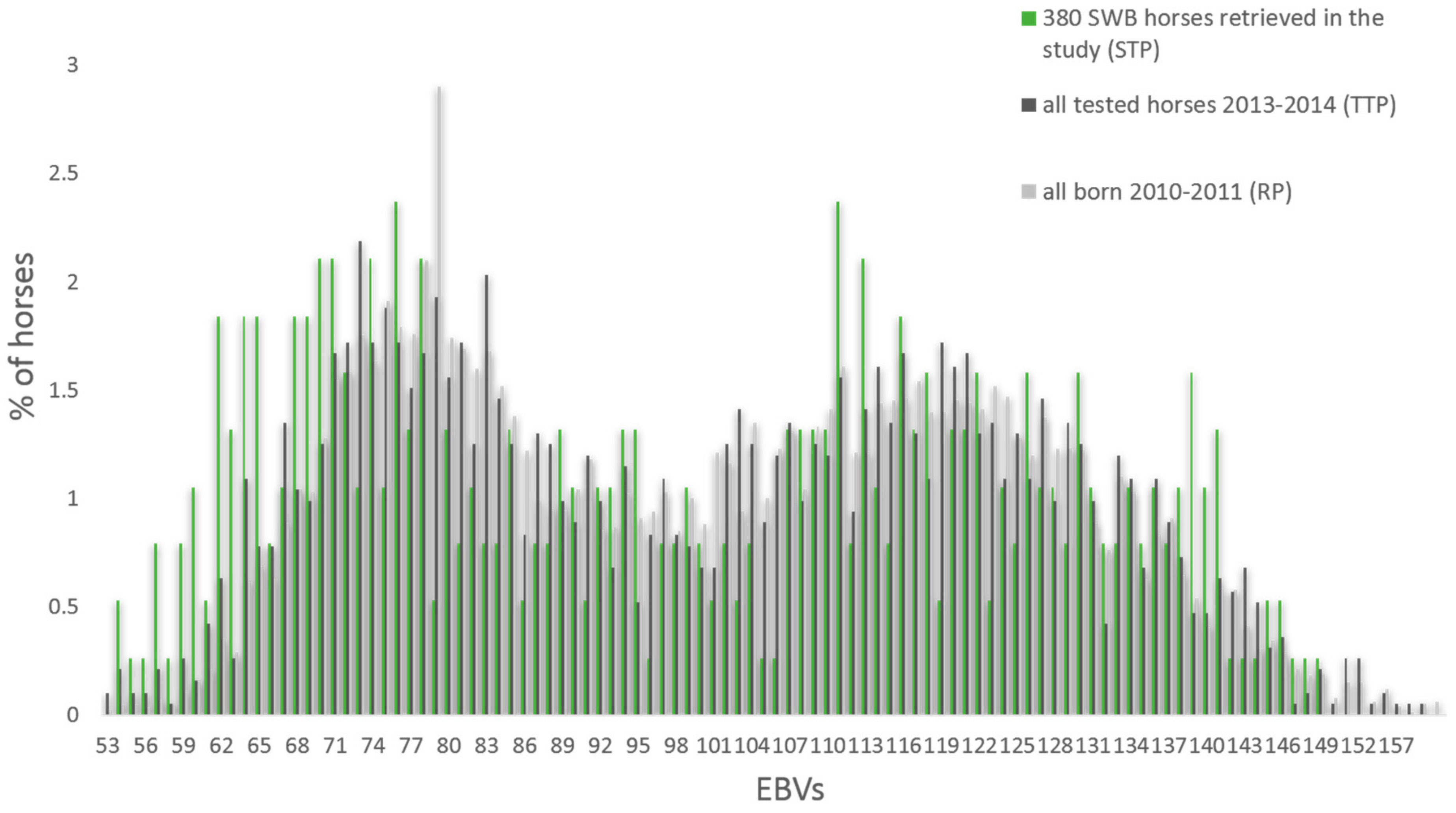
3.1. Genotypic and Phenotypic Population Structure
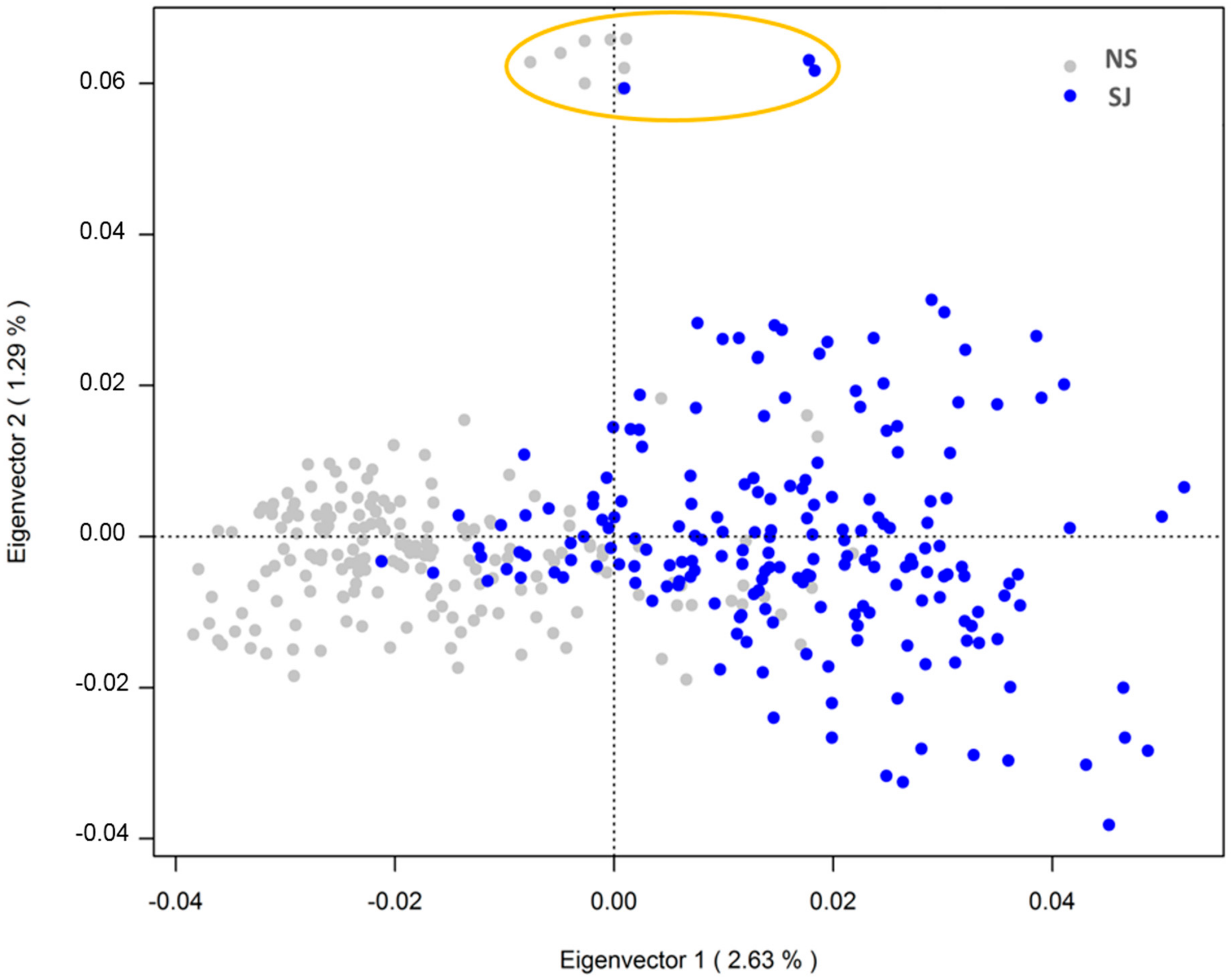
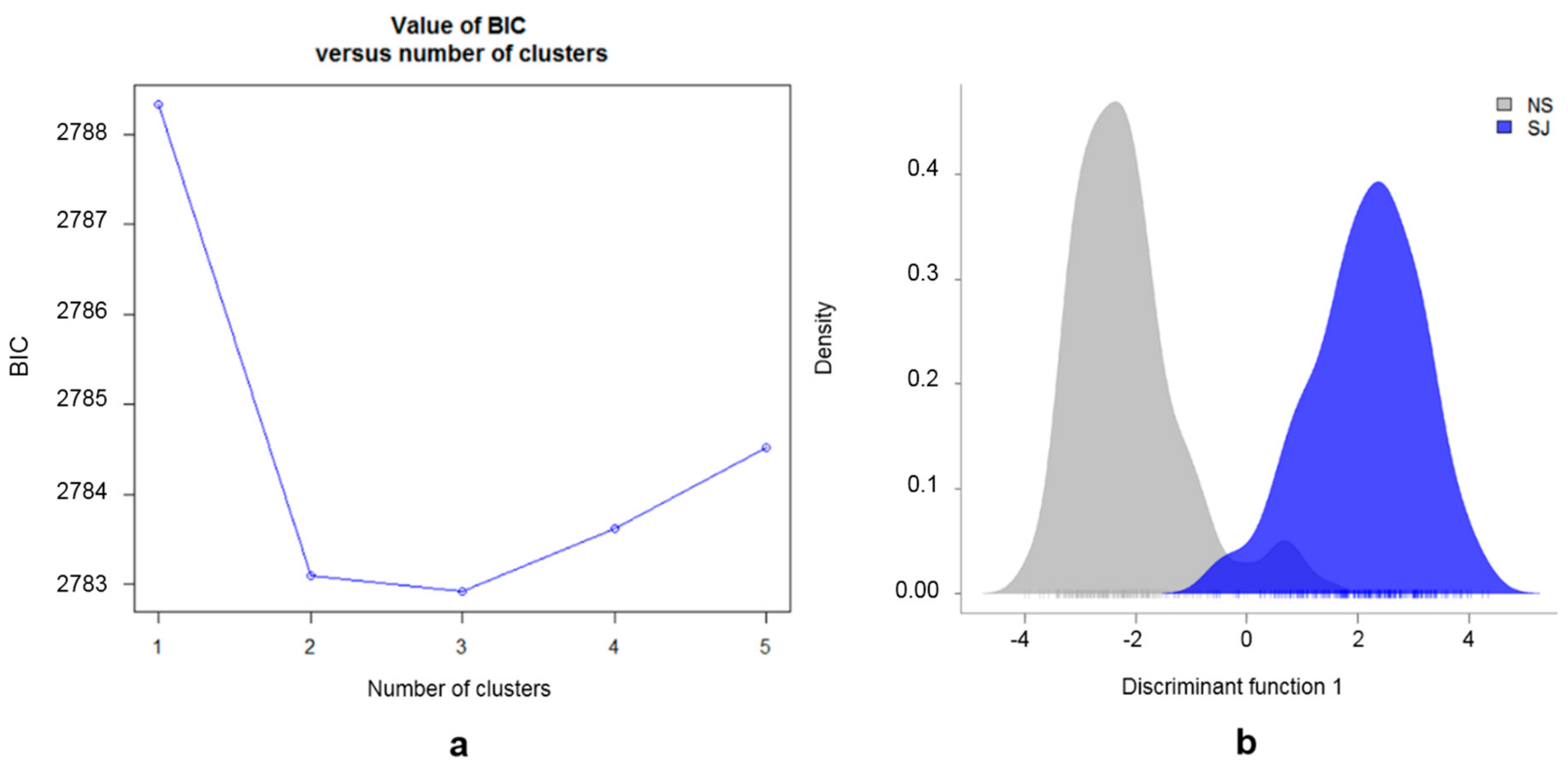
3.2. Population Substructure
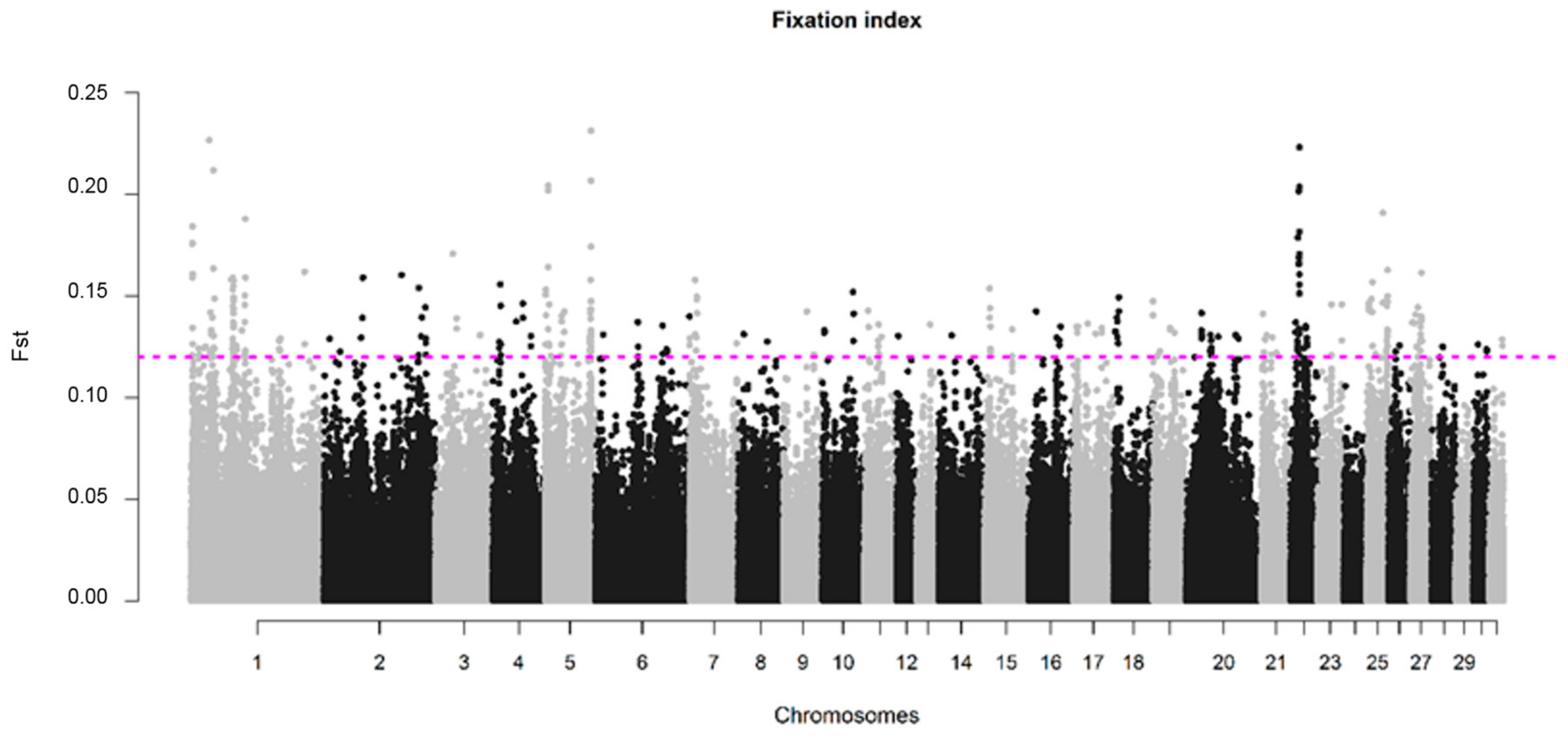
3.3. Genomic Divergence in SWB Subpopulations
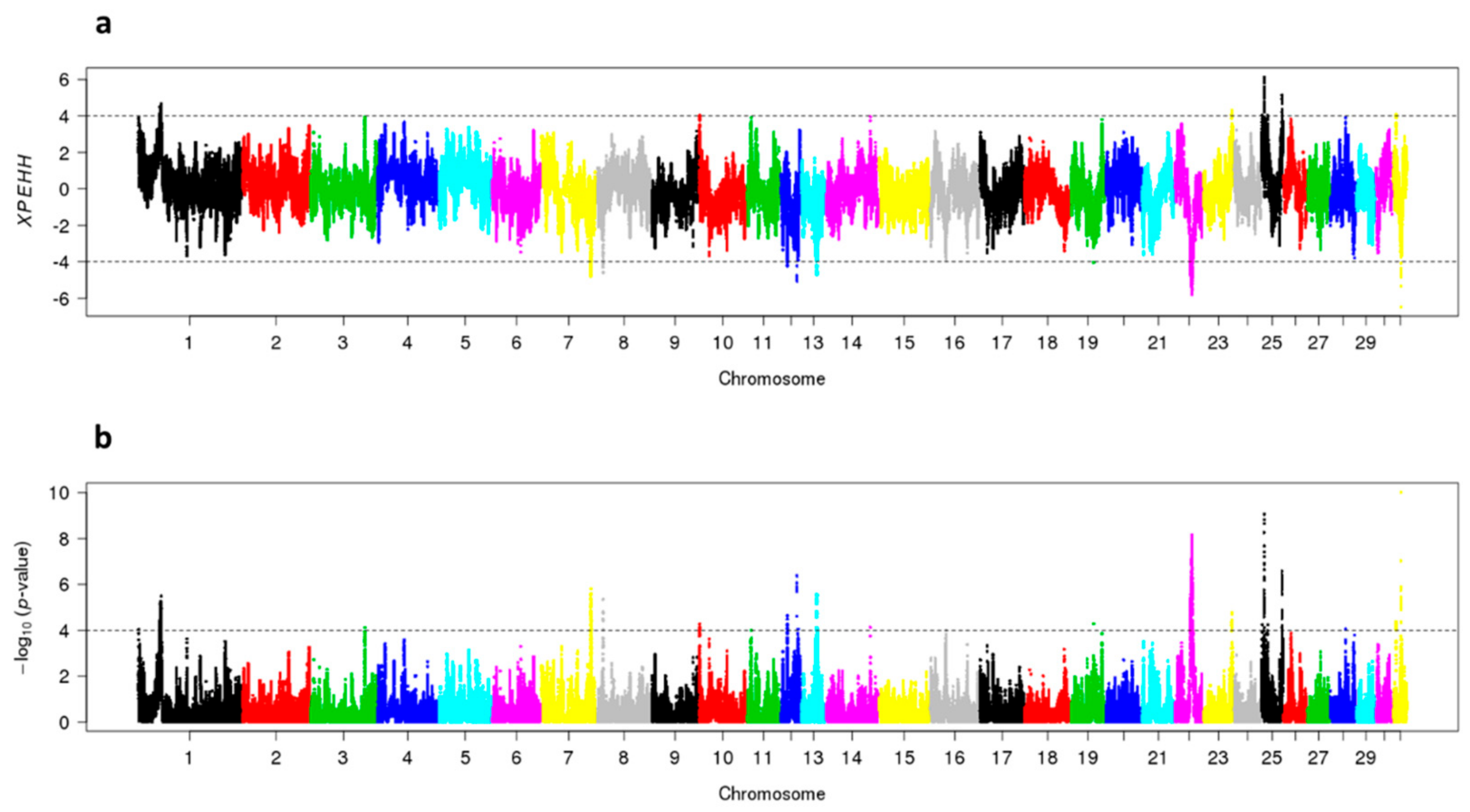
3.4. Signatures of Selection in SWB Subpopulations
4. Discussion
4.1. Horses Selected for This Study
4.2. SWB Breeding Program and Population Substructure
4.3. Genomic Divergence Between SJ and NS SWB Horses
4.4. Selection for High Mobility and Relaxed Locomotion in NS SWB Horses
4.5. Selection for Mentality and Postsynaptic Signaling in SJ SWB Horses
4.6. Selection for Growth and Muscle Function in SJ SWB Horses
4.7. Selection for CNS Reward System and Motoneuronal Control of Coordination in SJ SWB Horses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Rovere, G.; Madsen, P.; Norberg, E.; van Arendonk, J.A.M.; Ducro, B.J. Genetic connections between dressage and show-jumping horses in Dutch Warmblood horses. Acta Agric. Scand. A Anim. Sci. 2014, 64, 57–66. [Google Scholar] [CrossRef]
- SWB. SWBs Avelsplan 2015. Available online: https://swb.org/wp-content/uploads/2016/11/Avelsplan-fi%CC%82r-SWB.pdf (accessed on 28 October 2019).
- Viklund, Å.; Granberg, L.; Eriksson, S. Genetic analysis of data from Swedish stallion performance test. In Proceedings of the 69th Annual Meeting of the EAAP, Dubrovnik, Croatia, 27–31 August 2018; p. 660. [Google Scholar]
