The Farmed Atlantic Salmon (Salmo salar) Skin–Mucus Proteome and Its Nutrient Potential for the Resident Bacterial Community


 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish and Sampling Procedure
2.2. Sample Preparation for 16S rRNA Gene Sequencing
2.3. Analysis of 16s rRNA Gene Sequencing Data
2.4. Sample Preparation and Proteomic Analysis
2.5. Analysis of Proteomic Data
2.6. Mapping of Salmo salar Mucus Proteins to RNAseq Data
2.7. Mucin Quantification
2.8. Nucleotide Sequence Accession Numbers
2.9. Mass Spectrometry Proteomic Data
3. Results
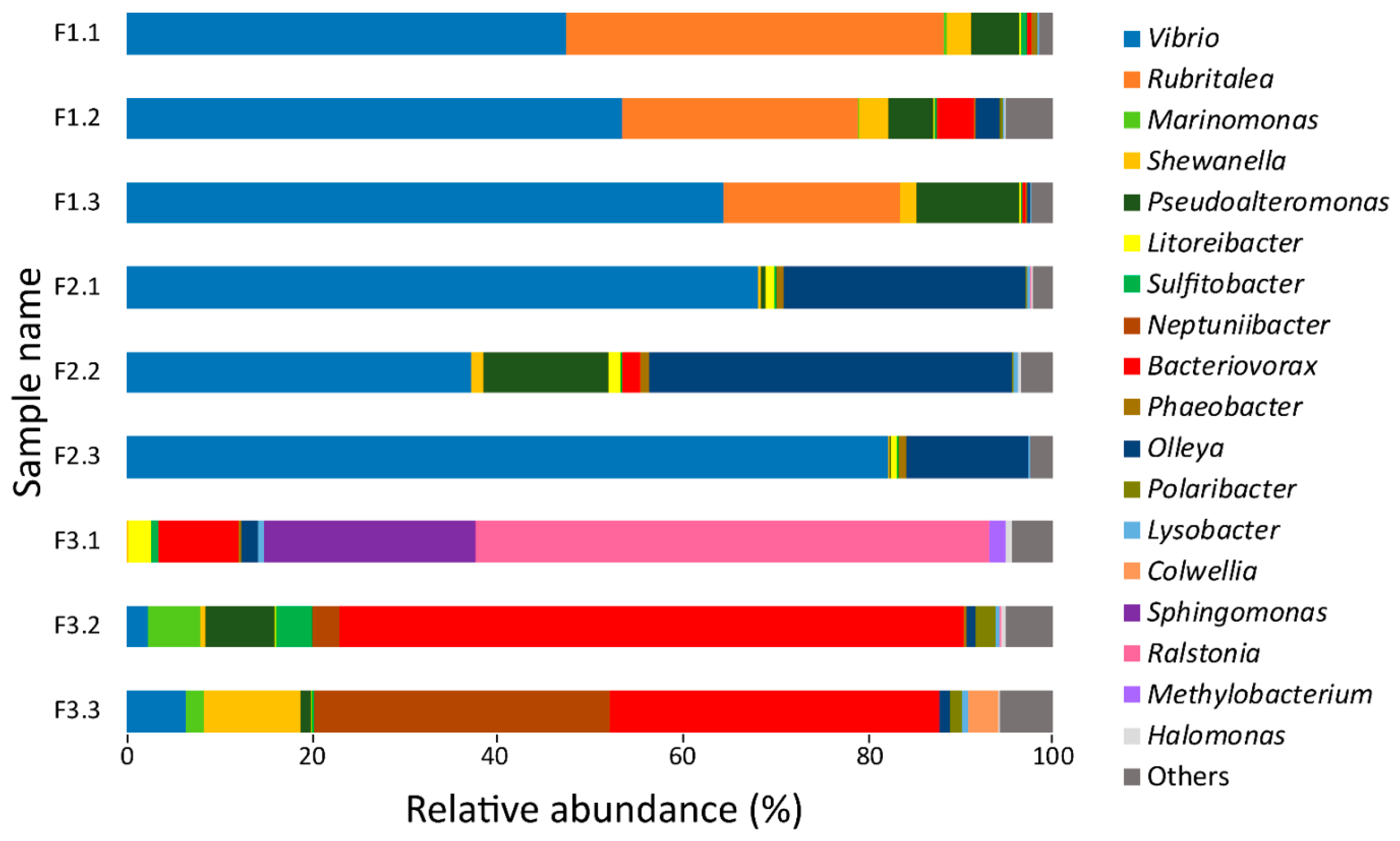
3.1. 16S rRNA Gene Sequencing Analysis of Bacteria Utilizing Salmon Skin–Mucus as Nutrient Source
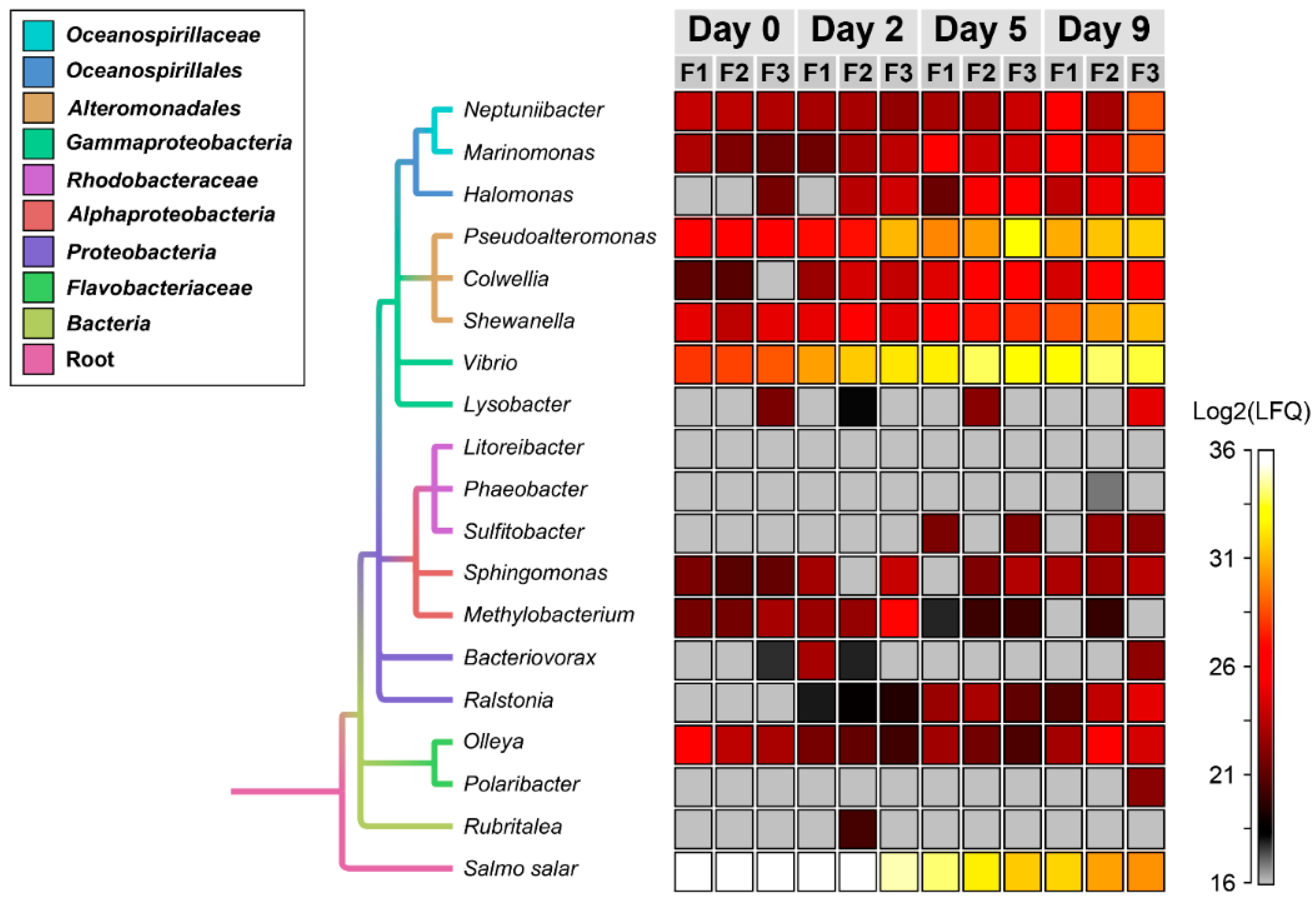
3.2. Temporal Quantitative Proteomic Analysis of the Salmon Skin Microbiome
3.3. Host Proteins Detected in the Salmo salar Skin–mucus
3.4. Quantification of Mucins
4. Discussion
4.1. Skin Mucus Can Be Used by Microorganisms as a Source of Nutrients
4.2. Proteases and Siderophore Transporters Dominate the Bacterial Exoproteome
4.3. The Salmo salar Skin–Mucus Proteome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Linden, S.K.; Sutton, P.; Karlsson, N.G.; Korolik, V.; McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; van Passel, M.W.; van de Bovenkamp, J.H.; Schipper, R.G.; de Vos, W.M.; Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Schroers, V.; van der Marel, M.; Neuhaus, H.; Steinhagen, D. Changes of intestinal mucus glycoproteins after peroral application of Aeromonas hydrophila to common carp (Cyprinus carpio). Aquaculture 2009, 288, 184–189. [Google Scholar] [CrossRef]
- Neuhaus, H.; van der Marel, M.; Caspari, N.; Meyer, W.; Enss, M.L.; Steinhagen, D. Biochemical and histochemical effects of perorally applied endotoxin on intestinal mucin glycoproteins of the common carp Cyprinus carpio. Dis. Aquat. Org. 2007, 77, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Linden, S.; Mahdavi, J.; Semino-Mora, C.; Olsen, C.; Carlstedt, I.; Boren, T.; Dubois, A. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PLoS Pathog. 2008, 4, e2. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.S.; Kissoon-Singh, V.; Gibson, D.L.; Ma, C.; Montero, M.; Sham, H.P.; Ryz, N.; Huang, T.; Velcich, A.; Finlay, B.B. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010, 6, e1000902. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265. [Google Scholar] [CrossRef]
- Szabady, R.L.; Yanta, J.H.; Halladin, D.K.; Schofield, M.J.; Welch, R.A. TagA is a secreted protease of Vibrio cholerae that specifically cleaves mucin glycoproteins. Microbiology 2011, 157, 516–525. [Google Scholar] [CrossRef]
- Budiarti, S.; Mubarik, N.R. Extracellular protease activity of enteropathogenic Escherechia coli on mucin substrate. HAYATI J. Biosci. 2007, 14, 36–38. [Google Scholar] [CrossRef][Green Version]
- Negus, V. Function of mucus—A hypothesis. Proc. R. Soc. Med. 1967, 60, 75. [Google Scholar] [PubMed]
- Park, J.Y.; Kim, I.S.; Kim, S.Y. Structure and histochemistry of the skin of a torrent catfish, Liobagrus mediadiposalis. Environ. Biol. Fish. 2003, 66, 3–8. [Google Scholar] [CrossRef]
