The Genome of Blue-Capped Cordon-Bleu Uncovers Hidden Diversity of LTR Retrotransposons in Zebra Finch


 ,
,
Abstract
1. Introduction
2. Materials and Methods
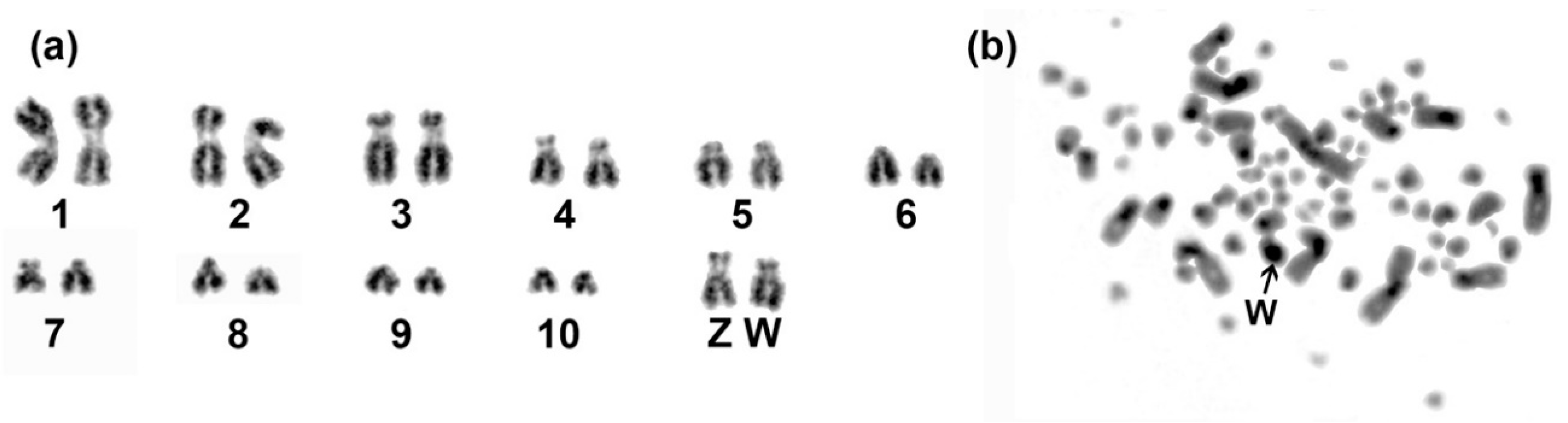
2.1. Sequencing, Genome Assembly and Karyotyping
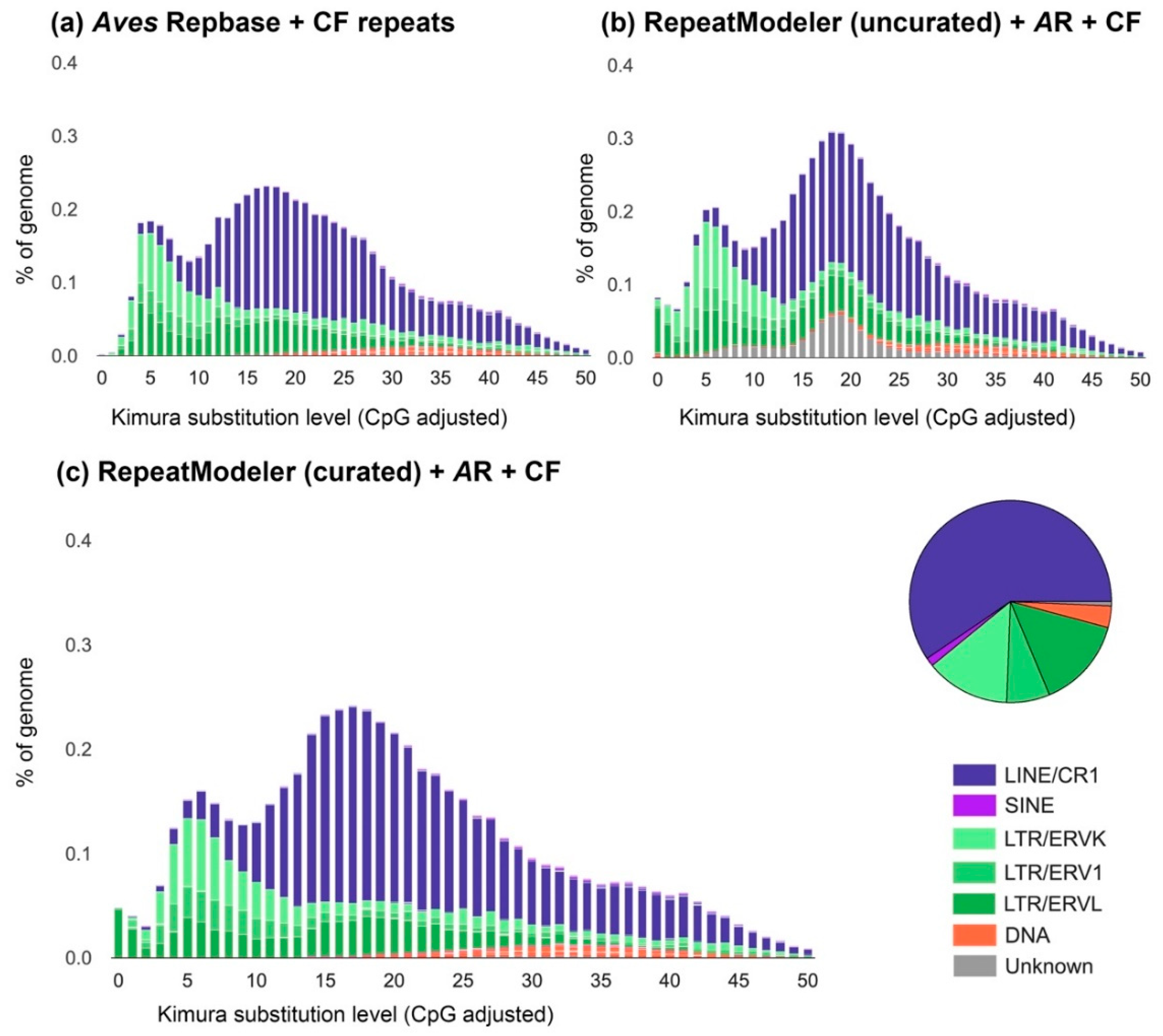
2.2. Computational and Manual Curation of Transposable Elements
2.3. Data Analysis
2.4. Data Deposition
3. Results
3.1. Genome Assembly and Karyotype of Blue-Capped Cordon-Bleu
3.2. The Transposable Element Landscape of Blue-Capped Cordon-Bleu
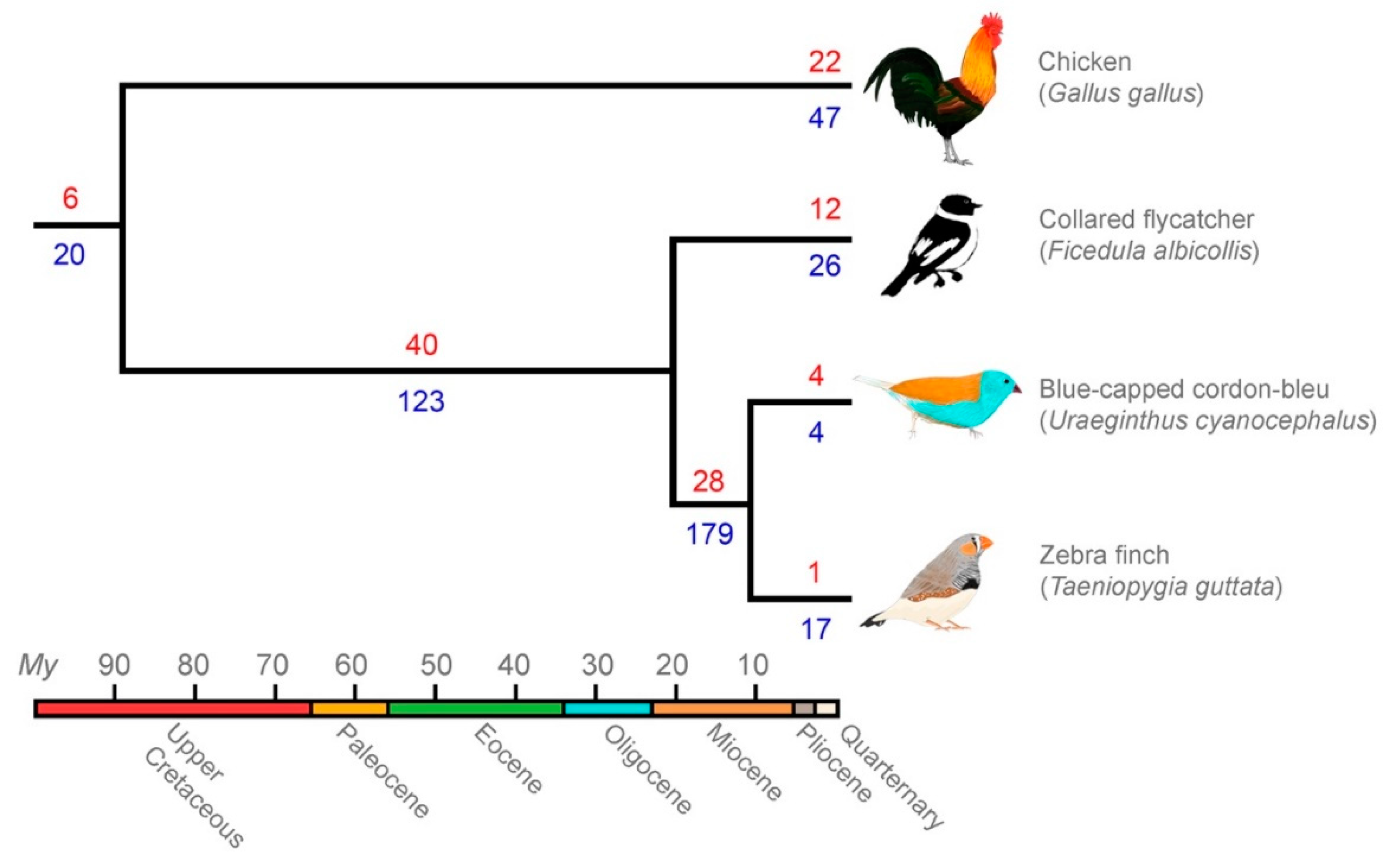
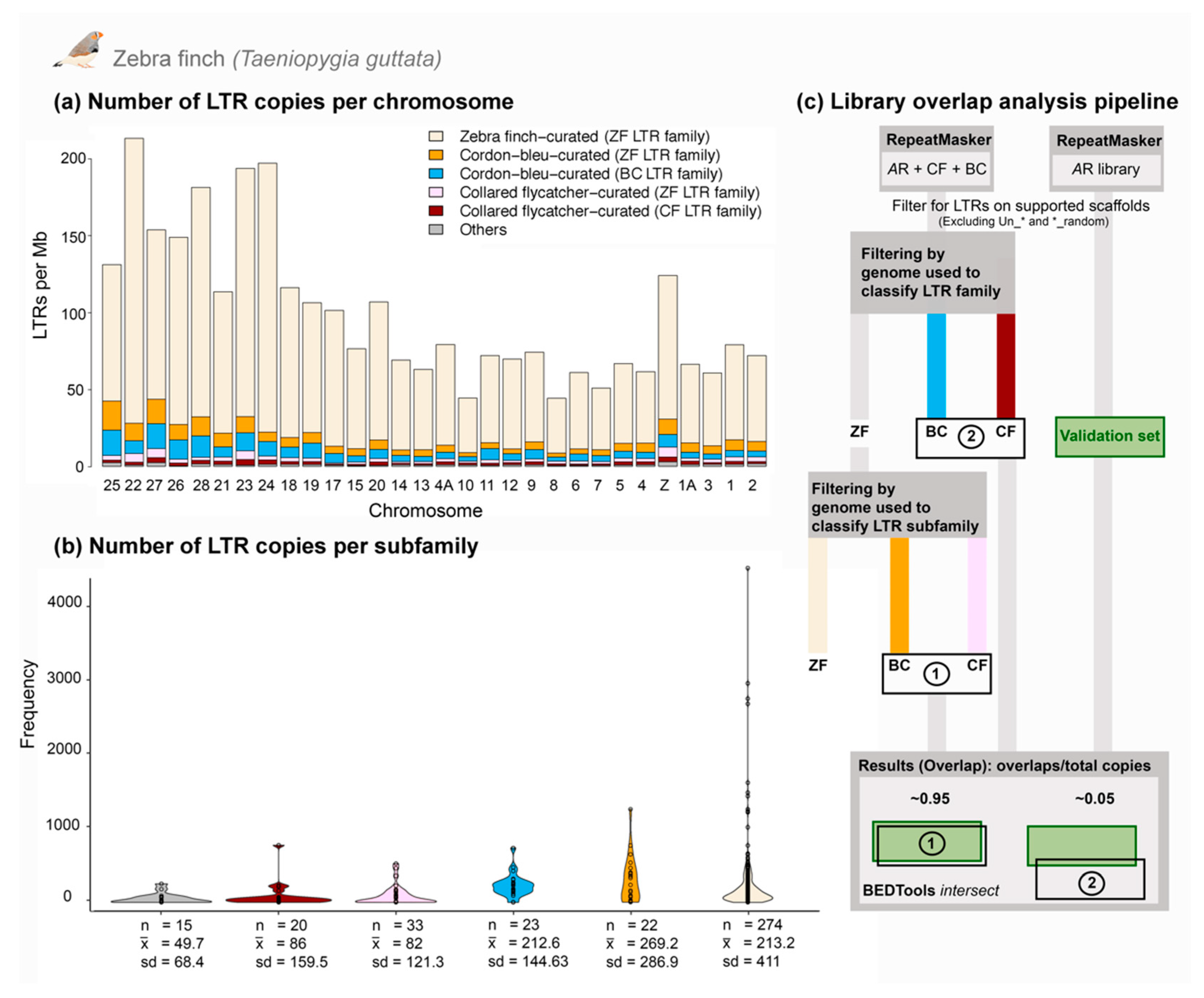
3.3. Comparative Genomics Revealed Extensive Shared Diversity of LTRs among Estrildid Finches
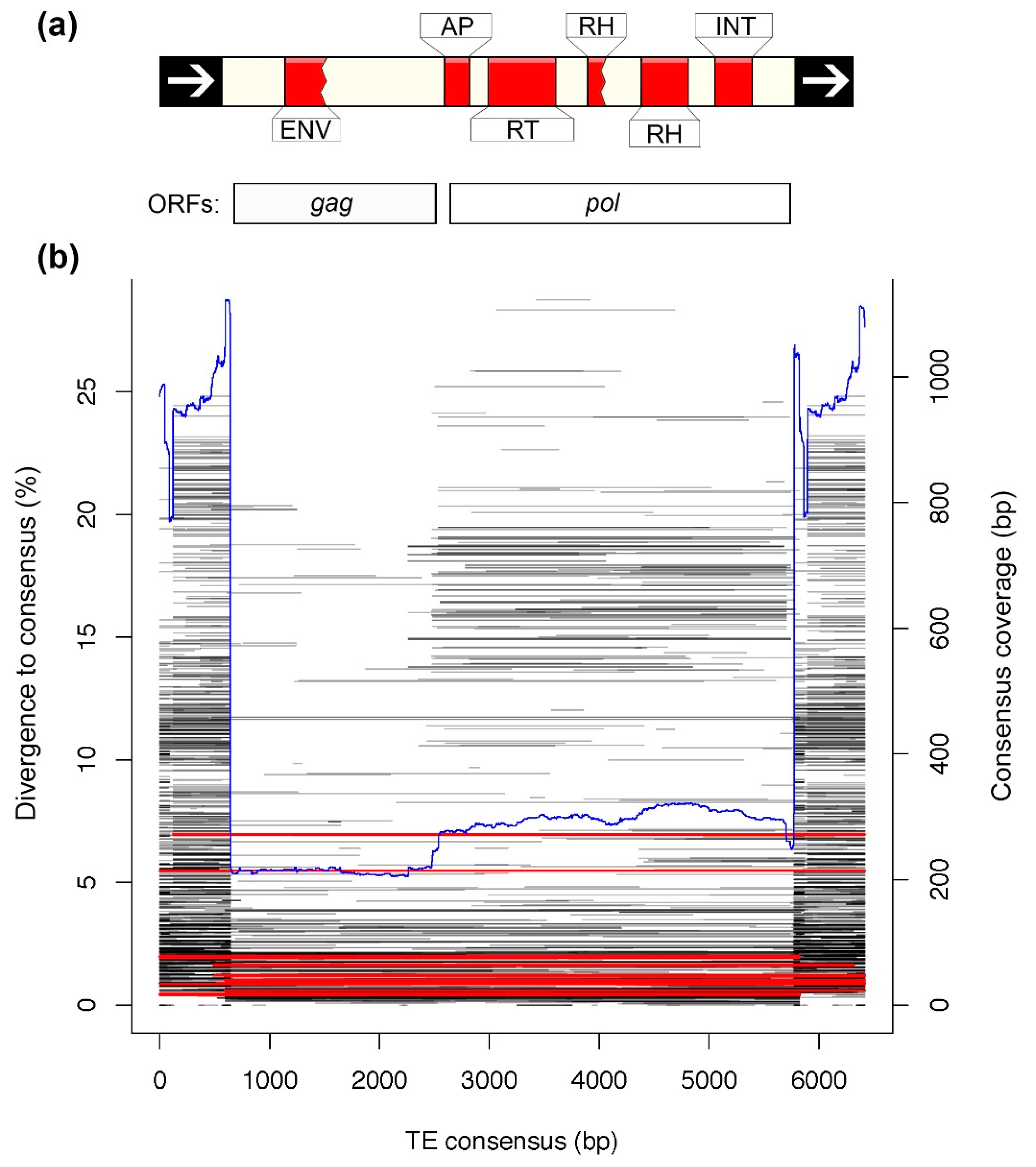
3.4. Analysis of a Recently Active TE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gregory, T.R. Animal Genome Size Database. Available online: http://www.genomesize.com (accessed on 7 December 2018).
- Kapusta, A.; Suh, A. Evolution of bird genomes—A transposon’s-eye view. Ann. N. Y. Acad. Sci. 2016, 1389, 164–185. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef]