- Viklund, Å.; Näsholm, A.; Strandberg, E.; Philipsson, J. Genetic trends for performance of Swedish Warmblood horses. Livest. Sci. 2011, 141, 113–122. [Google Scholar] [CrossRef]
- Maiorano, A.M.; Lourenco, D.L.; Tsuruta, S.; Toro Ospina, A.M.; Stafuzza, N.B.; Masuda, Y.; Filho, A.E.V.; Dos Santos Goncalves Cyrillo, J.N.; Curi, R.A.; De Vasconcelos Silva, J.A. Assessing genetic architecture and signatures of selection of dual purpose Gir cattle populations using genomic information. PLoS ONE 2018, 13, e0200694. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, C.M.; Pereira, G.L.; Maiorano, A.M.; Rogatto, G.M.; Assoni, A.D.; Augusto, V., II; Silva, J.; Chardulo, L.A.L.; Curi, R.A. Linkage disequilibrium and population structure characterization in the cutting and racing lines of Quarter Horses bred in Brazil. Livest. Sci. 2019, 219, 45–51. [Google Scholar] [CrossRef]
- Bomba, L.; Nicolazzi, E.L.; Milanesi, M.; Negrini, R.; Mancini, G.; Biscarini, F.; Stella, A.; Valentini, A.; Ajmone-Marsan, P. Relative extended haplotype homozygosity signals across breeds reveal dairy and beef specific signatures of selection. Genet. Sel. Evol. 2015, 47, 25. [Google Scholar] [CrossRef]
- Petersen, J.L.; Mickelson, J.R.; Cleary, K.D.; McCue, M.E. The american quarter horse: Population structure and relationship to the thoroughbred. J. Hered. 2014, 105, 148–162. [Google Scholar] [CrossRef][Green Version]
- Avila, F.; Mickelson, J.R.; Schaefer, R.J.; McCue, M.E. Genome-Wide Signatures of Selection Reveal Genes Associated With Performance in American Quarter Horse Subpopulations. Front. Genet. 2018, 9, 249. [Google Scholar] [CrossRef]