- Rosen, M.W.; Cornford, N.E. Fluid friction of fish slimes. Nature 1971, 234, 49. [Google Scholar] [CrossRef]
- Shepard, K.L. Functions of fish mucus. Rev. Fish. Biol. Fish. 1994, 4, 401–429. [Google Scholar] [CrossRef]
- Lowrey, L.; Woodhams, D.C.; Tacchi, L.; Salinas, I. Topographical mapping of the rainbow trout (Oncorhynchus mykiss) microbiome reveals a diverse bacterial community with antifungal properties in the skin. Appl. Environ. Microbiol. 2015, 81, 6915–6925. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, M.; Villéger, S.; Bouvier, C.; Bettarel, Y.; Bouvier, T. High diversity of skin-associated bacterial communities of marine fishes is promoted by their high variability among body parts, individuals and species. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef]
- Gómez, G.D.; Balcázar, J.L. A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol. Med. Microbiol. 2007, 52, 145–154. [Google Scholar] [CrossRef]
- Padra, J.T.; Sundh, H.; Jin, C.; Karlsson, N.G.; Sundell, K.; Lindén, S.K. Aeromonas salmonicida binds differentially to mucins isolated from skin and intestinal regions of Atlantic salmon in an N-acetylneuraminic acid-dependent manner. Infect. Immun. 2014, 82, 5235–5245. [Google Scholar] [CrossRef]
- Sveen, L.R.; Grammes, F.T.; Ytteborg, E.; Takle, H.; Jørgensen, S.M. Genome-wide analysis of Atlantic salmon (Salmo salar) mucin genes and their role as biomarkers. PloS ONE 2017, 12, e0189103. [Google Scholar] [CrossRef]
- Cordero, H.; Cuesta, A.; Meseguer, J.; Esteban, M.A. Changes in the levels of humoral immune activities after storage of gilthead seabream (Sparus aurata) skin mucus. Fish Shellfish Immun. 2016, 58, 500–507. [Google Scholar] [CrossRef]
- Du, Y.; Yi, M.; Xiao, P.; Meng, L.; Li, X.; Sun, G.; Liu, Y. The impact of Aeromonas salmonicida infection on innate immune parameters of Atlantic salmon (Salmo salar L). Fish Shellfish Immunol. 2015, 44, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Easy, R.H.; Ross, N.W. Changes in Atlantic salmon Salmo salar mucus components following short- and long-term handling stress. J. Fish. Biol. 2010, 77, 1616–1631. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, F.A.; Cuartero, M.; Collado-Gonzalez, M.D.; Arizcun, M.; Banos, F.G.D.; Meseguer, J.; Cuesta, A.; Esteban, M.A. Description and comparative study of physico-chemical parameters of the teleost fish skin mucus. Biorheology 2015, 52, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, F.A.; Cuartero, M.; Del Mar Collado-Gonzalez, M.; Diaz Banos, F.G.; Cuesta, A.; Morinigo, M.A.; Esteban, M.A. Terminal carbohydrates abundance, immune related enzymes, bactericidal activity and physico-chemical parameters of the Senegalese sole (Solea senegalensis, Kaup) skin mucus. Fish Shellfish Immunol. 2017, 60, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, F.A.; Cuesta, A.; Abellan, E.; Meseguer, J.; Esteban, M.A. Comparative analysis of the humoral immunity of skin mucus from several marine teleost fish. Fish Shellfish Immun. 