A Novel Role for Polycystin-2 (Pkd2) in P. tetraurelia as a Probable Mg2+ Channel Necessary for Mg2+-Induced Behavior

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Solutions, and Statistics
2.2. RNAi Construct Design and mRNA Depletion Using RNAi by Feeding
2.3. Swimming Behavior and Heavy Metal Resistance
2.4. Creation of Epitope-Tagged Proteins, Microinjection, and Immunofluorescence
2.5. Cell Membrane and Cilia Isolations
2.6. Immunoprecipitations (IP) and Western Blots
2.7. Electrophysiology
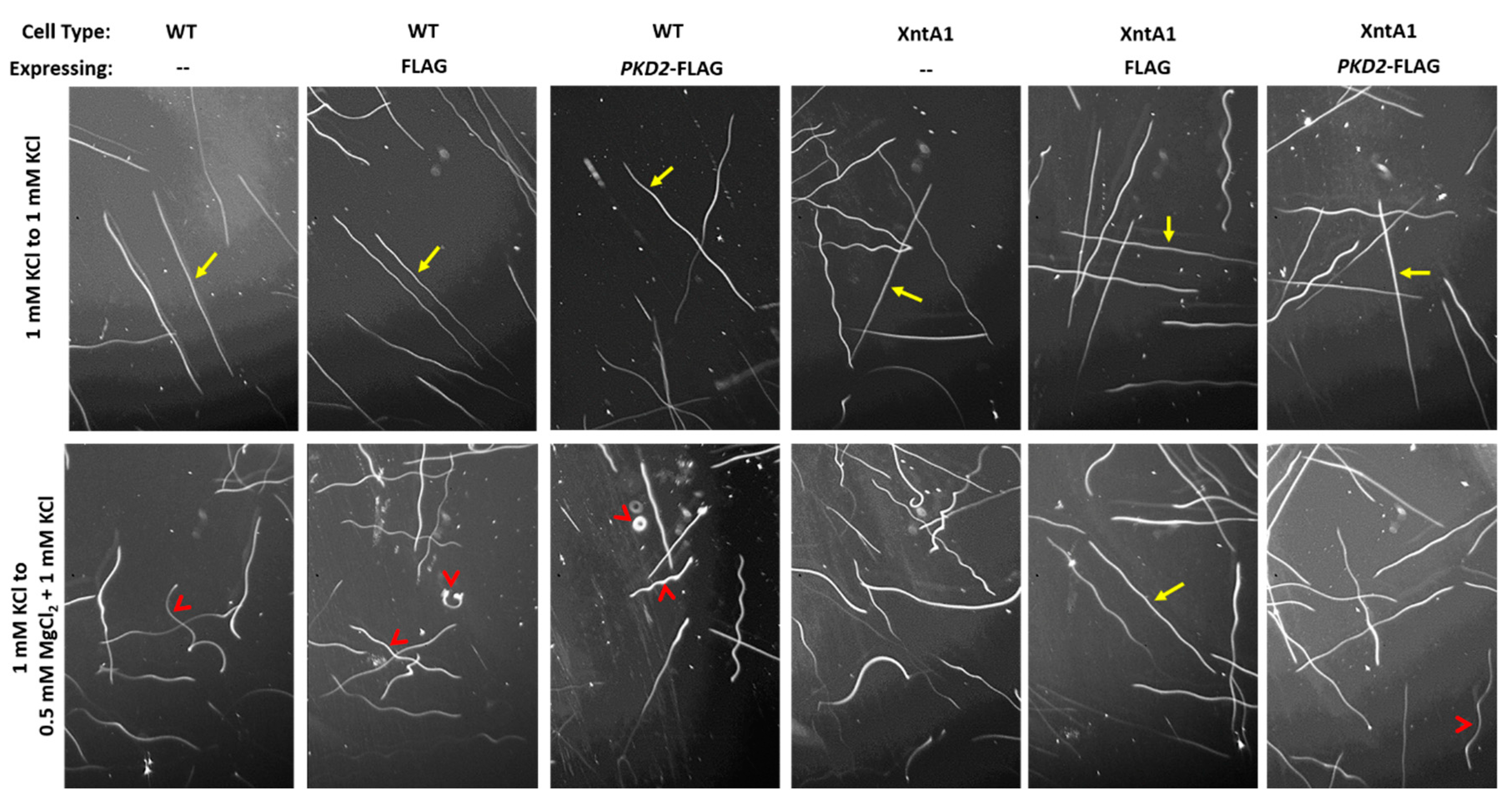
2.8. Swimming Traces
3. Result
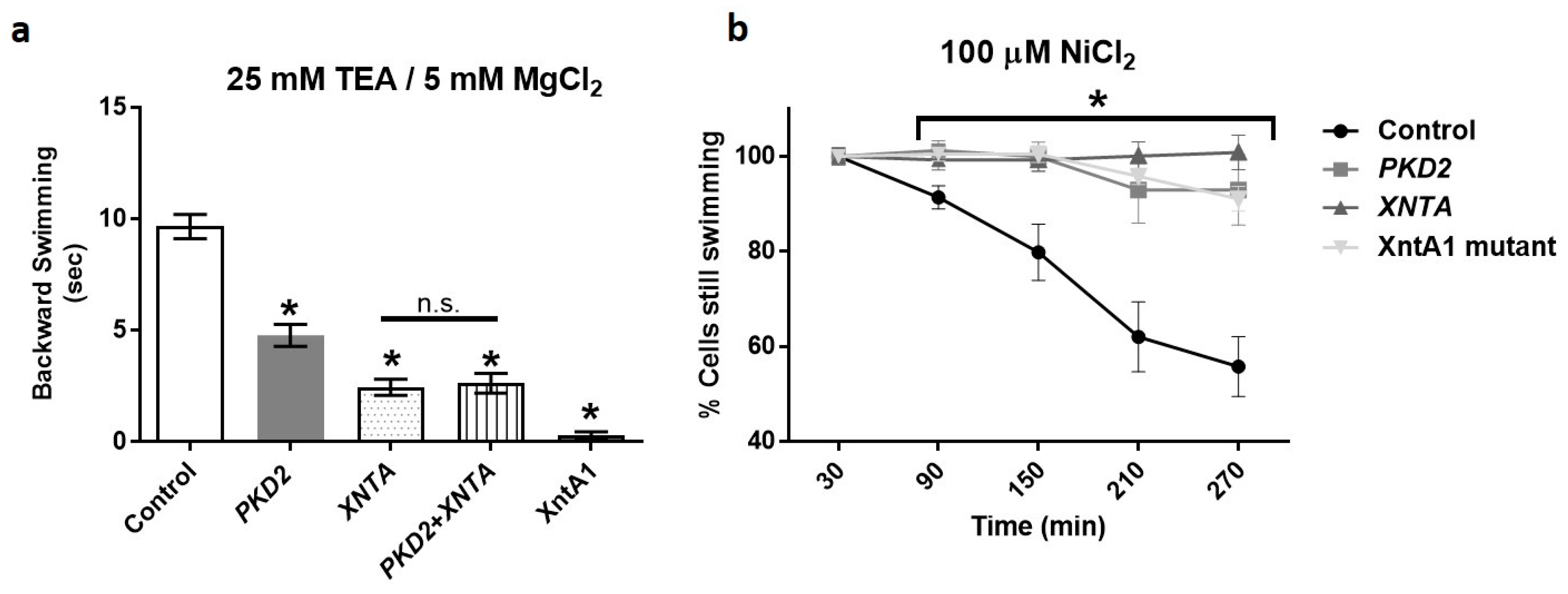
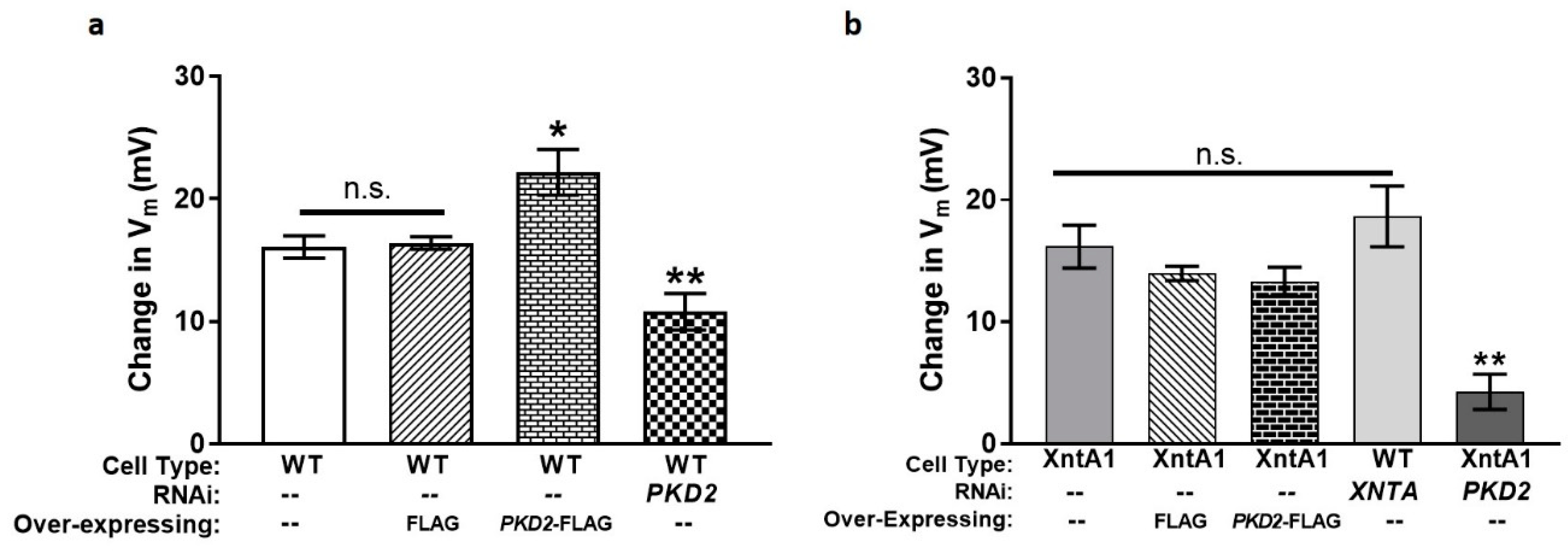
3.1. Depletion of PKD2 Leads to Short Backward Swimming in Mg2+ and Resistance to Heavy Metal Paralysis
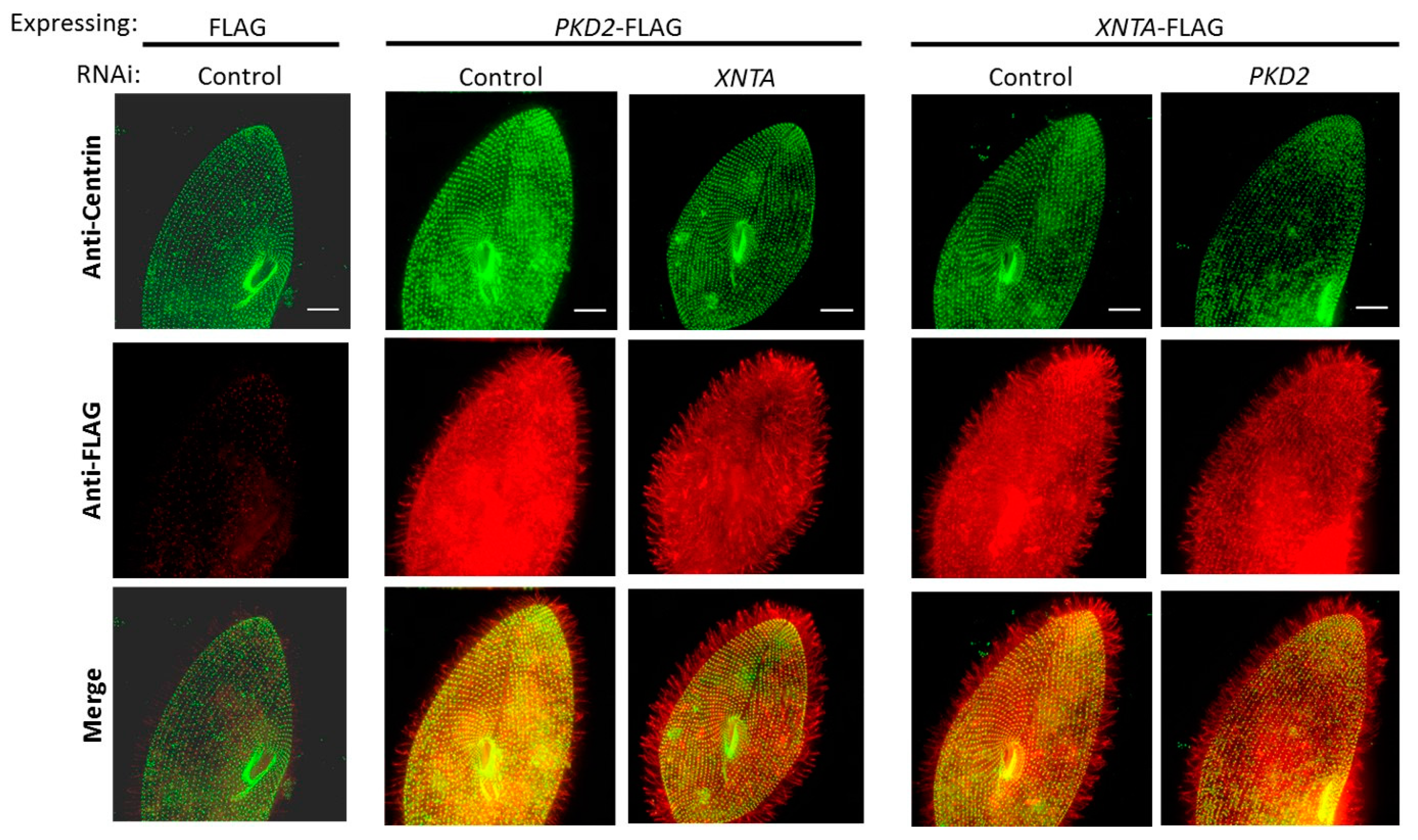
3.2. Pkd2 and XntA Do Not Require Each Other for Trafficking
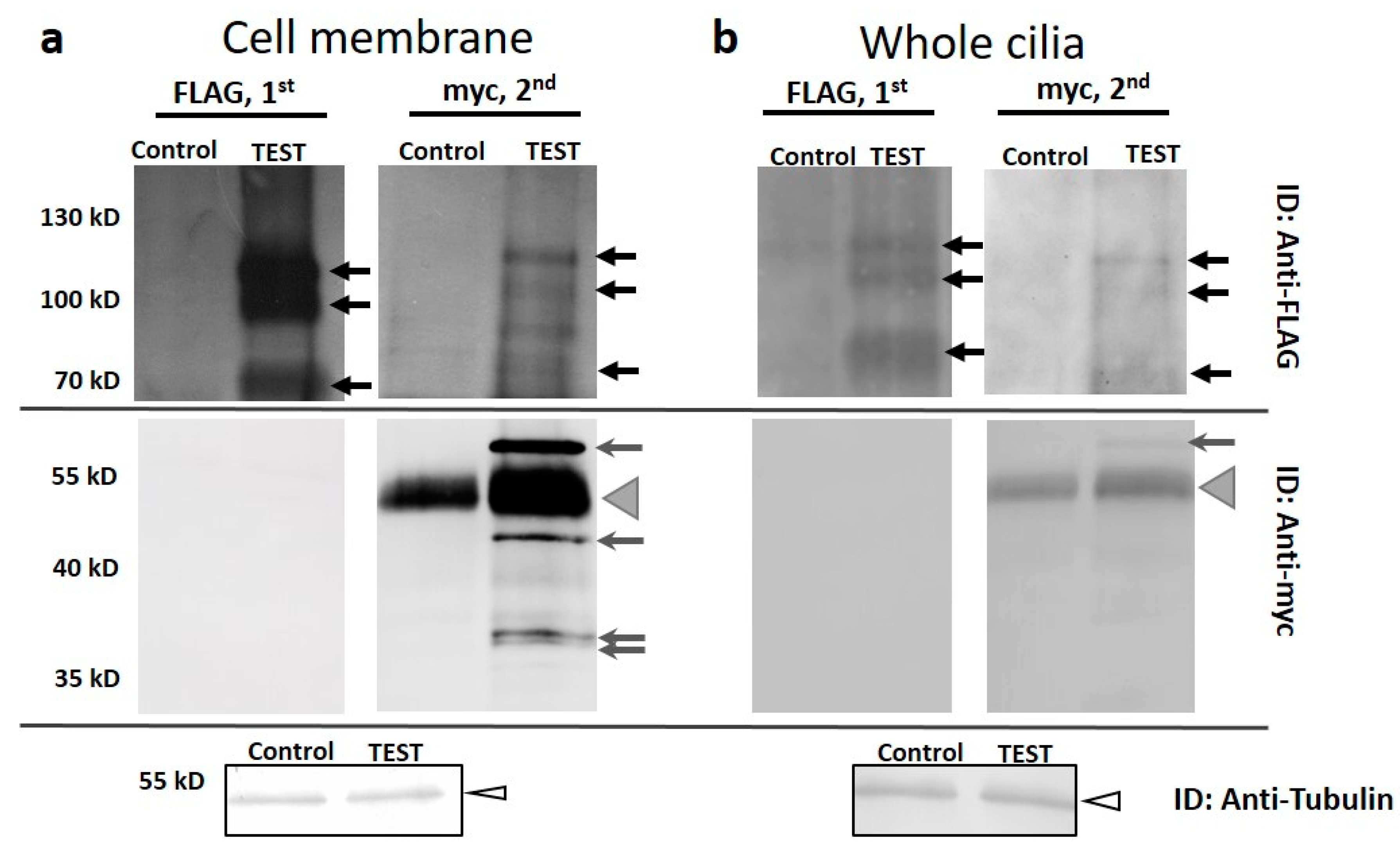
3.3. XntA-myc Co-Immunoprecipitates (co-IPs) Pkd2-FLAG
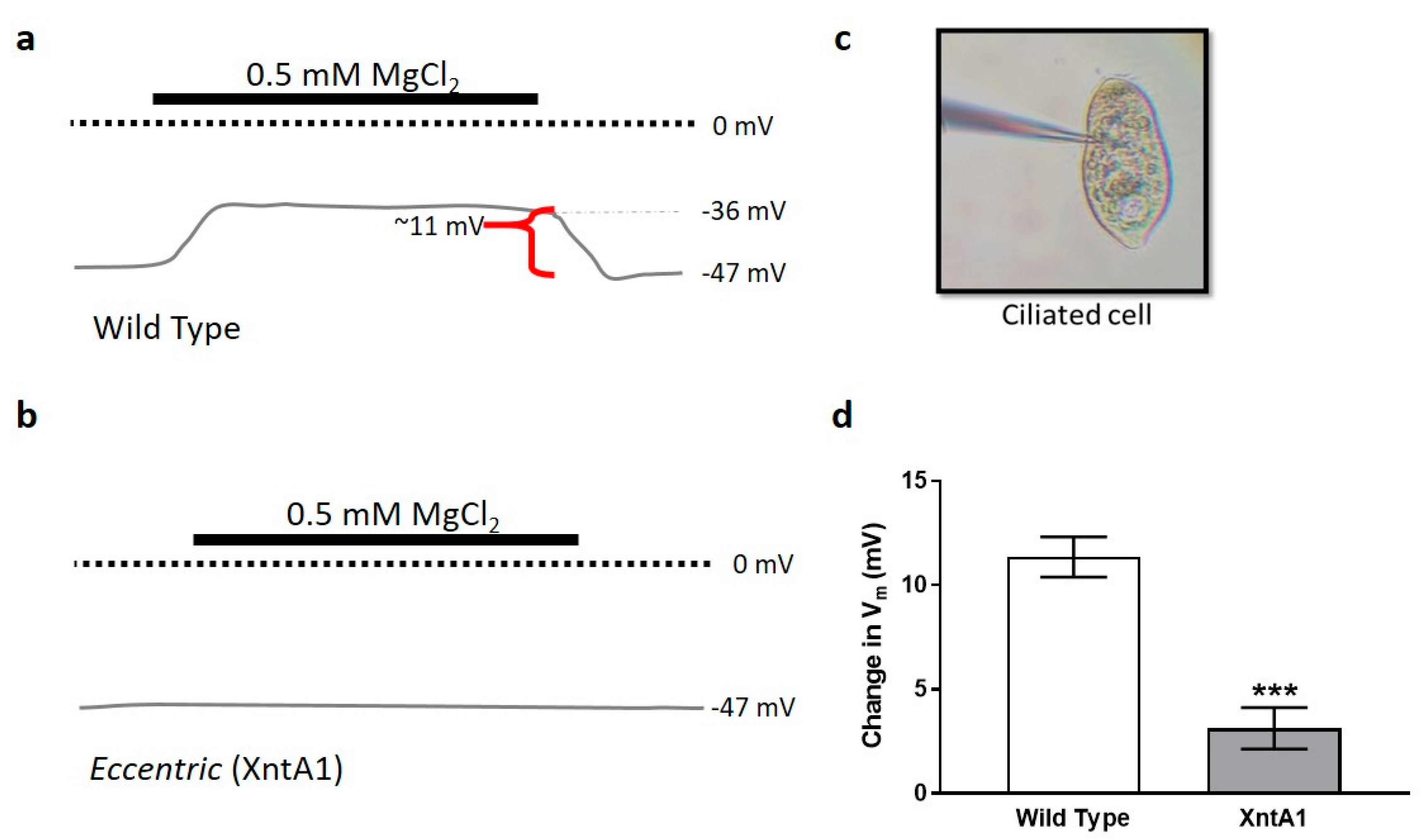
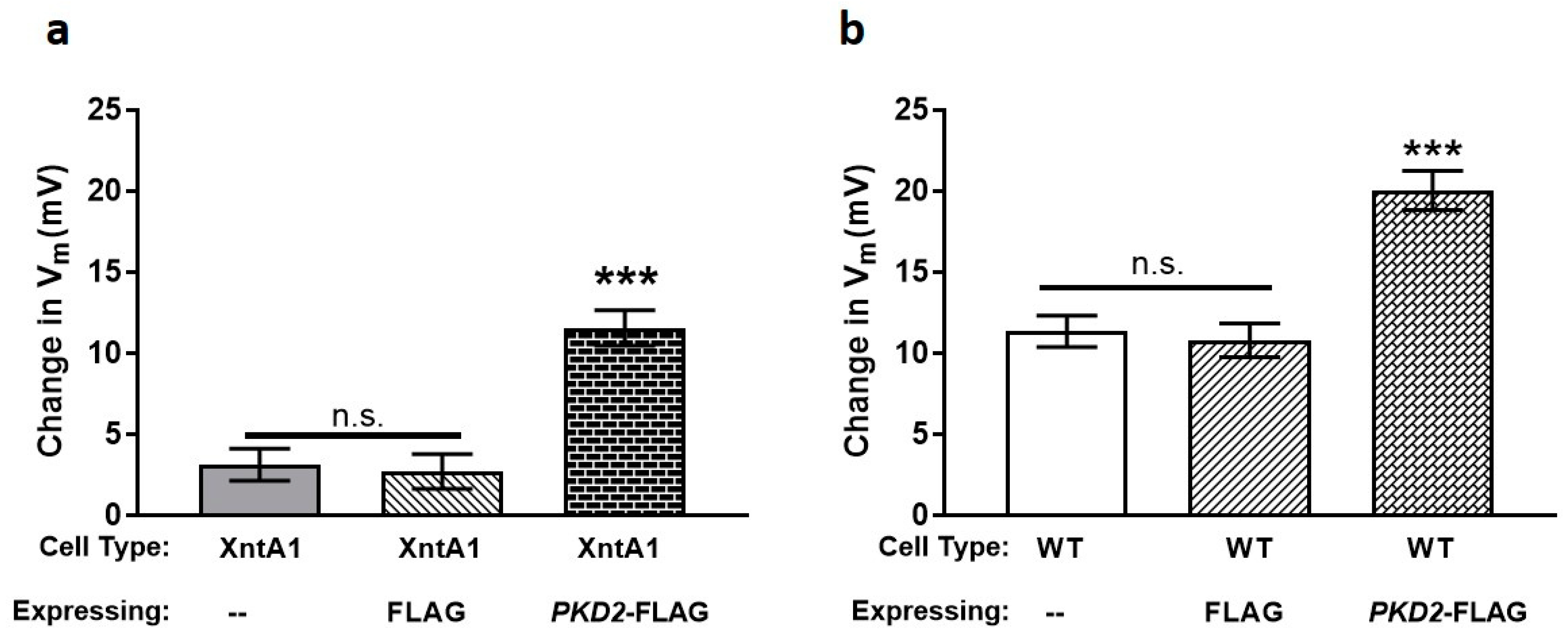
3.4. Wild Type Cells Are Permeable to Mg2+ and XntA1 Mutants Are Not
3.5. The Amount of Pkd2 Is Important to Mg2+ Permeability
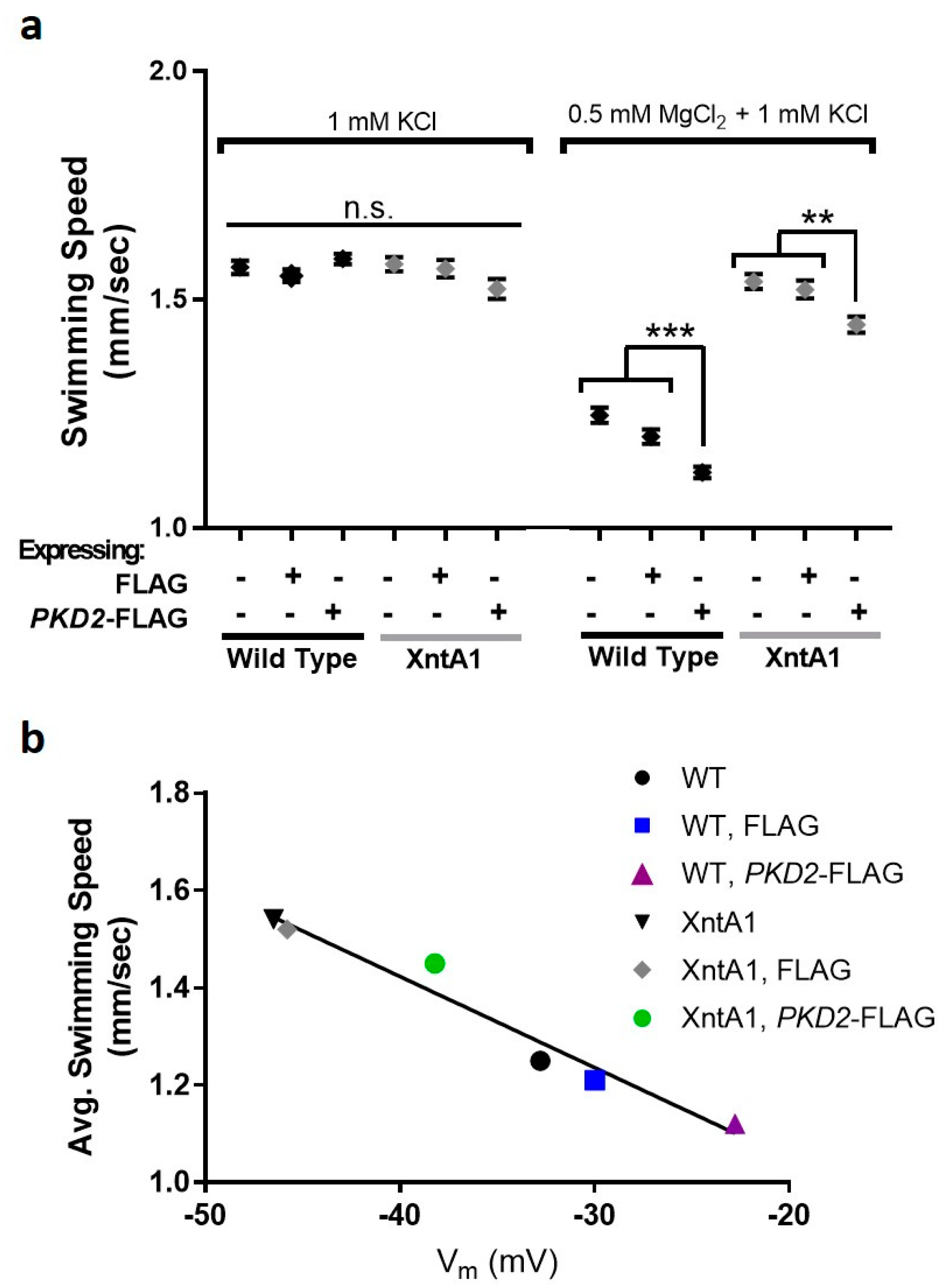
3.6. Swimming Speed Is Slowed by Over-Expression of PKD2
3.7. Deciliated Cells Suggest Pkd2 Plays an Important Role in the Cell Body Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giamarchi, A.; Delmas, P. Activation mechanisms and functional roles of TRPP2 cation channels. In Trp Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, 3. [Google Scholar] [CrossRef] [PubMed]