- Lopes, M.S.; Mendonça, D.; Rojer, H.; Cabral, V.; Bettencourt, S.X.; da Câmara Machado, A. Morphological and genetic characterization of an emerging Azorean horse breed: The Terceira Pony. Front. Genet. 2015, 6, 62. [Google Scholar] [CrossRef]
- Ovchinnikov, I.V.; Dahms, T.; Herauf, B.; McCann, B.; Juras, R.; Castaneda, C.; Cothran, E.G. Genetic diversity and origin of the feral horses in Theodore Roosevelt National Park. PLoS ONE 2018, 13, e0200795. [Google Scholar] [CrossRef]
- Velie, B.D.; Shrestha, M.; Francois, L.; Schurink, A.; Tesfayonas, Y.G.; Stinckens, A.; Blott, S.; Ducro, B.J.; Mikko, S.; Thomas, R.; et al. Using an Inbred Horse Breed in a High Density Genome-Wide Scan for Genetic Risk Factors of Insect Bite Hypersensitivity (IBH). PLoS ONE 2016, 11, e0152966. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Dobretsberger, M.; Horna, M.; Brem, G. High-resolution population structure and runs of homozygosity reveal the genetic architecture of complex traits in the Lipizzan horse. BMC Genom. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Metzger, J.; Karwath, M.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Gut, I.G.; Distl, O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genom. 2015, 16, 764. [Google Scholar] [CrossRef] [PubMed]
- Grilz-seger, G.; Neuditschko, M.; Mesaric, M.; Cotman, M.; Brem, G.; Druml, T. Changes in breeding objectives of the Haflinger horse breed from a genome—wide perspective. Züchtungskunde 2019, 91, 296–311. [Google Scholar]