- Hughes, A.L.; Hughes, M.K. Small genomes for better flyers. Nature 1995, 377, 391. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R. A Bird’s-Eye View of the C-Value Enigma: Genome Size, Cell Size, and Metabolic Rate in the Class Aves. Evolution 2002, 56, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J. Cell Sci. 1978, 34, 247–278. [Google Scholar]
- Burton, D.W.; Bickham, J.W.; Genoways, H.H. Flow-Cytometric Analyses of Nuclear DNA Content in Four Families of Neotropical Bats. Evolution 1989, 43, 756–765. [Google Scholar] [CrossRef]
- Van den Bussche, R.A.; Longmire, J.L.; Baker, R.J. How bats achieve a small C-value: Frequency of repetitive DNA in Macrotus. Mamm. Genome 1995, 6, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.; Peona, V.; Guichard, E.; Taccioli, C.; Boattini, A. Transposable Elements Activity is Positively Related to Rate of Speciation in Mammals. J. Mol. Evol. 2018, 86, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Lazar, N.H.; Nevonen, K.A.; O’Connell, B.; McCann, C.; O’Neill, R.J.; Green, R.E.; Meyer, T.J.; Okhovat, M.; Carbone, L. Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res. 2018, 28, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.E.; Kolodner, R.D. A Genetic and Structural Study of Genome Rearrangements Mediated by High Copy Repeat Ty1 Elements. PLoS Genet. 2011, 7, e1002089. [Google Scholar] [CrossRef] [PubMed]