- Giamarchi, A.; Padilla, F.; Coste, B.; Raoux, M.; Crest, M.; Honore, E.; Delmas, P. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006, 7, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Ward, C.J.; Peral, B.; Aspinwall, R.; Clark, K.; San Millan, J.L.; Gamble, V.; Harris, P.C. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 1995, 10, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Sweet, T.B.; Clapham, D.E. International union of basic and clinical pharmacology. LXXVI. Current progress in the mammalian trp ion channel family. Pharmacol. Rev. 2010, 62, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J. Polycystins and primary cilia: Primers for cell cycle progression. Annu. Rev. Physiol. 2009, 71, 83–113. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Germino, F.J.; Cai, Y.; Zhang, X.; Somlo, S.; Germino, G.G. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 1997, 16, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [CrossRef]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef]
- Delmas, P.; Nauli, S.M.; Li, X.; Coste, B.; Osorio, N.; Crest, M.; Brown, D.A.; Zhou, J. Gating of the polycystin ion channel signaling complex in neurons and kidney cells. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 740–742. [Google Scholar] [CrossRef]
- Tsiokas, L.; Kim, E.; Arnould, T.; Sukhatme, V.P.; Walz, G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. USA 1997, 94, 6965–6970. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Okuhara, D.; Yu, Z.; Tian, X.; Cai, Y.; Shibazaki, S.; Somlo, S. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J. Cell Sci. 2006, 119, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Hoefele, J.; Mayer, K.; Scholz, M.; Klein, H.G. Novel PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transpl. 2011, 26, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D. Polycystic kidney disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.C.; Harris, P.C. Molecular pathogenesis of ADPKD: The polycystin complex gets complex. Kidney Int. 2005, 67, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Kleene, S.J.; Kleene, N.K. The native TRPP2-dependent channel of murine renal primary cilia. Am. J. Physiol. Ren. Physiol. 2017, 312, F96–F108. [Google Scholar] [CrossRef]