- Ablondi, M.; Viklund, Å.; Lindgren, G.; Eriksson, S.; Mikko, S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genom. 2019, 20, 717. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Frischknecht, M.; Flury, C.; Leeb, T.; Rieder, S.; Neuditschko, M. Selection signatures in Shetland ponies. Anim. Genet. 2016, 47, 370–372. [Google Scholar] [CrossRef]
- Moon, S.; Lee, J.W.; Shin, D.; Shin, K.Y.; Kim, J.; Choi, I.Y.; Kim, J.; Kim, H. A Genome-wide scan for selective sweeps in racing horses. Asian-Australas. J. Anim. Sci. 2015, 28, 1525–1531. [Google Scholar] [CrossRef]
- Metzger, J.; Philipp, U.; Lopes, M.S.; da Camara Machado, A.; Felicetti, M.; Silvestrelli, M.; Distl, O. Analysis of copy number variants by three detection algorithms and their association with body size in horses. BMC Genom. 2013, 14, 487. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Hou, C.; Xing, Y.; Cao, J.; Wu, K.; Liu, C.; Zhang, D.; Zhang, L.; Zhang, Y.; et al. Genome-wide detection of copy number variations among diverse horse breeds by array CGH. PLoS ONE 2014, 9, e86860. [Google Scholar] [CrossRef]
- Schurink, A.; da Silva, V.H.; Velie, B.D.; Dibbits, B.W.; Crooijmans, R.P.M.A.; François, L.; Janssens, S.; Stinckens, A.; Blott, S.; Buys, N.; et al. Copy number variations in Friesian horses and genetic risk factors for insect bite hypersensitivity. BMC Genet. 2018, 19, 49. [Google Scholar] [CrossRef]
- SAS® 9.4 Fourth Edition; SAS Institute Inc.: Cary, NC, USA, 2015.
