Genome Assembly and Annotation of the Trichoplusia ni Tni-FNL Insect Cell Line Enabled by Long-Read Technologies

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions
2.2. PacBio Library Preparation and Sequencing
2.3. Bionano Optical Mapping
2.4. 10X Genomics Linked Reads Sequencing
2.5. Transcriptome Sequencing of Tni-FNL Cell Line
2.6. Propidium Iodide (PI) Staining for Ploidy Determination
2.7. Flow Cytometry Analysis
2.8. Karyotype Analysis
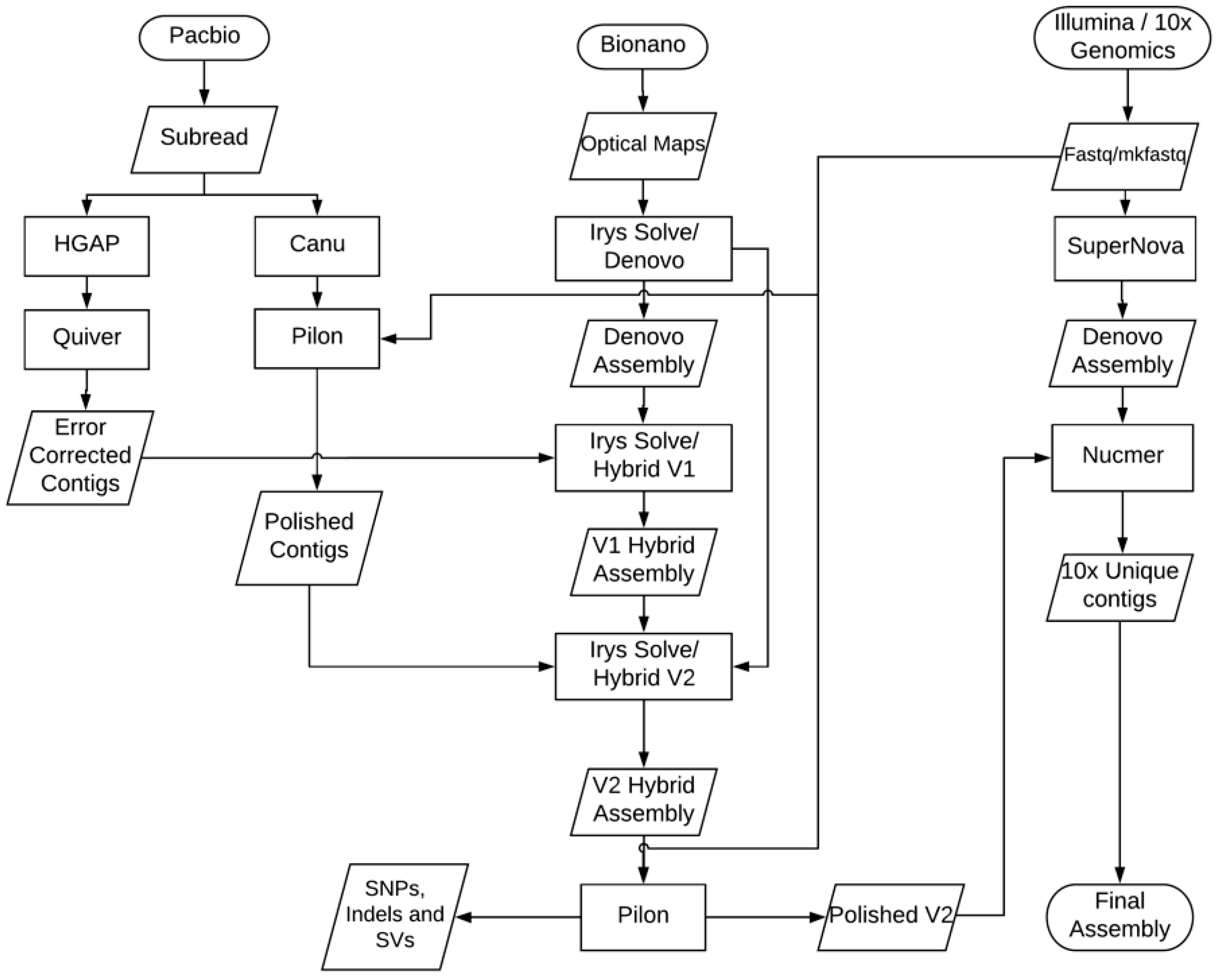
2.9. NGS De Novo Assembly Methods
2.10. Bionano De Novo Assembly
2.11. Bionano Hybrid Assembly
2.12. Assembly Error Correction
2.13. Transcriptome Assembly
2.14. Gene Predictions and Repeat Annotations
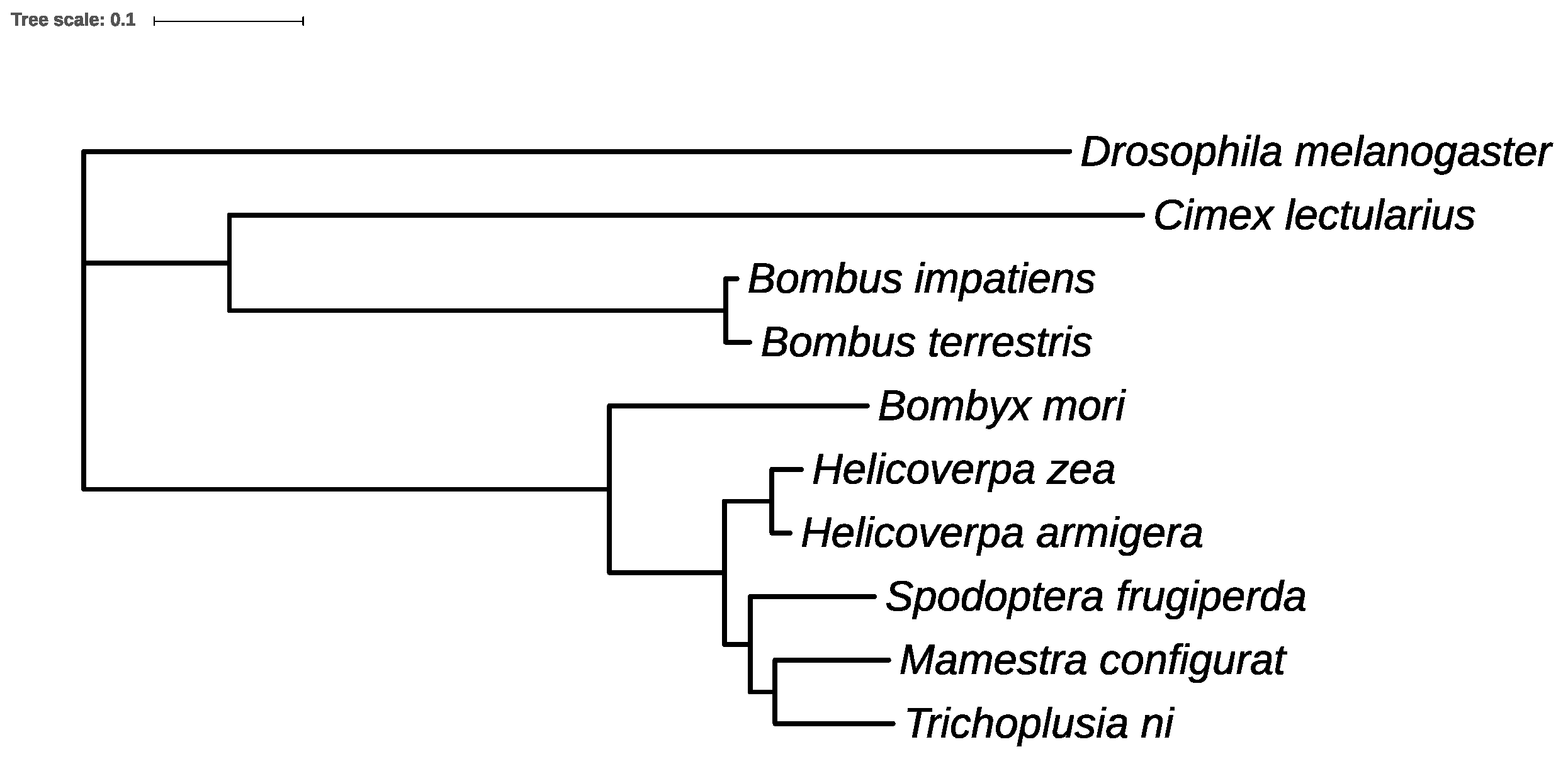
2.15. Phylogeny Analysis for Ten Insect Genomes
3. Results
3.1. Genome Sequencing and Assembly
3.2. Improved Genome Assembly Using Bionano Optical Mapping Data
3.3. Assembly Conflict Resolution
3.4. Error Correction of Genome Assembly
3.5. Genome Size Estimation Based on K-Mers
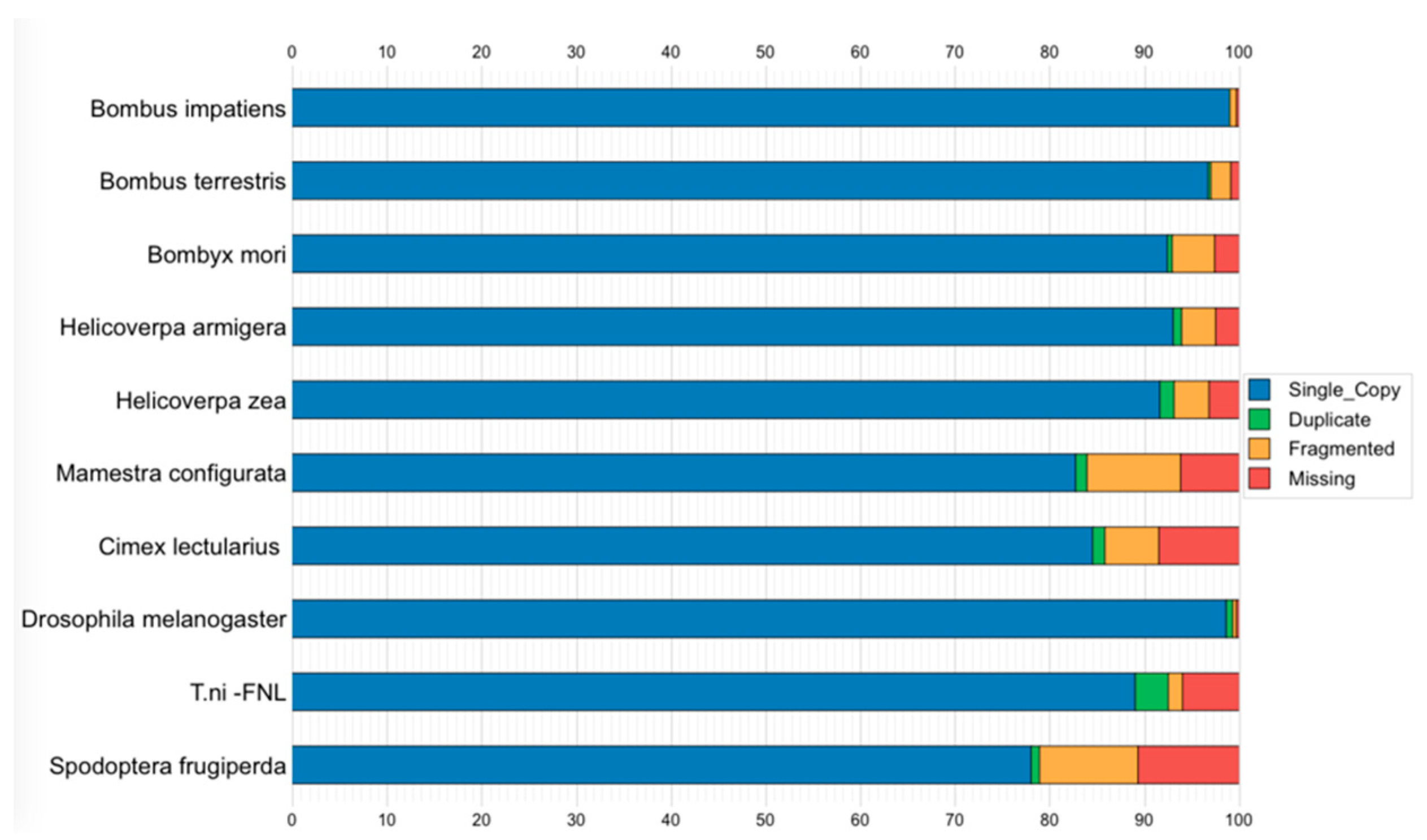