- Gonzalez-Perrett, S.; Kim, K.; Ibarra, C.; Damiano, A.E.; Zotta, E.; Batelli, M.; Harris, P.C.; Reisin, I.L.; Arnaout, M.A.; Cantiello, H.F. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc. Natl. Acad. Sci. USA 2001, 98, 1182–1187. [Google Scholar] [CrossRef]
- Anyatonwu, G.I.; Ehrlich, B.E. Organic cation permeation through the channel formed by polycystin-2. J. Biol. Chem. 2005, 280, 29488–29493. [Google Scholar] [CrossRef]
- Koulen, P.; Cai, Y.; Geng, L.; Maeda, Y.; Nishimura, S.; Witzgall, R.; Ehrlich, B.E.; Somlo, S. Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 2002, 4, 191–197. [Google Scholar] [CrossRef]
- Mangolini, A.; de Stephanis, L.; Aguiari, G. Role of calcium in polycystic kidney disease: From signaling to pathology. World J. Nephrol. 2016, 5, 76–83. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hempson, S.J.; Reif, G.A.; Hedge, A.M.; Wallace, D.P. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. JASN 2006, 17, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Nishii, K.; Yoshihara, D.; Kurahashi, H.; Nagaoka, K.; Yamashita, T.; Takahashi, H.; Yamaguchi, T.; Calvet, J.P.; Wallace, D.P. Calcium channel inhibition accelerates polycystic kidney disease progression in the Cy/+ rat. Kidney Int. 2008, 73, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Somlo, S.; Ehrlich, B. Human disease: Calcium signaling in polycystic kidney disease. Curr. Biol. CB 2001, 11, R356–R360. [Google Scholar] [CrossRef]