- Farré, M.; Narayan, J.; Slavov, G.T.; Damas, J.; Auvil, L.; Li, C.; Jarvis, E.D.; Burt, D.W.; Griffin, D.K.; Larkin, D.M. Novel Insights into Chromosome Evolution in Birds, Archosaurs, and Reptiles. Genome Biol. Evol. 2016, 8, 2442–2451. [Google Scholar] [CrossRef]
- Wolf, J.B.W.; Ellegren, H. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet. 2017, 18, 87–100. [Google Scholar] [CrossRef]
- Hooper, D.M.; Price, T.D. Rates of karyotypic evolution in Estrildid finches differ between island and continental clades. Evolution 2015, 69, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Romanov, M.N.; Farré, M.; Lithgow, P.E.; Fowler, K.E.; Skinner, B.M.; O’Connor, R.; Fonseka, G.; Backström, N.; Matsuda, Y.; Nishida, C.; et al. Reconstruction of gross avian genome structure, organization and evolution suggests that the chicken lineage most closely resembles the dinosaur avian ancestor. BMC Genom. 2014, 15, 1060. [Google Scholar] [CrossRef]
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S.; et al. The genome of a songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef]
- Garfinkel, D.J.; Boeke, J.D.; Fink, G.R. Ty element transposition: Reverse transcriptase and virus-like particles. Cell 1985, 42, 507–517. [Google Scholar] [CrossRef]
- Ota, N.; Gahr, M.; Soma, M. Tap dancing birds: The multimodal mutual courtship display of males and females in a socially monogamous songbird. Sci. Rep. 2015, 5, 16614. [Google Scholar] [CrossRef]
- Ota, N.; Gahr, M.; Soma, M. Songbird tap dancing produces non-vocal sounds. Bioacoustics 2017, 26, 161–168. [Google Scholar] [CrossRef]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- Suh, A.; Smeds, L.; Ellegren, H. Abundant recent activity of retrovirus-like retrotransposons within and among flycatcher species implies a rich source of structural variation in songbird genomes. Mol. Ecol. 2017, 27, 99–111. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A.; et al. Resolving the Full Spectrum of Human Genome Variation using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef]
- Kinsella, C.M.; Ruiz-Ruano, F.J.; Dion-Côté, A.-M.; Charles, A.J.; Gossmann, T.I.; Cabrero, J.; Kappei, D.; Hemmings, N.; Simons, M.J.P.; Camacho, J.P.M.; et al. Programmed DNA elimination of germline development genes in songbirds. bioRxiv 2018, 444364. [Google Scholar]