2014, 40, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Liu, B.L.; Feng, W.R.; Han, C.; Huang, B.; Lei, J.L. Stress and immune responses in skin of turbot (Scophthalmus maximus) under different stocking densities. Fish Shellfish Immun. 2016, 55, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, E.; Berendsen, J.; Guzman, F.; Gallardo, J.A.; Mercado, L. An immunological method for quantifying antibacterial activity in Salmo salar (Linnaeus, 1758) skin mucus. Fish Shellfish Immun. 2010, 28, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.D.; Powell, M.D. The viscosity and glycoprotein biochemistry of salmonid mucus varies with species, salinity and the presence of amoebic gill disease. J. Comp. Physiol. B 2005, 175, 1–11. [Google Scholar] [CrossRef]
- Ross, N.W.; Firth, K.J.; Wang, A.; Burka, J.F.; Johnson, S.C. Changes in hydrolytic enzyme activities of naive Atlantic salmon Salmo salar skin mucus due to infection with the salmon louse Lepeophtheirus salmonis and cortisol implantation. Dis. Aquat. Org. 2000, 41, 43–51. [Google Scholar] [CrossRef]
- Subramanian, S.; Ross, N.W.; MacKinnon, S.L. Comparison of antimicrobial activity in the epidermal mucus extracts of fish. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 85–92. [Google Scholar] [CrossRef]
- Cordero, H.; Brinchmann, M.F.; Cuesta, A.; Meseguer, J.; Esteban, M.A. Skin mucus proteome map of European sea bass (Dicentrarchus labrax). Proteomics 2015, 15, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Cordero, H.; Morcillo, P.; Cuesta, A.; Brinchmann, M.F.; Esteban, M.A. Differential proteome profile of skin mucus of gilthead seabream (Sparus aurata) after probiotic intake and/or overcrowding stress. J. Proteomics 2016, 132, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Jurado, J.; Fuentes-Almagro, C.A.; Guardiola, F.A.; Cuesta, A.; Esteban, M.A.; Prieto-Alamo, M.J. Proteomic profile of the skin mucus of farmed gilthead seabream (Sparus aurata). J. Proteomics 2015, 120, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Micallef, G.; Cash, P.; Fernandes, J.M.O.; Rajan, B.; Tinsley, J.W.; Bickerdike, R.; Martin, S.A.M.; Bowman, A.S. Dietary yeast cell wall extract alters the proteome of the skin mucous barrier in Atlantic salmon (Salmo salar): Increased abundance and expression of a calreticulin-like protein. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Brinchmann, M.F. Skin mucus proteins of lumpsucker (Cyclopterus lumpus). Biochem. Biophys. Rep. 2017, 9, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Raeder, I.L.; Paulsen, S.M.; Smalas, A.O.; Willassen, N.P. Effect of fish skin mucus on the soluble proteome of Vibrio salmonicida analysed by 2-D gel electrophoresis and tandem mass spectrometry. Microb. Pathog. 2007, 42, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Valdenegro-Vega, V.A.; Crosbie, P.; Bridle, A.; Leef, M.; Wilson, R.; Nowak, B.F. Differentially expressed proteins in gill and skin mucus of Atlantic salmon (Salmo salar) affected by amoebic gill disease. Fish Shellfish Immunol. 