- Giamarchi, A.; Feng, S.; Rodat-Despoix, L.; Xu, Y.; Bubenshchikova, E.; Newby, L.J.; Hao, J.; Gaudioso, C.; Crest, M.; Lupas, A.N.; et al. A polycystin-2 (TRPP2) dimerization domain essential for the function of heteromeric polycystin complexes. EMBO J. 2010, 29, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Tsiokas, L.; Arnould, T.; Zhu, C.; Kim, E.; Walz, G.; Sukhatme, V.P. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc. Natl. Acad. Sci. USA 1999, 96, 3934–3939. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.X.; Giamarchi, A.; Rodat-Despoix, L.; Padilla, F.; Downs, T.; Tsiokas, L.; Delmas, P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008, 9, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Luo, Y.; Chasan, B.; Gonzalez-Perrett, S.; Montalbetti, N.; Timpanaro, G.A.; Cantero Mdel, R.; Ramos, A.J.; Goldmann, W.H.; Zhou, J.; et al. The multimeric structure of polycystin-2 (TRPP2): Structural-functional correlates of homo-and hetero-multimers with TRPC1. Hum. Mol. Genet. 2009, 18, 1238–1251. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ding, M.; Sours-Brothers, S.; Graham, S.; Ma, R. Mediation of angiotensin II-induced Ca2+ signaling by polycystin 2 in glomerular mesangial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F909–F918. [Google Scholar] [CrossRef]
- Kottgen, M.; Buchholz, B.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T.; et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef]
- Zhang, Z.R.; Chu, W.F.; Song, B.; Gooz, M.; Zhang, J.N.; Yu, C.J.; Jiang, S.; Baldys, A.; Gooz, P.; Steele, S.; et al. TRPP2 and TRPV4 form an EGF-activated calcium permeable channel at the apical membrane of renal collecting duct cells. PLoS ONE 2013, 8, e73424. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Rodat-Despoix, L.; Delmas, P.; Ong, A.C. A single amino acid residue constitutes the third dimerization domain essential for the assembly and function of the tetrameric polycystin-2 (TRPP2) channel. J. Biol. Chem. 2011, 286, 18994–19000. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wright, J.M.; Qian, F.; Germino, G.G.; Guggino, W.B. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J. Biol. Chem. 2005, 280, 41298–41306. [Google Scholar] [CrossRef] [PubMed]
- Anyatonwu, G.I.; Estrada, M.; Tian, X.; Somlo, S.; Ehrlich, B.E. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc. Natl. Acad. Sci. USA 2007, 104, 6454–6459. [Google Scholar] [CrossRef] [PubMed]
- Kobori, T.; Smith, G.D.; Sandford, R.; Edwardson, J.M. The transient receptor potential channels TRPP2 and TRPC1 form a heterotetramer with a 2:2 stoichiometry and an alternating subunit arrangement. J. Biol. Chem. 2009, 284, 35507–35513. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.P.; Smith, G.D.; Sandford, R.N.; Edwardson, J.M. Atomic force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4 heterotetramer. Biophys. J. 2010, 99, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Behn, D.; Bosk, S.; Hoffmeister, H.; Janshoff, A.; Witzgall, R.; Steinem, C. Quantifying the interaction of the C-terminal regions of polycystin-2 and polycystin-1 attached to a lipid bilayer by means of QCM. Biol. Phys. Chem. 2010, 150, 47–53. [Google Scholar] [CrossRef]
- Yu, Y.; Ulbrich, M.H.; Li, M.H.; Buraei, Z.; Chen, X.Z.; Ong, A.C.; Tong, L.; Isacoff, E.Y.; Yang, J. Structural and molecular basis of the assembly of the TRPP2/PKD1 complex. Proc. Natl. Acad. Sci. USA 2009, 106, 11558–11563. [Google Scholar] [CrossRef]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, eaat9819. [Google Scholar] [CrossRef]
- Luo, Y.; Vassilev, P.M.; Li, X.; Kawanabe, Y.; Zhou, J. Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol. Cell. Biol. 2003, 23, 2600–2607. [Google Scholar] [CrossRef]