- Torgasheva, A.A.; Malinovskaya, L.P.; Zadesenets, K.S.; Karamysheva, T.V.; Kizilova, E.A.; Pristyazhnyuk, I.E.; Shnaider, E.P.; Volodkina, V.A.; Saifutdinova, A.F.; Galkina, S.A.; et al. Germline-Restricted Chromosome (GRC) is Widespread among Songbirds. bioRxiv 2018, 414276. [Google Scholar]
- Biederman, M.K.; Nelson, M.M.; Asalone, K.C.; Pedersen, A.L.; Saldanha, C.J.; Bracht, J.R. Discovery of the First Germline-Restricted Gene by Subtractive Transcriptomic Analysis in the Zebra Finch, Taeniopygia guttata. Curr. Biol. 2018, 28, 1620–1627. [Google Scholar] [CrossRef]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2013, 2, 10. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Sumner, A.T. A simple technique for demonstrating centromeric heterochromatin. Exp. Cell Res. 1972, 75, 304–306. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeuchi, T.; Makino, S. A feather pulp culture technique for avian chromosomes, with notes on the chromosomes of the peafowl and the ostrich. Experientia 1968, 24, 1292–1293. [Google Scholar] [CrossRef]
- Dos Santos, M.D.S.; Kretschmer, R.; Frankl-Vilches, C.; Bakker, A.; Gahr, M.; Ferguson-Smith, M.A.; De Oliveira, E.H. Comparative Cytogenetics between Two Important Songbird, Models: The Zebra Finch and the Canary. PLoS ONE 2017, 12, e0170997. [Google Scholar]
- de Oliveira Furo, I.; Kretschmer, R.; dos Santos, M.S.; de Lima Carvalho, C.A.; Gunski, R.J.; O’Brien, P.C.; Ferguson-Smith, M.A.; Cioffi, M.B.; de Oliveira, E.H. Chromosomal Mapping of Repetitive DNAs in Myiopsitta monachus and Amazona aestiva (Psittaciformes, Psittacidae) with Emphasis on the Sex Chromosomes. Cytogenet. Genome Res. 2017, 151, 151–160. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R. RepeatModeler. Available online: http://www.repeatmasker.org/RepeatModeler/ (accessed on 21 August 2018).
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0 2013–2015. Available online: http://www.repeatmasker.org (accessed on 6 December 2018).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Advanced Consensus Maker. Available online: https://www.hiv.lanl.gov/content/sequence/CONSENSUS/AdvCon.html (accessed on 30 October 2018).
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Suh, A. The Specific Requirements for CR1 Retrotransposition Explain the Scarcity of Retrogenes in Birds. J. Mol. Evol. 2015, 81, 18–20. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 30 December 2017).