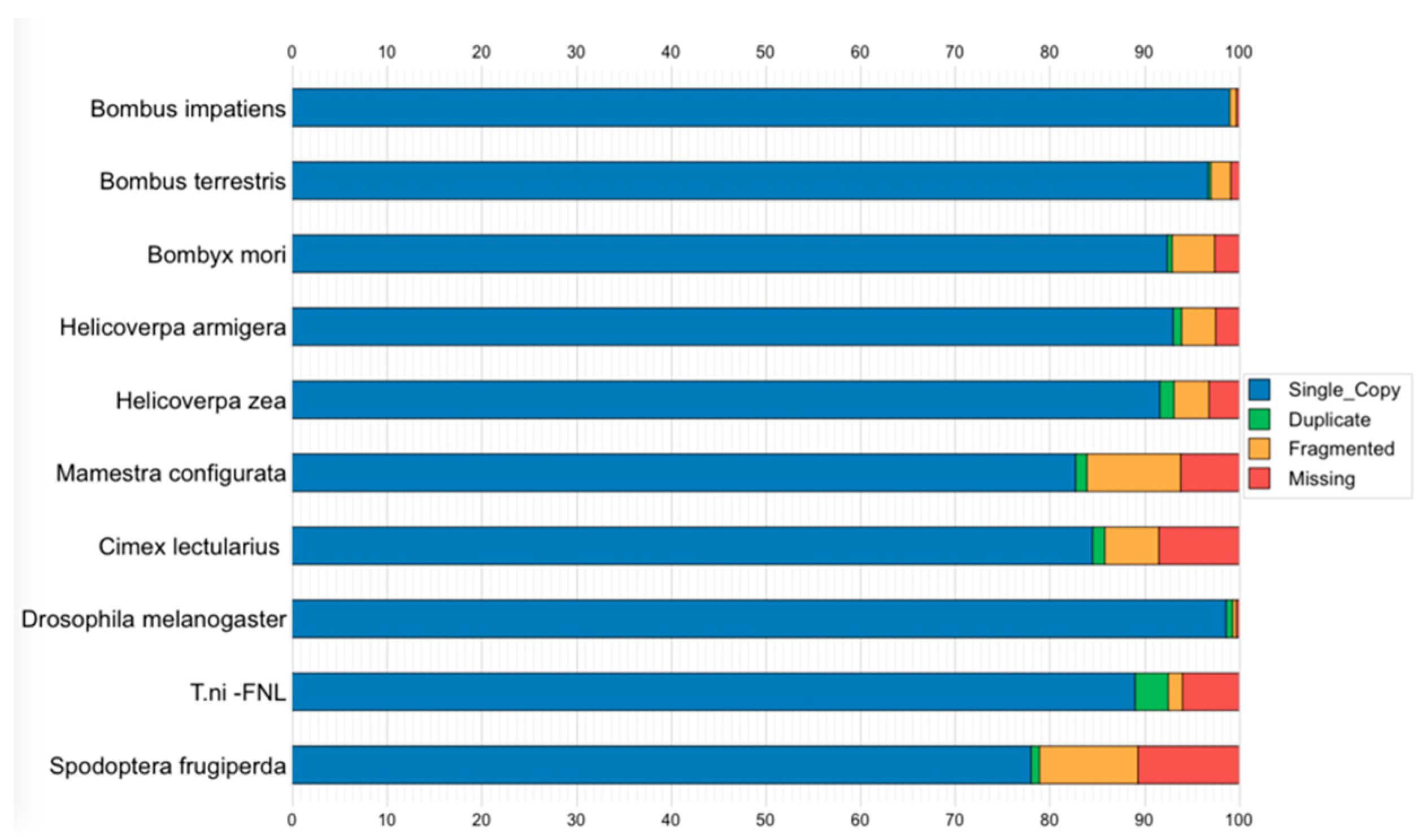
3.6. Genome Assembly Quality and Completeness Assessment
3.7. Determination of Cell Line Ploidy and Karyotype Analysis
3.8. Analysis of GC Content and CpG Islands
3.9. Analysis of Repeat Elements Including Endogenous Viral Elements
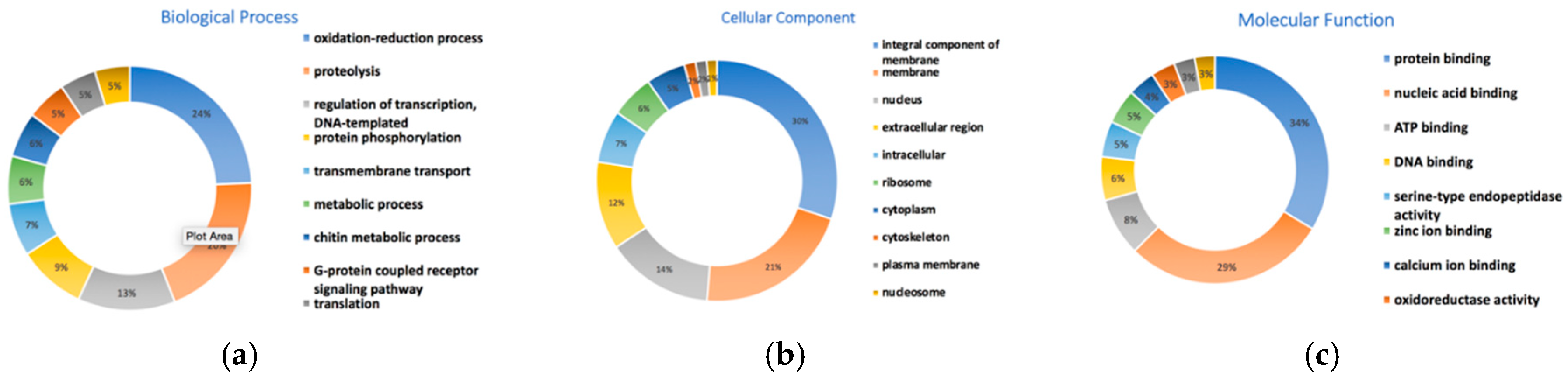
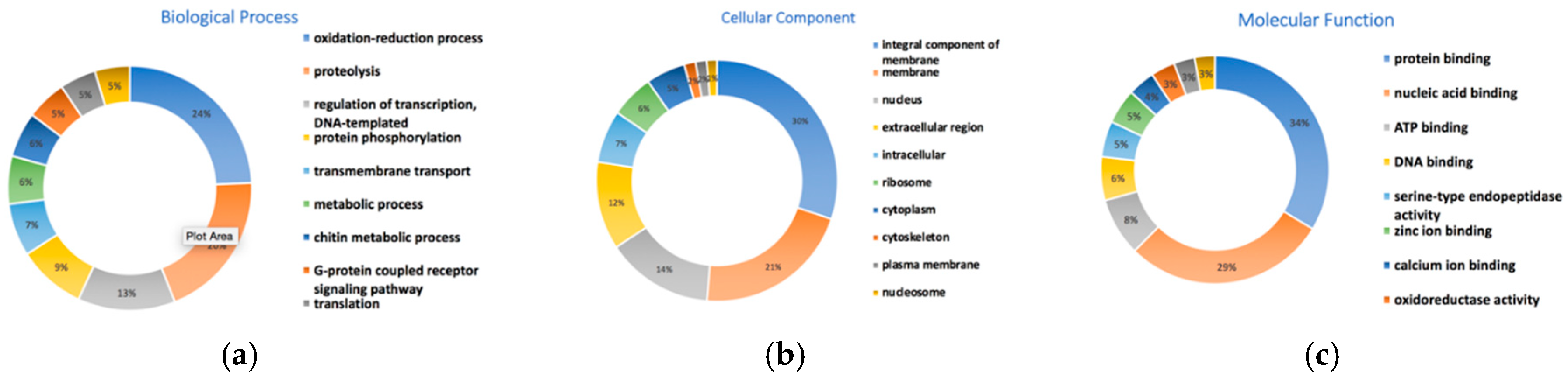
3.10. Gene Prediction and Functional Annotation
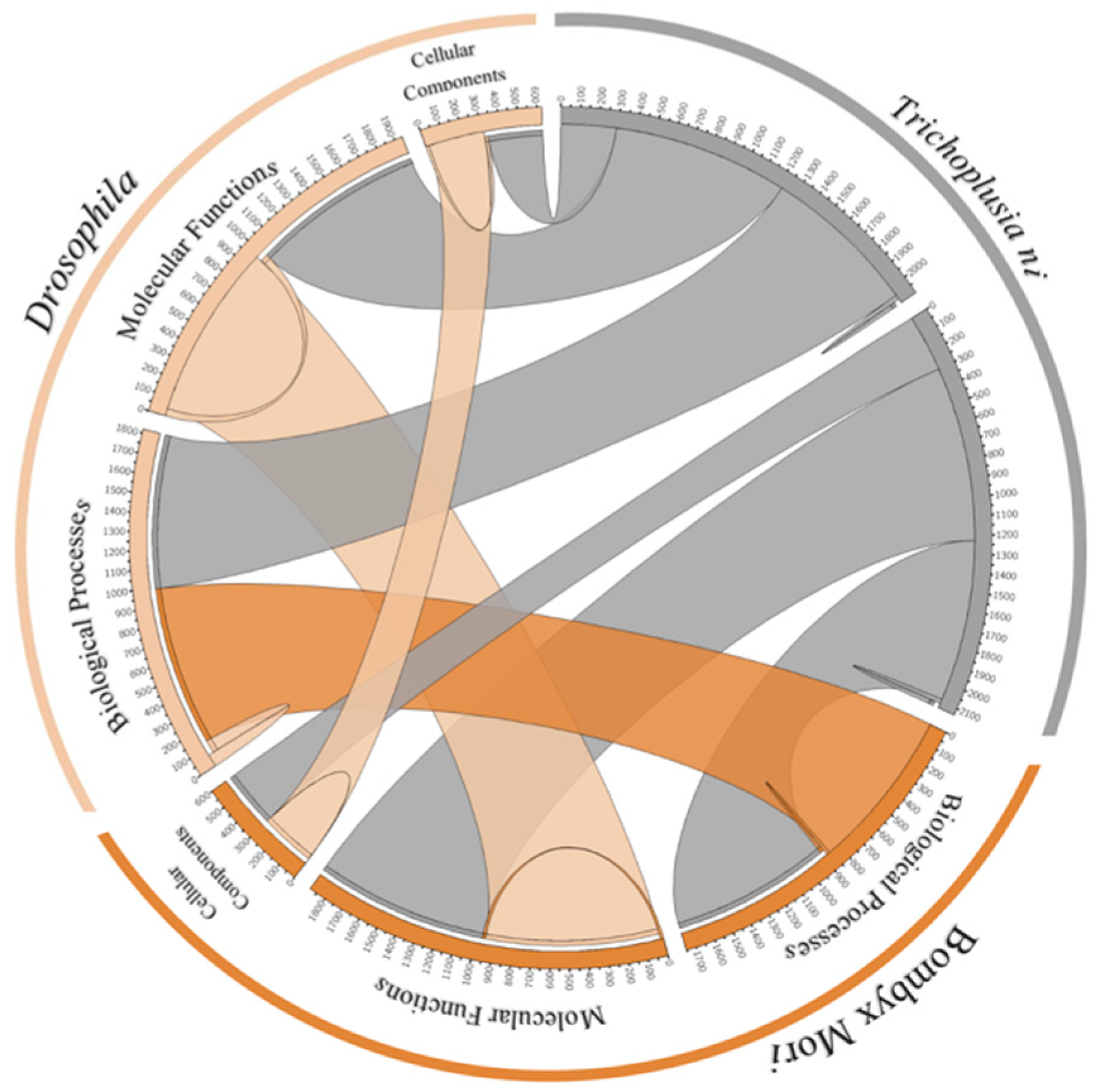
3.11. Orthologs and Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davis, T.R.; Wickham, T.J.; McKenna, K.A.; Granados, R.R.; Shuler, M.L.; Wood, H.A. Comparative recombinant protein production of eight insect cell lines. In Vitro Cell Dev. Biol. Anim. 1993, 29A, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M. Recombinant protein vaccines produced in insect cells. Vaccine 2012, 30, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Felberbaum, R.S. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors. Biotechnol. J. 2015, 10, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Mita, K. The genome sequence of silkworm, bombyx mori. DNA Res. 2004, 11, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kakumani, P.K.; Malhotra, P.; Mukherjee, S.K.; Bhatnagar, R.K. A draft genome assembly of the army worm, spodoptera frugiperda. Genomics 2014, 104, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zhong, S.; Fei, Z.; Gao, S.; Zhang, S.; Li, Z.; Wang, P.; Blissard, G.W. Transcriptome responses of the host trichoplusia ni to infection by the baculovirus autographa californica multiple nucleopolyhedrovirus. J. Virol. 2014, 88, 13781–13797. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Yu, Y.; Tang, X.; Chen, H.; Xiao, J.; Su, X.D. Transcriptome analyses of insect cells to facilitate baculovirus-insect expression. Protein Cell 2016, 7, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Davis, T.; Granados, R.R.; Shuler, M.L.; Wood, H.A. Screening of insect cell lines for the production of recombinant proteins and infectious virus in the baculovirus expression system. Biotechnol. Prog. 1992, 8, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Phillippy, A.M. New advances in sequence assembly. Genome Res. 2017, 27, xi–xiii. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yang, Y.; Zhang, H.; Farley, G.; Wang, J.; Quarles, K.A.; Weng, Z.; Zamore, P.D. The genome of the hi5 germ cell line from trichoplusia ni, an agricultural pest and novel model for small rna biology. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for illumina rna-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. Maker2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef]
- Mott, R. Est_genome: A program to align spliced DNA sequences to unspliced genomic DNA. Bioinformatics 1997, 13, 477–478. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. Augustus: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using maker and maker-p. Curr. Protoc. Bioinform. 2014, 48, 1–39. [Google Scholar]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml-vi-hpc: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (itol) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. Quast: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Shelton, J.M.; Coleman, M.C.; Herndon, N.; Lu, N.; Lam, E.T.; Anantharaman, T.; Sheth, P.; Brown, S.J. Tools