- Yoshiba, S.; Shiratori, H.; Kuo, I.Y.; Kawasumi, A.; Shinohara, K.; Nonaka, S.; Asai, Y.; Sasaki, G.; Belo, J.A.; Sasaki, H.; et al. Cilia at the node of mouse embryos sense fluid flow for left-right determination via PKD2. Science 2012, 338, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Maeda, Y.; Cedzich, A.; Torres, V.E.; Wu, G.; Hayashi, T.; Mochizuki, T.; Park, J.H.; Witzgall, R.; Somlo, S. Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 1999, 274, 28557–28565. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Qin, H.; Knobel, K.M.; Hu, J.; Rosenbaum, J.L.; Barr, M.M. General and cell-type specific mechanisms target TRPP2/PKD-2 to cilia. Development 2006, 133, 3859–3870. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.M.; DeModena, J.; Braun, D.; Nguyen, C.Q.; Hall, D.H.; Sternberg, P.W. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr. Biol. CB 2001, 11, 1341–1346. [Google Scholar] [CrossRef]
- Huang, K.; Diener, D.R.; Mitchell, A.; Pazour, G.J.; Witman, G.B.; Rosenbaum, J.L. Function and dynamics of PKD2 in Chlamydomonas reinhardtii flagella. J. Cell Biol. 2007, 179, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Kleene, S.J.; Van Houten, J.L. Electrical signaling in motile and primary cilia. Bioscience 2014, 64, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, J. Membrane potential changes during chemokinesis in paramecium. Science 1979, 204, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, J.L.; Yang, W.Q.; Bergeron, A. Chemosensory signal transduction in Paramecium. J. Nutr. 2000, 130, 946S–949S. [Google Scholar] [CrossRef]
- Machemer, H.; Eckert, R. Electrophysiological control of reversed ciliary beating in Paramecium. J. Gen. Physiol. 1973, 61, 572–587. [Google Scholar] [CrossRef]