2014, 40, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Provan, F.; Jensen, L.B.; Uleberg, K.E.; Larssen, E.; Rajalahti, T.; Mullins, J.; Obach, A. Proteomic analysis of epidermal mucus from sea liceinfected Atlantic salmon, Salmo salar L. J. Fish Dis. 2013, 36, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Brinchmann, M.F. Immune relevant molecules identified in the skin mucus of fish using-omics technologies. Mol. Biosyst. 2016, 12, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Ángeles Esteban, M. An overview of the immunological defenses in fish skin. ISRN Immunol. 2012. [Google Scholar] [CrossRef]
- Saurabh, S.; Sahoo, P.K. Lysozyme: An important defence molecule of fish innate immune system. Aquac. Res. 2008, 39, 223–239. [Google Scholar] [CrossRef]
- Luders, T.; Birkemo, G.A.; Nissen-Meyer, J.; Andersen, O.; Nes, I.F. Proline conformation-dependent antimicrobial activity of a proline-rich histone H1N-terminal peptide fragment isolated from the skin mucus of Atlantic Salmon. Antimicrob. Agents Chemother. 2005, 49, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Hatten, F.; Fredriksen, A.; Hordvik, I.; Endresen, C. Presence of IgM in cutaneous mucus, but not in gut mucus of Atlantic salmon, Salmo salar. Serum IgM is rapidly degraded when added to gut mucus. Fish Shellfish Immunol. 2001, 11, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.B.; Cheung, R.C.F.; Ng, C.C.W.; Fang, E.F.; Wong, J.H. A review of fish lectins. Curr. Protein Pept. Sci. 2015, 16, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Firth, K.J.; Johnson, S.C.; Ross, N.W. Characterization of proteases in the skin mucus of Atlantic salmon (Salmo salar) infected with the salmon louse (Lepeophtheirus salmonis) and in whole-body louse homogenate. J. Parasitol. 2000, 86, 1199–1205. [Google Scholar] [CrossRef]
- Ezaki, T.; Suzuki, S. Achromopeptidase for lysis of anaerobic gram-positive cocci. J. Clin. Microbiol. 1982, 16, 844–846. [Google Scholar] [PubMed]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PloS ONE 2014, 9, e105592. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Microbiol. 2012, 10.7.1–10.7.20. [Google Scholar]
- Arntzen, M.Ø.; Karlskås, I.L.; Skaugen, M.; Eijsink, V.G.; Mathiesen, G. Proteomic investigation of the response of Enterococcus faecalis V583 when cultivated in urine. PloS ONE 2015, 10, e0126694. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Juncker, A.S.; Willenbrock, H.; Von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’donovan, C.; Apweiler, R. QuickGO: A web-based tool for gene ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Jin, C.; Padra, J.N.T.S.; Sundell, K.; Sundh, H.; Karlsson, N.G.; Lindén, S.K. Atlantic salmon carries a range of novel O-glycan structures differentially localized on skin and intestinal mucins. J. Proteome Res. 2015, 14, 3239–3251. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2015, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Finn, R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2015, 44, D343–D350. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Hagen, L.H.; Porcellato, D.; Jørgensen, S.M.; Pope, P.B.; Vaaje-Kolstad, G. The skin-mucus microbial community of farmed Atlantic salmon (Salmo salar). Front. Microbiol. 2017, 8, 2043. [Google Scholar] [CrossRef] [PubMed]