- Schaefer, R.J.; Schubert, M.; Bailey, E.; Bannasch, D.L.; Barrey, E.; Bar-Gal, G.K.; Brem, G.; Brooks, S.A.; Distl, O.; Fries, R.; et al. Developing a 670 k genotyping array to tag~2 M SNPs across 24 horse breeds. BMC Genom. 2017, 18, 565. [Google Scholar] [CrossRef] [PubMed]
- Kalbfleisch, T.S.; Rice, E.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O’Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Petersen, J.L.; et al. EquCab3, an Updated Reference Genome for the Domestic Horse. bioRxiv 2018. [Google Scholar] [CrossRef]
- Beeson, S.K.; Schaefer, R.J.; Mason, V.C.; McCue, M.E. Robust remapping of equine SNP array coordinates to EquCab3. Anim. Genet. 2019, 50, 114–115. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome Sequence, Comparative Analysis, and Population Genetics of the Domestic Horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef]
- Mardia, K.V. Some properties of clasical multi-dimesional scaling. Commun. Stat. Theory Methods 1978, 7, 1233–1241. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Akey, J.M. Interrogating a High-Density SNP Map for Signatures of Natural Selection. Genome Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations, Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4. [Google Scholar]
- Makina, S.O.; Muchadeyi, F.C.; Van Marle-Köster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 2015, 47, 1–14. [Google Scholar] [CrossRef]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Coulonges, C.; Zagury, J.-F. Shape-IT: New rapid and accurate algorithm for haplotype inference. BMC Bioinform. 2008, 9, 540. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Vitalis, R. Rehh An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 2012, 28, 1176–1177. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Naves, M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 2011, 20, 3128–3143. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate—A Practical And Powerful Approach To Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; García Girón, C.; Hourlier, T.; et al. The Ensembl gene annotation system. Database 2016, 2016, 93. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef]
- Viklund, Å.; Braam, Å.; Näsholm, A.; Strandberg, E.; Philipsson, J. Genetic variation in competition traits at different ages and time periods and correlations with traits at field tests of 4-year-old Swedish Warmblood horses. Animal 2010, 4, 682. [Google Scholar] [CrossRef]
- Hellsten, E.T.; Näsholm, A.; Jorjani, H.; Strandberg, E.; Philipsson, J. Influence of foreign stallions on the Swedish Warmblood breed and its genetic evaluation. Livest. Sci. 2009, 121, 207–214. [Google Scholar] [CrossRef]
- Storz, J.F. Invited review: Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol. Ecol. 2005, 14, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Pan, D.; Ren, H.; Fu, J.; Li, J.; Su, G.; Wang, A.; Jiang, L.; Zhang, Q.; Liu, J.F. Identification of selective sweeps reveals divergent selection between Chinese Holstein and Simmental cattle populations. Genet. Sel. Evol. 2016, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Doan, R.; Cohen, N.; Harrington, J.; Veazy, K.; Juras, R.; Cothran, G.; McCue, M.E.; Skow, L.; Dindot, S.V. Identification of copy number variants in horses. Genome Res. 2012, 22, 899–907. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, W.; Han, Q.; Liu, Z.; Zheou, W.; Cao, Q.; Zhou, W. Exome sequencing reveals a de novo PRKG1 mutation in a sporadic patient with aortic dissection. BMC Med. Genet. 2018, 19, 1–5. [Google Scholar] [CrossRef]
- Lee, W.; Park, K.-D.; Taye, M.; Lee, C.; Kim, H.; Lee, H.-K.; Shin, D. Analysis of cross-population differentiation between Thoroughbred and Jeju horses. Asian-Australas. J. Anim. Sci. 2018, 31, 1110–1118. [Google Scholar] [CrossRef]
- Nekrasova, T.; Jobes, M.L.; Ting, J.H.; Wagner, G.C.; Minden, A. Targeted disruption of the Pak5 and Pak6 genes in mice leads to deficits in learning and locomotion. Dev. Biol. 2008, 322, 95–108. [Google Scholar] [CrossRef]
- Carlisle, H.J.; Luong, T.N.; Medina-Marino, A.; Schenker, L.; Khorosheva, E.; Indersmitten, T.; Gunapala, K.M.; Steele, A.D.; O’Dell, T.J.; Patterson, P.H.; et al. Deletion of Densin-180 Results in Abnormal Behaviors Associated with Mental Illness and Reduces mGluR5 and DISC1 in the Postsynaptic Density Fraction. J. Neurosci. 2011, 31, 16194–16207. [Google Scholar] [CrossRef]
- Udawela, M.; Scarr, E.; Hannan, A.J.; Thomas, E.A.; Dean, B. Phospholipase C beta 1 expression in the dorsolateral prefrontal cortex from patients with schizophrenia at different stages of illness. Aust. N. Z. J. Psychiatry 2011, 45, 140–147. [Google Scholar] [CrossRef]