- Machemer, H.; Ogura, A. Ionic conductances of membranes in ciliated and deciliated Paramecium. J. Physiol. 1979, 296, 49–60. [Google Scholar] [CrossRef]
- Kung, C.; Chang, S.Y.; Satow, Y.; Houten, J.V.; Hansma, H. Genetic dissection of behavior in Paramecium. Science 1975, 188, 898–904. [Google Scholar] [PubMed]
- Valentine, M.S.; Rajendran, A.; Yano, J.; Weeraratne, S.D.; Beisson, J.; Cohen, J.; Koll, F.; Van Houten, J. Paramecium BBS genes are key to presence of channels in cilia. Cilia 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.J.; Kung, C.; Saimi, Y.; Preston, R.R. An exchanger-like protein underlies the large Mg2+ current in Paramecium. Proc. Natl. Acad. Sci. USA 2002, 99, 15717–15722. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.R.; Kung, C. Isolation and characterization of paramecium mutants defective in their response to magnesium. Genetics 1994, 137, 759–769. [Google Scholar] [PubMed]
- Preston, R.R.; Kung, C. Inhibition of Mg2+ current by single-gene mutation in Paramecium. J. Membr. Biol. 1994, 139, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Sasner, J.M.; Van Houten, J.L. Evidence for a Paramecium folate chemoreceptor. Chem. Sens. 1989, 14, 587–595. [Google Scholar] [CrossRef]
- Satow, Y.; Kung, C. A ’TEA+-insensitive’ mutant with increased potassium conductance in Paramecium aurelia. J. Exp. Biol. 1976, 65, 51–63. [Google Scholar]
- ParameciumDB. Available online: https://paramecium.i2bc.paris-saclay.fr/ (accessed on 15 December 2005).
- Picariello, T.; Valentine, M.S.; Yano, J.; Van Houten, J. Reduction of meckelin leads to general loss of cilia, ciliary microtubule misalignment and distorted cell surface organization. Cilia 2014, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- ParameciumDB Off-target. Available online: https://paramecium.i2bc.paris-saclay.fr/cgi/tool/rnai_off_target (accessed on 22 May 2019).