- Colwell, R.; Grimes, D. Vibrio diseases of marine fish populations. Helgoländer Meeresuntersuchungen 1984, 37, 265. [Google Scholar] [CrossRef]
- Austin, B. The bacterial microflora of fish, revised. Sci. World J. 2006, 6, 931–945. [Google Scholar] [CrossRef]
- Benediktsdottir, E.; Helgason, S.; Sigurjónsdóttir, H. Vibrio spp. isolated from salmonids with shallow skin lesions and reared at low temperature. J. Fish Dis. 1998, 21, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Strom, M.S.; Paranjpye, R.N. Epidemiology and pathogenesis of Vibrio vulnificus. Microb. Infect. 2000, 2, 177–188. [Google Scholar] [CrossRef]
- Frans, I.; Michiels, C.; Bossier, P.; Willems, K.; Lievens, B.; Rediers, H. Vibrio anguillarum as a fish pathogen: Virulence factors, diagnosis and prevention. J. Fish Dis. 2011, 34, 643–661. [Google Scholar] [CrossRef]
- Jensen, S.; Samuelsen, O.B.; Andersen, K.; Torkildsen, L.; Lambert, C.; Choquet, G.; Paillard, C.; Bergh, Ø. Characterization of strains of Vibrio splendidus and V. tapetis isolated from corkwing wrasse Symphodus melops suffering vibriosis. Dis. Aquat. Org. 2003, 53, 25–31. [Google Scholar] [CrossRef]
- Gatesoupe, F.; Lambert, C.; Nicolas, J.-L. Pathogenicity of Vibrio splendidus strains associated with turbot larvae, Scophthalmus maximus. J. Appl. Microbiol. 1999, 87, 757–763. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lopez, J.; Balboa, S.; Nunez, S.; de La Roca, E.; de La Herran, R.; Navas, J.; Toranzo, A.; Romalde, J. Characterization of Vibrio tapetis strains isolated from diseased cultured Wedge sole (Dicologoglossa cuneata Moreau). Res. Vet. Sci. 2011, 90, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.H.; Peatman, E. Mucosal Health in Aquaculture; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Cahill, M.M. Bacterial flora of fishes: A review. Microb. Ecol. 1990, 19, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Fraumene, C.; Pagnozzi, D.; Manghina, V.; Deligios, M.; Muth, T.; Rapp, E.; Martens, L.; Addis, M.F. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Renard, B.Y.; Martens, L. Metaproteomic data analysis at a glance: Advances in computational microbial community proteomics. Expert Rev. Proteomics 2016, 13, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Drake, D.; Montie, T. Flagella, motility and invasive virulence of Pseudomonas aeruginosa. J. Gen. Microbiol. 1988, 134, 43. [Google Scholar] [CrossRef] [PubMed]