- Suh, A.; Churakov, G.; Ramakodi, M.P.; Platt, R.N.; Jurka, J.; Kojima, K.K.; Caballero, J.; Smit, A.F.; Vliet, K.A.; Hoffmann, F.G.; et al. Multiple Lineages of Ancient CR1 Retroposons Shaped the Early Genome Evolution of Amniotes. Genome Biol. Evol. 2015, 7, 205–217. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018, 46, D762–D769. [Google Scholar] [PubMed]
- Smit, A.; Hubley, R.; Wheeler, T. dfam_consensus. Available online: https://dfam.org/home (accessed on 10 April 2019).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Church, D.M.; Federhen, S.; Lash, A.E.; Madden, T.L.; Pontius, J.U.; Schuler, G.D.; Schriml, L.M.; Sequeira, E.; Tatusova, T.A.; et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003, 31, 28–33. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Goubert, C. consensus2genome. Available online: https://github.com/clemgoub/consensus2genome (accessed on 8 September 2018).
- Christidis, L. Chromosomal evolution within the family Estrildidae (Aves) I. The Poephilae. Genetica 1986, 71, 81–97. [Google Scholar] [CrossRef]
- Ladjali-Mohammedi, K.; Bitgood, J.J.; Tixier-Boichard, M.; de Leon, F.A.P. International System for Standardized Avian Karyotypes (ISSAK): Standardized banded karyotypes of the domestic fowl (Gallus domesticus). Cytogenet. Genome Res. 1999, 86, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Moyle, R.G.; Oliveros, C.H.; Andersen, M.J.; Hosner, P.A.; Benz, B.W.; Manthey, J.D.; Travers, S.L.; Brown, R.M.; Faircloth, B.C. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat. Commun. 2016, 7, 12709. [Google Scholar] [CrossRef]
- Arkhipova, I.R. Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mob. DNA 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S. Ribonuclease H evolution in retrotransposable elements. Cytogenet. Genome Res. 2005, 110, 392–401. [Google Scholar] [CrossRef]
- Schmidt, J.; Klupp, B.G.; Karger, A.; Mettenleiter, T.C. Adaptability in herpesviruses: Glycoprotein D-independent infectivity of pseudorabies virus. J. Virol. 1997, 71, 17–24. [Google Scholar]
- Churchill, A.E.; Biggs, P.M. Agent of Marek’s Disease in Tissue Culture. Nature 1967, 215, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Wu, P.; Sui, D.; Ren, D.; Kamil, J.; Kung, H.J.; Witter, R.L. The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc. Natl. Acad. Sci. USA 2000, 97, 6091–6096. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Henikoff, S.; Eickbush, T.H. Poised for Contagion: Evolutionary Origins of the Infectious Abilities of Invertebrate Retroviruses. Genome Res. 2000, 10, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Devos, K.M.; Brown, J.K.M.; Bennetzen, J.L. Genome Size Reduction through Illegitimate Recombination Counteracts Genome Expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef]
- Schweizer, M.; Warmuth, V.; Kakhki, N.A.; Aliabadian, M.; Förschler, M.; Shirihai, H.; Suh, A.; Burri, R. Parallel plumage colour evolution and introgressive hybridization in wheatears. J. Evol. Biol. 2019, 32, 100–110. [Google Scholar] [CrossRef]
- Toomey, M.B.; Marques, C.I.; Andrade, P.; Araújo, P.M.; Sabatino, S.; Gazda, M.A.; Afonso, S.; Lopes, R.J.; Corbo, J.C.; Carneiro, M. A non-coding region near Follistatin controls head colour polymorphism in the Gouldian finch. Proc. R. Soc. B Biol. Sci. 2018, 285, 20181788. [Google Scholar] [CrossRef]
- Mason, A.S.; Fulton, J.E.; Hocking, P.M.; Burt, D.W. A new look at the LTR retrotransposon content of the chicken genome. BMC Genom. 2016, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, M.D.; Balakrishnan, C.N.; Payne, R.B. Clade-Limited Colonization in Brood Parasitic Finches (Vidua spp.). Syst. Biol. 2004, 53, 140–153. [Google Scholar] [CrossRef]