- Weng, Y.T.; Chien, T.; Kuan, I.I.; Chern, Y. The TRAX, DISC1, and GSK3 complex in mental disorders and therapeutic interventions 06 Biological Sciences 0604 Genetics 11 Medical and Health Sciences 1103 Clinical Sciences. J. Biomed. Sci. 2018, 25, 1–14. [Google Scholar]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long non-coding RNAs in the regulation of gene expression: Physiology and disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.; Rubin, C.-J.; Di Palma, F.; Albert, F.W.; Alfoldi, J.; Barrio, A.M.; Pielberg, G.; Rafati, N.; Sayyab, S.; Turner-Maier, J.; et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Scince 2014, 345, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Schroder, W.; Klostermann, A.; Stock, K.F.; Distl, O. A genome-wide association study for quantitative trait loci of show-jumping in Hanoverian warmblood horses. Anim. Genet. 2012, 43, 92–400. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Hung, C.; Fujisawa, Y.; Sakaguchi, D.; Angelastro, J.; Omanska-Klusek, A.; Schoenfeld, R.; Giulivi, C. Mitochondrial dysfunction in Pten Haplo-insufficient mice with social deficits and repetitive behavior: Interplay between Pten and p53. PLoS ONE 2012, 7, e42504. [Google Scholar] [CrossRef]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef]
- Rankinen, T.; Roth, S.M.; Bray, M.S.; Loos, R.; Pérusse, L.; Wolfarth, B.; Hagberg, J.M.; Bouchard, C. Advances in Exercise, Fitness, and Performance Genomics. Med. Sci. Sports Exerc. 2010, 42, 835–846. [Google Scholar] [CrossRef]
- Coricor, G.; Serra, R. TGF-β regulates phosphorylation and stabilization of Sox9 protein in chondrocytes through p38 and Smad dependent mechanisms. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Chavez, R.D.; Coricor, G.; Perez, J.; Seo, H.S.; Serra, R. SOX9 protein is stabilized by TGF-β and regulates PAPSS2 mRNA expression in chondrocytes. Osteoarthr. Cart. 2017, 25, 332–340. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Borza, C.M.; Su, Y.; Tran, T.-L.; Yu, L.; Steyns, N.; Temple, K.J.; Skwark, M.J.; Meiler, J.; Lindsley, C.W.; Hicks, B.R.; et al. Discoidin domain receptor 1 kinase activity is required for regulating collagen IV synthesis. Matrix Biol. 2017, 57–58, 258–271. [Google Scholar] [CrossRef]
- Chiusa, M.; Hu, W.; Liao, H.-J.; Su, Y.; Borza, C.M.; de Caestecker, M.P.; Skrypnyk, N.I.; Fogo, A.B.; Pedchenko, V.; Li, X.; et al. The Extracellular Matrix Receptor Discoidin Domain Receptor 1 Regulates Collagen Transcription by Translocating to the Nucleus. J. Am. Soc. Nephrol. 2019, 30, 1605–1624. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Piuhola, J.; Komulainen, J.; Sormunen, R.; Ongvarrasopone, C.; Fässler, R.; Muona, A.; Ilves, M.; Ruskoaho, H.; Takala, T.E.S.; et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.J.; Lindert, U.; Opitz, L.; Hausser, I.; Rohrbach, M.; Giunta, C. Transcriptome Profiling of Primary Skin Fibroblasts Reveal Distinct Molecular Features Between PLOD1- and FKBP14-Kyphoscoliotic Ehlers–Danlos Syndrome. Genes 2019, 10, 517. [Google Scholar] [CrossRef]
- Castori, M.; Tinkle, B.; Levy, H.; Grahame, R.; Malfait, F.; Hakim, A. A framework for the classification of joint hypermobility and related conditions. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Monthoux, C.; de Brot, S.; Jackson, M.; Bleul, U.; Walter, J. Skin malformations in a neonatal foal tested homozygous positive for Warmblood Fragile Foal Syndrome. BMC Vet. Res. 2015, 11, 1–8. [Google Scholar] [CrossRef]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.A.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro Dysplastic Form of the Ehlers-Danlos Syndrome-An Autosomal-Recessive Entity Caused by Mutations in the Zinc Transporter Gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef]
- Mushtaq, M.; Gaza, H.V.; Kashuba, E.V. Role of the RB-Interacting Proteins in Stem Cell Biology. Adv. Cancer Res. 2016, 131, 133–157. [Google Scholar]
- Minocherhomji, S.; Hansen, C.; Kim, H.G.; Mang, Y.; Bak, M.; Guldberg, P.; Papadopoulos, N.; Eiberg, H.; Doh, G.D.; Møllgård, K.; et al. Epigenetic remodelling and dysregulation of DLGAP4 is linked with early-onset cerebellar ataxia. Hum. Mol. Genet. 2014, 23, 6163–6176. [Google Scholar] [CrossRef]
- Rasmussen, A.H.; Rasmussen, H.B.; Silahtaroglu, A. The DLGAP family: Neuronal expression, function and role in brain disorders. Mol. Brain 2017, 10, 1–13. [Google Scholar] [CrossRef]
- Banerjee, A.; Wang, H.Y.; Borgmann-Winter, K.E.; MacDonald, M.L.; Kaprielian, H.; Stucky, A.; Kvasic, J.; Egbujo, C.; Ray, R.; Talbot, K.; et al. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol. Psychiatry 2015, 20, 1091–1100. [Google Scholar] [CrossRef]
- Schob, C.; Morellini, F.; Ohana, O.; Bakota, L.; Hrynchak, M.V.; Brandt, R.; Brockmann, M.D.; Cichon, N.; Hartung, H.; Hanganu-Opatz, I.L.; et al. Cognitive impairment and autistic-like behaviour in SAPAP4-deficient mice. Transl. Psychiatry 2019, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, S.I.J.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.; Yoneda, T.; Boycet, B.F.; Chent, H.; Mundy, G.R.; Sorianot, P. Osteopetrosis in Src-deficient mice is due to an autonomous defect of osteoclasts. Proc. Natl. Acad. Sci. USA 1993, 90, 4485–4489. [Google Scholar] [CrossRef] [PubMed]