- Attridge, S.; Rowley, D. The role of the flagellum in the adherence of Vibrio cholerae. J. Infect. Dis. 1983, 147, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Chaban, B.; Hughes, H.V.; Beeby, M. The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 2015, 46, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Tuveng, T.R.; Arntzen, M.Ø.; Bengtsson, O.; Gardner, J.G.; Vaaje-Kolstad, G.; Eijsink, V.G. Proteomic investigation of the secretome of Cellvibrio japonicus during growth on chitin. Proteomics 2016, 16, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Bjornsdottir, B.; Fridjonsson, O.H.; Magnusdottir, S.; Andresdottir, V.; Hreggvidsson, G.O.; Gudmundsdottir, B.K. Characterisation of an extracellular vibriolysin of the fish pathogen Moritella viscosa. Vet. Microbiol. 2009, 136, 326–334. [Google Scholar] [CrossRef]
- Ghosh, A.; Koley, H.; Pal, A. The role of Vibrio cholerae haemagglutinin protease (HAP) in extra-intestinal infection. J. Clin. Diagn. Res. JCDR 2016, 10, DC10. [Google Scholar] [CrossRef] [PubMed]
- Lövgren, A.; Zhang, M.; Engström, A.; Dalhammar, G.; Landén, R. Molecular characterization of immune inhibitor A, a secreted virulence protease from Bacillus thuringiensis. Mol. Microbiol. 1990, 4, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Milton, D.L.; Norqvist, A.; Wolf-Watz, H. Cloning of a metalloprotease gene involved in the virulence mechanism of Vibrio anguillarum. J. Bacteriol. 1992, 174, 7235–7244. [Google Scholar] [CrossRef] [PubMed]
- Mengaud, J.; Geoffroy, C.; Cossart, P. Identification of a new operon involved in Listeria monocytogenes virulence: Its first gene encodes a protein homologous to bacterial metalloproteases. Infect. Immun. 1991, 59, 1043–1049. [Google Scholar] [PubMed]
- Cao, D.; Wang, N.; Lu, C.; Liu, Y. Identification and characterization of a novel virulence—Associated metalloprotease from aeromonas hydrophila. Pak. Vet. J. 2015, 35, 38–42. [Google Scholar]
- Tang, W.J.; Fernandez, J.G.; Sohn, J.J.; Amemiya, C.T. Chitin is endogenously produced in vertebrates. Curr. Biol. 2015, 25, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Rogers, M.; Shakarian, A.; Yamage, M.; Al-Harthi, S.; Bates, P.; Dwyer, D. Molecular characterization, expression, and in vivo analysis of LmexCht1: The chitinase of the human pathogen, Leishmania mexicana. J. Biol. Chem. 2005, 280, 3847–3861. [Google Scholar] [CrossRef]
- DebRoy, S.; Dao, J.; Söderberg, M.; Rossier, O.; Cianciotto, N.P. Legionella pneumophila type II secretome reveals unique exoproteins and a chitinase that promotes bacterial persistence in the lung. Proc. Nat. Acad. Sci. USA 2006, 103, 19146–19151. [Google Scholar] [CrossRef]
- Frederiksen, R.F.; Paspaliari, D.K.; Larsen, T.; Storgaard, B.G.; Larsen, M.H.; Ingmer, H.; Palcic, M.M.; Leisner, J.J. Bacterial chitinases and chitin-binding proteins as virulence factors. Microbiology 2013, 159, 833–847. [Google Scholar] [CrossRef]
- Mondal, M.; Nag, D.; Koley, H.; Saha, D.R.; Chatterjee, N.S. The Vibrio cholerae extracellular chitinase ChiA2 is important for survival and pathogenesis in the host intestine. PloS ONE 2014, 9, e103119. [Google Scholar] [CrossRef]
- Crosa, J.H.; Mey, A.R.; Payne, S.M. Iron Transport in Bacteria; ASM Press: Washington, DC, USA, 2004; Volume 22. [Google Scholar]