- Wright, M.V.; van Houten, J.L. Characterization of a putative Ca2+-transporting Ca2+-ATPase in the pellicles of Paramecium tetraurelia. Biochim. Biophys. Acta 1990, 1029, 241–251. [Google Scholar] [CrossRef]
- Adoutte, A.; Ramanathan, R.; Lewis, R.M.; Dute, R.R.; Ling, K.Y.; Kung, C.; Nelson, D.L. Biochemical studies of the excitable membrane of Paramecium tetraurelia. III. Proteins of cilia and ciliary membranes. J. Cell Biol. 1980, 84, 717–738. [Google Scholar] [CrossRef]
- Yano, J.; Rajendran, A.; Valentine, M.S.; Saha, M.; Ballif, B.A.; Van Houten, J.L. Proteomic analysis of the cilia membrane of Paramecium tetraurelia. J. Proteom. 2013, 78, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Lodh, S.; Yano, J.; Valentine, M.S.; Van Houten, J.L. Voltage-gated calcium channels of Paramecium cilia. J. Exp. Biol. 2016, 219, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Satow, Y.; Kung, C. A mutant of Paramecium with increased relative resting potassium permeability. J. Neurobiol. 1976, 7, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.R.; Van Houten, J.L. Localization of the chemoreceptive properties of the surface membrane of Paramecium tetraurelia. J. Comp. Physiol. A Sens. Neural Behav. Physiol. 1987, 160, 537–541. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Naitoh, Y. Ionic control of the reversal response of cilia in Paramecium caudatum. A calcium hypothesis. J. Gen. Physiol. 1968, 51, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Hansma, H.G. Sodium uptake and membrane excitation in Paramecium. J. Cell Biol. 1979, 81, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Machemer-Rohnisch, S.; Machemer, H. A Ca paradox: Electric and behavioural responses of Paramecium following changes in external ion concentration. Eur. J. Protistol. 1989, 25, 45–59. [Google Scholar] [CrossRef]
- Naitoh, Y.; Eckert, R.; Friedman, K. A regenerative calcium response in Paramecium. J. Exp. Biol. 1972, 56, 667–681. [Google Scholar] [PubMed]
- Eckert, R. Bioelectric control of ciliary activity. Science 1972, 176, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Brehm, P.; Dunlap, K.; Eckert, R. Calcium-dependent repolarization in Paramecium. J. Physiol. 1978, 274, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.R. Transmembrane Mg2+ currents and intracellular free Mg2+ concentration in Paramecium tetraurelia. J. Membr. Biol. 1998, 164, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Naitoh, Y. Reversal response elicited in nonbeating cilia of Paramecium by membrane depolarizatin. Science 1966, 154, 660–662. [Google Scholar] [CrossRef] [PubMed]
- Gabridge, M.G.; Dougherty, E.P.; Gladd, M.F.; Meccoli, R.A. Effects of heavy metals on structure, function, and metabolism of ciliated respiratory epithelium in vitro. Vitro 1982, 18, 1023–1032. [Google Scholar] [CrossRef]
- Nakaoka, Y.; Machemer, H. Effects of cyclic-nucleotides and intracellular Ca on voltage-activated ciliary beating in Paramecium. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 1990, 166, 401–406. [Google Scholar] [CrossRef]
- Voets, T.; Nilius, B.; Hoefs, S.; van der Kemp, A.W.; Droogmans, G.; Bindels, R.J.; Hoenderop, J.G. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J. Biol. Chem. 2004, 279, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Monteilh-Zoller, M.K.; Hermosura, M.C.; Nadler, M.J.; Scharenberg, A.M.; Penner, R.; Fleig, A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 2003, 121, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Topala, C.N.; Groenestege, W.T.; Thebault, S.; van den Berg, D.; Nilius, B.; Hoenderop, J.G.; Bindels, R.J. Molecular determinants of permeation through the cation channel TRPM6. Cell Calcium 2007, 41, 513–523. [Google Scholar] [CrossRef]
- Schmitz, C.; Dorovkov, M.V.; Zhao, X.; Davenport, B.J.; Ryazanov, A.G.; Perraud, A.L. The channel kinases TRPM6 and TRPM7 are functionally nonredundant. J. Biol. Chem. 2005, 280, 37763–37771. [Google Scholar] [CrossRef]
- Ryazanova, L.V.; Rondon, L.J.; Zierler, S.; Hu, Z.; Galli, J.; Yamaguchi, T.P.; Mazur, A.; Fleig, A.; Ryazanov, A.G. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat. Commun. 2010, 1, 109. [Google Scholar] [CrossRef] [PubMed]
- Chubanov, V.; Gudermann, T.; Schlingmann, K.P. Essential role for TRPM6 in epithelial magnesium transport and body magnesium homeostasis. Pflug. Arch. Eur. J. Physiol. 2005, 451, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Anyatonwu, G.; Okuhara, D.; Lee, K.B.; Yu, Z.; Onoe, T.; Mei, C.L.; Qian, Q.; Geng, L.; Wiztgall, R.; et al. Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser812. J. Biol. Chem. 2004, 279, 19987–19995. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.M.; Gonzalez-Perrett, S.; Essafi, M.; Timpanaro, G.A.; Montalbetti, N.; Arnaout, M.A.; Cantiello, H.F. Polycystin-1 activates and stabilizes the polycystin-2 channel. J. Biol. Chem. 2003, 278, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Montalbetti, N.; Shen, P.Y.; Dai, X.Q.; Cheeseman, C.I.; Karpinski, E.; Wu, G.; Cantiello, H.F.; Chen, X.Z. Alpha-actinin associates with polycystin-2 and regulates its channel activity. Hum. Mol. Genet. 2005, 14, 1587–1603. [Google Scholar] [CrossRef] [PubMed]
- Roitbak, T.; Surviladze, Z.; Tikkanen, R.; Wandinger-Ness, A. A polycystin multiprotein complex constitutes a cholesterol-containing signalling microdomain in human kidney epithelia. Biochem. J. 2005, 392, 29–38. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentine, M.S.; Yano, J.; Van Houten, J. A Novel Role for Polycystin-2 (Pkd2) in P. tetraurelia as a Probable Mg2+ Channel Necessary for Mg2+-Induced Behavior. Genes 2019, 10, 455. https://doi.org/10.3390/genes10060455
Valentine MS, Yano J, Van Houten J. A Novel Role for Polycystin-2 (Pkd2) in P. tetraurelia as a Probable Mg2+ Channel Necessary for Mg2+-Induced Behavior. Genes. 2019; 10(6):455. https://doi.org/10.3390/genes10060455
Chicago/Turabian StyleValentine, Megan S., Junji Yano, and Judith Van Houten. 2019. "A Novel Role for Polycystin-2 (Pkd2) in P. tetraurelia as a Probable Mg2+ Channel Necessary for Mg2+-Induced Behavior" Genes 10, no. 6: 455. https://doi.org/10.3390/genes10060455
APA StyleValentine, M. S., Yano, J., & Van Houten, J. (2019). A Novel Role for Polycystin-2 (Pkd2) in P. tetraurelia as a Probable Mg2+ Channel Necessary for Mg2+-Induced Behavior. Genes, 10(6), 455. https://doi.org/10.3390/genes10060455