- Skujina, I.; Winton, C.L.; Hegarty, M.J.; McMahon, R.; Nash, D.M.; Davies Morel, M.C.G.; McEwan, N.R. Detecting genetic regions associated with height in the native ponies of the British Isles by using high density SNP genotyping. Genome 2018, 61, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.B.; Sanchez-Lara, P.A.; Randolph, L.M.; Krieger, M.D.; Wu, S.Q.; Panigrahy, A.; Shimada, H.; Erdreich-Epstein, A. Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): A case report and review of the literature. BMC Med. Genet. 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.Q.; Fertig, N.; Medsger, T.A.; Wright, T.M. Molecular Recognition Patterns of Serum Anti-DNA Topoisomerase I Antibody in Systemic Sclerosis. J. Immunol. 2014, 173, 2834–2841. [Google Scholar] [CrossRef]
- Nevitt, C.; Tooley, J.G.; Schaner Tooley, C.E. N-terminal acetylation and methylation differentially affect the function of MYL9. Biochem. J. 2018, 475, 3201–3219. [Google Scholar] [CrossRef]
- Rylatt, D.B.; Aitken, A.; Bilham, T.; Condon, G.D.; Embi, N.; Cohen, P. Glycogen Synthase from Rabbit Skeletal Muscle. Eur. J. Biochem. 2005, 107, 529–537. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Liu, X.; Ding, X.; Xin, X.; Jin, C.; Zhang, S.; Li, G.; Guo, H. Parallel comparative proteomics and phosphoproteomics reveal that cattle myostatin regulates phosphorylation of key enzymes in glycogen metabolism and glycolysis pathway. Oncotarget 2018, 9, 11352–11370. [Google Scholar] [CrossRef]
- Lewis, S.S.; Nicholson, A.M.; Williams, Z.J.; Valberg, S.J. Clinical characteristics and muscle glycogen concentrations in warmblood horses with polysaccharide storage myopathy. Am. J. Vet. Res. 2017, 78, 1305–1312. [Google Scholar] [CrossRef]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.H.; Zhang, W.; Sinton, C.; Powell, C.M.; Parada, L.F. Pharmacological inhibition of mTORCl suppresses anatomical, cellular, and behavioral abnormalities in neural-specific PTEN knock-out mice. J. Neurosci. 2009, 29, 1773–1783. [Google Scholar] [CrossRef]
- Hassed, S.; Li, S.; Mulvihill, J.; Aston, C.; Palmer, S. Adams–Oliver syndrome review of the literature: Refining the diagnostic phenotype. Am. J. Med. Genet. Part A 2017, 173, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.F.R.; Scotton, C.; Zhang, J.; Passarelli, C.; Ortiz-Bonnin, B.; Simrick, S.; Schwerte, T.; Poon, K.-L.; Fang, M.; Rinné, S.; et al. POPDC1S201F causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Invest. 2015, 126, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Swan, A.H.; Gruscheski, L.; Boland, L.A.; Brand, T. The Popeye domain containing gene family encoding a family of cAMP-effector proteins with important functions in striated muscle and beyond. J. Muscle Res. Cell Motil. 2019, 40, 169–183. [Google Scholar] [CrossRef]
- Hendrickson, S.L. A genome wide study of genetic adaptation to high altitude in feral Andean Horses of the páramo. BMC Evol. Biol. 2013, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Adrover, M.F.; Wess, J.; Alvarez, V.A. Muscarinic regulation of dopamine and glutamate transmission in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 2015, 112, 8124–8129. [Google Scholar] [CrossRef]
- Kader, A.; Liu, X.; Dong, K.; Song, S.; Pan, J.; Yang, M.; Chen, X.; He, X.; Jiang, L.; Ma, Y. Identification of copy number variations in three Chinese horse breeds using 70 K single nucleotide polymorphism BeadChip array. Anim. Genet. 2016, 47, 560–569. [Google Scholar] [CrossRef]
- Ueda, H.; Sasaki, K.; Halder, S.K.; Deguchi, Y.; Takao, K.; Miyakawa, T.; Tajima, A. Prothymosin alpha-deficiency enhances anxiety-like behaviors and impairs learning/memory functions and neurogenesis. J. Neurochem. 2017, 141, 124–136. [Google Scholar] [CrossRef]
- George, E.M.; Brown, D.T. Prothymosin α is a component of a linker histone chaperone. FEBS Lett. 2010, 584, 2833–2836. [Google Scholar] [CrossRef]
- McNeill, E.M.; Klöckner-Bormann, M.; Roesler, E.C.; Talton, L.E.; Moechars, D.; Clagett-Dame, M. Nav2 hypomorphic mutant mice are ataxic and exhibit abnormalities in cerebellar development. Dev. Biol. 2011, 353, 331–343. [Google Scholar] [CrossRef]
- Lanuza, G.M.; Gosgnach, S.; Pierani, A.; Jessell, T.M.; Goulding, M. Genetic identification of spinal interneurons that coordinate left-right locomotor activity necessary for walking movements. Neuron 2004, 42, 375–386. [Google Scholar] [CrossRef]
- Griener, A.; Zhang, W.; Kao, H.; Haque, F.; Gosgnach, S. Anatomical and electrophysiological characterization of a population of dI6 interneurons in the neonatal mouse spinal cord. Neuroscience 2017, 362, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.S.; Larhammar, M.; Memic, F.; Wootz, H.; Schwochow, D.; Rubin, C.J.; Patra, K.; Arnason, T.; Wellbring, L.; Hjälm, G.; et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature 2012, 488, 642–646. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | SJ Horses | NS Horses |