- Sandy, M.; Butler, A. Microbial iron acquisition: Marine and terrestrial siderophores. Chem. Rev. 2009, 109, 4580. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J. Siderophores: Structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010, 6, e1000949. [Google Scholar] [CrossRef] [PubMed]
- Biosca, E.G.; Fouz, B.; Alcaide, E.; Amaro, C. Siderophore-mediated iron acquisition mechanisms in Vibrio vulnificus biotype 2. Appl. Environ. Microbiol. 1996, 62, 928–935. [Google Scholar] [PubMed]
- Lamont, I.L.; Beare, P.A.; Ochsner, U.; Vasil, A.I.; Vasil, M.L. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc. Nat. Acad. Sci. USA 2002, 99, 7072–7077. [Google Scholar] [CrossRef] [PubMed]
- Renzi, F.; Manfredi, P.; Dol, M.; Fu, J.; Vincent, S.; Cornelis, G.R. Glycan-foraging systems reveal the adaptation of Capnocytophaga canimorsus to the dog mouth. MBio 2015, 6, e02507–14. [Google Scholar] [CrossRef]
- Noinaj, N.; Guillier, M.; Barnard, T.J.; Buchanan, S.K. TonB-dependent transporters: Regulation, structure, and function. Ann. Rev. Microbiol. 2010, 64, 43–60. [Google Scholar] [CrossRef]
- Harder, J.; Gläser, R.; Schröder, J.-M. Human antimicrobial proteins—Effectors of innate immunity. J. Endotoxin Res. 2007, 13, 317–338. [Google Scholar] [CrossRef]
- Antoni, L.; Nuding, S.; Weller, D.; Gersemann, M.; Ott, G.; Wehkamp, J.; Stange, E.F. Human colonic mucus is a reservoir for antimicrobial peptides. J. Crohn’s Colitis 2013, 7, e652–64. [Google Scholar] [CrossRef]
- Easy, R.H.; Ross, N.W. Changes in Atlantic salmon (Salmo salar) epidermal mucus protein composition profiles following infection with sea lice (Lepeophtheirus salmonis). Comp. Biochem. Physiol. Part D Genom. Proteomics 2009, 4, 159–167. [Google Scholar] [CrossRef]
- Rajan, B.; Fernandes, J.M.; Caipang, C.M.; Kiron, V.; Rombout, J.H.; Brinchmann, M.F. Proteome reference map of the skin mucus of Atlantic cod (Gadus morhua) revealing immune competent molecules. Fish Shellfish Immunol. 2011, 31, 224–231. [Google Scholar] [CrossRef]
- Rubin, B.K. Mucus structure and properties in cystic fibrosis. Paediatr. Respir. Rev. 2007, 8, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Eng. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [PubMed]
- Molle, V.; Campagna, S.; Bessin, Y.; Ebran, N.; Saint, N.; Molle, G. First evidence of the pore-forming properties of a keratin from skin mucus of rainbow trout (Oncorhynchus mykiss, formerly Salmo gairdneri). Biochem. J. 2008, 411, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.; Vosyliene, M.Z.; Golovkina, T. The effect of toxic and heliophysical factors on the biochemical parameters of the external mucus of carp, (Cyprinus carpio L.). Arch. Pol. Fish. Arch. Ryb. Pol. 2002, 10, 5–14. [Google Scholar]
- Liepke, C.; Baxmann, S.; Heine, C.; Breithaupt, N.; Ständker, L.; Forssmann, W.-G. Human hemoglobin-derived peptides exhibit antimicrobial activity: A class of host defense peptides. J. Chromatogr. B 2003, 791, 345–356. [Google Scholar] [CrossRef]
- Watson, A.J.; Duckworth, C.A.; Guan, Y.; Montrose, M.H. Mechanisms of epithelial cell shedding in the Mammalian intestine and maintenance of barrier function. Ann. N. Y. Acad. Sci. 2009, 1165, 135–142. [Google Scholar] [CrossRef]