- Löber, U.; Hobbs, M.; Dayaram, A.; Tsangaras, K.; Jones, K.; Alquezar-Planas, D.E.; Ishida, Y.; Meers, J.; Mayer, J.; Quedenau, C.; et al. Degradation and remobilization of endogenous retroviruses by recombination during the earliest stages of a germ-line invasion. Proc. Natl. Acad. Sci. USA 2018, 115, 8609–8614. [Google Scholar] [CrossRef] [PubMed]
- Stoye, J.P. Koala retrovirus: A genome invasion in real time. Genome Biol. 2006, 7, 241. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simmons, G.S.; Young, P.R.; Hanger, J.J.; Jones, K.; Clarke, D.; McKee, J.J.; Meers, J. Prevalence of koala retrovirus in geographically diverse populations in Australia. Aust. Vet. J. 2012, 90, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, R.; Meers, J.; Hanger, J.; Young, P. Real-time reverse transcriptase PCR for the endogenous koala retrovirus reveals an association between plasma viral load and neoplastic disease in koalas. J. Gen. Virol. 2005, 86, 783–787. [Google Scholar] [CrossRef]
- Platt, R.N.; Blanco-Berdugo, L.; Ray, D.A. Accurate Transposable Element Annotation Is Vital When Analyzing New Genome Assemblies. Genome Biol. Evol. 2016, 8, 403–410. [Google Scholar] [CrossRef]
- Knisbacher, B.A.; Levanon, E.Y. DNA Editing of LTR Retrotransposons Reveals the Impact of APOBECs on Vertebrate Genomes. Mol. Biol. Evol. 2016, 33, 554–567. [Google Scholar] [CrossRef]
- Peona, V.; Weissensteiner, M.H.; Suh, A. How complete are “complete” genome assemblies?—An avian perspective. Mol. Ecol. Resour. 2018, 18, 1188–1195. [Google Scholar] [CrossRef]
- Aswad, A.; Katzourakis, A. Convergent capture of retroviral superantigens by mammalian herpesviruses. Nat. Commun. 2015, 6, 8299. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistic | Quantity |
|---|---|
| Assembly size | 1099.6 Mb |
| “N” nucleotides | 105.6 Mb |
| Weighted mean molecule length | 42.4 kb |
| Number of reads | 254.2 million |
| Scaffolds | 26,389 |
| Scaffold N50 | 10.9 Mb |
| Contigs | 51,469 |
| Contig N50 | 66.3 kb |
| BUSCO (complete) | 90.1% |
| BUSCO (fragmented) | 5.9% |
| BUSCO (missing) | 4% |
| Repeat Type | Copies | Total bp | % of Genome |
|---|---|---|---|
| SINE | 7163 | 852,236 | 0.08 |
| LINE | 132,734 | 37,876,706 | 3.44 |
| LTR | 61,457 | 29,437,443 | 2.68 |
| DNA | 14,100 | 2,195,734 | 0.20 |
| Unclassified | 2367 | 416,198 | 0.04 |
| Total interspersed repeats | 217,821 | 70,778,317 | 6.44 |
| Small RNA | 1479 | 199,270 | 0.02 |
| Satellites | 1960 | 581,825 | 0.05 |
| Simple repeats | 211,440 | 9,408,016 | 0.86 |
| Low complexity | 43,325 | 2,238,772 | 0.20 |
| Total tandem repeats | 258,204 | 12,427,883 | 1.17 |
| Total repeats | 746,059 | 83,206,200 | 7.61 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boman, J.; Frankl-Vilches, C.; da Silva dos Santos, M.; de Oliveira, E.H.C.; Gahr, M.; Suh, A. The Genome of Blue-Capped Cordon-Bleu Uncovers Hidden Diversity of LTR Retrotransposons in Zebra Finch. Genes 2019, 10, 301. https://doi.org/10.3390/genes10040301
Boman J, Frankl-Vilches C, da Silva dos Santos M, de Oliveira EHC, Gahr M, Suh A. The Genome of Blue-Capped Cordon-Bleu Uncovers Hidden Diversity of LTR Retrotransposons in Zebra Finch. Genes. 2019; 10(4):301. https://doi.org/10.3390/genes10040301
Chicago/Turabian StyleBoman, Jesper, Carolina Frankl-Vilches, Michelly da Silva dos Santos, Edivaldo H. C. de Oliveira, Manfred Gahr, and Alexander Suh. 2019. "The Genome of Blue-Capped Cordon-Bleu Uncovers Hidden Diversity of LTR Retrotransposons in Zebra Finch" Genes 10, no. 4: 301. https://doi.org/10.3390/genes10040301
APA StyleBoman, J., Frankl-Vilches, C., da Silva dos Santos, M., de Oliveira, E. H. C., Gahr, M., & Suh, A. (2019). The Genome of Blue-Capped Cordon-Bleu Uncovers Hidden Diversity of LTR Retrotransposons in Zebra Finch. Genes, 10(4), 301. https://doi.org/10.3390/genes10040301