|---|---|---|
| Mean EBV for show jumping performance | 125 (13.4) * | 77 (11.3) * |
| Mean EBV for dressage performance | 94 (10.2) * | 119 (21.0) * |
| Mean score for jumping technique | 8.3 (1.2) * | 5.6 (1.2) * |
| Mean score for gaits ** | 7.2 (0.6) * | 7.3 (0.8) * |
| Mean score for the conformation trait “type” | 7.9 (0.7) * | 7.8 (0.7) * |
| Mean score for the conformation trait “head–neck–body” | 7.7 (0.6) * | 7.7 (0.7) * |
| Percentage of horses competing in show jumping at least at regional level | 71% | 11% |
| Percentage of horses competing in dressage at least at regional level | 10% | 38% |
| Chromosome | Start Position (bp) | End Position (bp) | Length (kb) | Lowest Adjusted p–Value | Highest FST |
|---|---|---|---|---|---|
| 7 | 89,669,652 | 91,203,503 | 2918 | 0.0022 | 0.16 |
| 8 | 10,891,925 | 11,400,093 | 508 | 0.0040 | 0.13 |
| 12 | 11,601,991 | 12,104,261 | 502 | 0.0129 | na |
| 12 | 28,794,818 | 29,306,185 | 511 | 0.0009 | 0.13 |
| 13 | 27,280,931 | 27,830,947 | 550 | 0.0031 | 0.14 |
| 19 | 41,459,599 | 41,959,599 | 500 | 0.0247 | 0.13 |
| 22 | 27,935,550 | 31,789,708 | 3854 | 0.0008 | 0.14 |
| 31 | 13,778,365 | 14,321,247 | 543 | 0.00002 | na |
| PANTHER GOs | Genes | Fold Enrichment | Raw p–Value |
|---|---|---|---|
| Slim Biological Process | |||
| Cell differentiation (GO:0030154) | RBL1, DBX1, SRC, NR1I2 | 5.68 | <0.001 |
| Multicellular organism development (GO:0007275) | DBX1, DLGAP4, NAV2, MYL9, KIAA1755, VSTM2L, GSK3B, NR1I2, POPDC2, DPF2 | 3.33 | 0.002 |
| Small GTPase mediated signal transduction (GO:0007264) | ARHGAP40, ARHGAP31, CDC42EP2 | 6.25 | 0.004 |
| Cellular response to lipopolysaccharide (GO:1901700.) | NR1I2, CD80 | 9.25 | 0.005 |
| DNA conformation change (GO:0071103) | CDCA5, TOP1 | 19.6 | 0.005 |
| Regulation of protein complex assembly (GO:0043254) | ARHGAP40 | 13.1 | 0.011 |
| Purine nucleoside triphosphate metabolic process (GO:0009144) | SAMHD1 | >100 | 0.014 |
| Regulation of sister chromatid segregation (GO:0033045) | CDCA5 | >100 | 0.014 |
| Regulation of exit from mitosis (GO:0007096) | CDCA5 | >100 | 0.014 |
| Protein K48-linked ubiquitination (GO:0070936) | SYVN1 | 71.9 | 0.018 |
| Pathways | |||
| De novo pyrimidine deoxyribonucleotide biosynthesis (P02739) | VSTM2L | 37.7 | <0.001 |
| CCKR signaling map (P06959) | GSK3B, SRC | 6.78 | <0.001 |
| DNA replication (P00017) | TOP1 | 17.3 | <0.001 |
| Angiogenesis (P00005) | GSK3B, SRC, NR1I2 | 4.78 | <0.001 |
| Chromosome | Start Position (bp) | End Position (bp) | Length (kb) | Lowest Adjusted p-Value | Highest FST |
|---|---|---|---|---|---|
| 1 | 40,592,555 | 43,510,660 | 2918 | 0.0230 | 0.21 |
| 23 | 51,321,303 | 51,844,470 | 523 | 0.0501 | 0.15 |
| 25 | 5,277,465 | 5,984,107 | 707 | 0.0002 | 0.15 |
| 25 | 37,351,887 | 37,886,254 | 534 | 0.0071 | 0.15 |
| PANTHER GOs | Genes | Fold Enrichment | Raw p-Value |
|---|---|---|---|
| Slim Biological Process | |||
| SRP-dependent translational protein targeting to membrane (GO:0006614) | SEC61B | 100.0 | 0.007 |
| Posttranslational protein targeting to endoplasmic reticulum membrane (GO:0006620) | SEC61B | 100.0 | 0.009 |
| Intracellular protein transmembrane transport (GO:0065002) | SEC61B | 88.7 | 0.012 |
| Activin receptor signaling pathway (GO:0032924) | TGFBR1 | 81.3 | 0.013 |
| Phospholipid biosynthetic process (GO:0008654) | SGMS1 | 75.0 | 0.014 |
| Pathways | |||
| Insulin/IGF pathway–protein kinase B signaling cascade (P00033) | PTEN | 35.0 | <0.001 |
| p53 pathway feedback loops 2 (P04398) | PTEN | 27.1 | <0.001 |
| Sulfate assimilation (P02778) | PAPSS2 | 100.0 | <0.001 |
| TGF-beta signaling pathway (P00052) | TGFBR1 | 14.5 | <0.001 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ablondi, M.; Eriksson, S.; Tetu, S.; Sabbioni, A.; Viklund, Å.; Mikko, S. Genomic Divergence in Swedish Warmblood Horses Selected for Equestrian Disciplines. Genes 2019, 10, 976. https://doi.org/10.3390/genes10120976
Ablondi M, Eriksson S, Tetu S, Sabbioni A, Viklund Å, Mikko S. Genomic Divergence in Swedish Warmblood Horses Selected for Equestrian Disciplines. Genes. 2019; 10(12):976. https://doi.org/10.3390/genes10120976
Chicago/Turabian StyleAblondi, Michela, Susanne Eriksson, Sasha Tetu, Alberto Sabbioni, Åsa Viklund, and Sofia Mikko. 2019. "Genomic Divergence in Swedish Warmblood Horses Selected for Equestrian Disciplines" Genes 10, no. 12: 976. https://doi.org/10.3390/genes10120976
APA StyleAblondi, M., Eriksson, S., Tetu, S., Sabbioni, A., Viklund, Å., & Mikko, S. (2019). Genomic Divergence in Swedish Warmblood Horses Selected for Equestrian Disciplines. Genes, 10(12), 976. https://doi.org/10.3390/genes10120976