- Williams, J.; Duckworth, C.; Burkitt, M.; Watson, A.; Campbell, B.; Pritchard, D. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef]
- Rudi, K.; Angell, I.L.; Pope, P.B.; Vik, J.O.; Sandve, S.R.; Snipen, L.-G. Stable core gut microbiota across the freshwater-to-saltwater transition for farmed Atlantic salmon. Appl. Environ. Microbiol. 2018, 84, e01974–17. [Google Scholar] [CrossRef]
- Severi, E.; Hood, D.W.; Thomas, G.H. Sialic acid utilization by bacterial pathogens. Microbiology 2007, 153, 2817–2822. [Google Scholar] [CrossRef]
- Venkatakrishnan, V.; Quintana-Hayashi, M.P.; Mahu, M.; Haesebrouck, F.; Pasmans, F.; Lindén, S.K. Brachyspira hyodysenteriae infection regulates mucin glycosylation synthesis inducing an increased expression of core-2 O-glycans in porcine colon. J. Proteome Res. 2017, 16, 1728–1742. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Protein Count | Secreted a |
|---|---|---|
| Salmo | 3583 | 523 (15%) |
| Vibrio | 2017 | 390 (19%) |
| Pseudoalteromonas | 1621 | 427 (26%) |
| Shewanella | 519 | 101 (19%) |
| Marinomonas | 154 | 27 (18%) |
| Neptuniibacter | 146 | 24 (16%) |
| Colwellia | 33 | 12 (36%) |
| Olleya | 22 | 10 (45%) |
| Halomonas | 18 | 0 |
| Sphingomonas | 11 | 3 (27%) |
| Lysobacter | 6 | 0 |
| Methylobacterium | 6 | 2 (33%) |
| Ralstonia | 4 | 0 |
| Bacteriovorax | 2 | 0 |
| Phaeobacter | 1 | 0 |
| Polaribacter | 1 | 0 |
| Rubritalea | 1 | 0 |
| Sulfitobacter | 1 | 0 |
| Litoreibacter | 0 | 0 |
| Sample | Mucin (mg/mL) | |
| F4 | 9.85 | |
| F5 | 20.73 | |
| F6 | 22.18 | |
| F7 | 13.18 | |
| Sample | Time (h) | |
| F8 | 0 | 8.9 (3.6) * |
| F8 | 12 | 9.2 (2.9) * |
| F8 | 24 | 6.2 (1.1) * |
| F8 | 48 | 4.1 (0.4) * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minniti, G.; Rød Sandve, S.; Padra, J.T.; Heldal Hagen, L.; Lindén, S.; Pope, P.B.; Ø. Arntzen, M.; Vaaje-Kolstad, G. The Farmed Atlantic Salmon (Salmo salar) Skin–Mucus Proteome and Its Nutrient Potential for the Resident Bacterial Community. Genes 2019, 10, 515. https://doi.org/10.3390/genes10070515
Minniti G, Rød Sandve S, Padra JT, Heldal Hagen L, Lindén S, Pope PB, Ø. Arntzen M, Vaaje-Kolstad G. The Farmed Atlantic Salmon (Salmo salar) Skin–Mucus Proteome and Its Nutrient Potential for the Resident Bacterial Community. Genes. 2019; 10(7):515. https://doi.org/10.3390/genes10070515
Chicago/Turabian StyleMinniti, Giusi, Simen Rød Sandve, János Tamás Padra, Live Heldal Hagen, Sara Lindén, Phillip B. Pope, Magnus Ø. Arntzen, and Gustav Vaaje-Kolstad. 2019. "The Farmed Atlantic Salmon (Salmo salar) Skin–Mucus Proteome and Its Nutrient Potential for the Resident Bacterial Community" Genes 10, no. 7: 515. https://doi.org/10.3390/genes10070515
APA StyleMinniti, G., Rød Sandve, S., Padra, J. T., Heldal Hagen, L., Lindén, S., Pope, P. B., Ø. Arntzen, M., & Vaaje-Kolstad, G. (2019). The Farmed Atlantic Salmon (Salmo salar) Skin–Mucus Proteome and Its Nutrient Potential for the Resident Bacterial Community. Genes, 10(7), 515. https://doi.org/10.3390/genes10070515