and pipelines for bionano data: Molecule assembly pipeline and fasta super scaffolding tool. BMC Genom. 2015, 16, 734. [Google Scholar] [CrossRef]
- Schmutz, J.; Wheeler, J.; Grimwood, J.; Dickson, M.; Yang, J.; Caoile, C.; Bajorek, E.; Black, S.; Chan, Y.M.; Denys, M.; et al. Quality assessment of the human genome sequence. Nature 2004, 429, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. Genomescope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Jarman-Smith, R.F.; Mannix, C.; Al-Rubeai, M. Characterisation of tetraploid and diploid clones of spodoptera frugiperda cell line. Cytotechnology 2004, 44, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Lery, X.; Charpentier, G.; Belloncik, S. DNA content analysis of insect cell lines by flow cytometry. Cytotechnology 1999, 29, 103–113. [Google Scholar] [CrossRef] [PubMed]
- d’Alencon, E.; Sezutsu, H.; Legeai, F.; Permal, E.; Bernard-Samain, S.; Gimenez, S.; Gagneur, C.; Cousserans, F.; Shimomura, M.; Brun-Barale, A.; et al. Extensive synteny conservation of holocentric chromosomes in lepidoptera despite high rates of local genome rearrangements. Proc. Natl. Acad. Sci. USA 2010, 107, 7680–7685. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.E. Development and characterization of insect cell lines. Cytotechnology 1996, 20, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lequime, S.; Lambrechts, L. Discovery of flavivirus-derived endogenous viral elements inanophelesmosquito genomes supports the existence ofanopheles-associated insect-specific flaviviruses. Virus Evol. 2017, 3, vew035. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Han, L.; Su, B.; Li, W.-H.; Zhao, Z. Cpg island density and its correlations with genomic features in mammalian genomes. Genome Biol. 2008, 9, R79. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J. Repeats in genomic DNA: Mining and meaning. Curr. Opin. Struct. Biol. 1998, 8, 333–337. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Reeves, D.; Brugler, M.R.; Narechania, A.; Simon, S.; Durrett, R.; Foox, J.; Shianna, K.; Schatz, M.C.; Gandara, J.; et al. Genome assembly and geospatial phylogenomics of the bed bug cimex lectularius. Nat. Commun. 2016, 7, 10164. [Google Scholar] [CrossRef] [PubMed]
- Sadd, B.M.; Barribeau, S.M.; Bloch, G.; de Graaf, D.C.; Dearden, P.; Elsik, C.G.; Gadau, J.; Grimmelikhuijzen, C.J.; Hasselmann, M.; Lozier, J.D.; et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 2015, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Flegel, T.W. Hypothesis for heritable, anti-viral immunity in crustaceans and insects. Biol. Direct 2009, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. The evolution of endogenous viral elements. Cell Host Microbe 2011, 10, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. Interpro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2016, 45, D190–D199. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simao, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. Orthodb v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef]
- Mabashi-Asazuma, H.; Jarvis, D.L. Crispr-cas9 vectors for genome editing and host engineering in the baculovirus-insect cell system. Proc. Natl. Acad. Sci. USA 2017, 114, 9068–9073. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | HGAP Contigs | Canu Contigs | Supernova Contigs |
|---|---|---|---|
| Total contigs | 1428 | 2101 | 18,923 |
| Contigs (≥1000 bp) | 1418 | 2101 | 18,196 |
| Contigs (≥10,000 bp) | 1323 | 2097 | 6467 |
| Contigs (≥25,000 bp) | 1101 | 1780 | 3592 |
| Contigs (≥50,000 bp) | 706 | 1041 | 1828 |
| Largest contig (bp) | 4,352,893 | 6,104,320 | 445,812 |
| Total length (bp) | 366,261,337 | 408,408,011 | 316,721,011 |
| GC (%) | 35.66 | 35.74 | 35.33 |
| N50 (bp) | 939,843 | 737,233 | 54,240 |
| N75 (bp) | 421,565 | 250,244 | 22,505 |
| L50 (bp) | 115 | 158 | 1653 |
| L75 (bp) | 259 | 399 | 3894 |
| Types | Supernova Scaffolds | Hybrid Scaffolds (V1) | Polished Hybrid Scaffolds (V2) |
|---|---|---|---|
| Total scaffolds | 12,875 | 301 | 280 |
| Scaffolds (≥1000 bp) | 12,875 | 301 | 280 |
| Scaffolds (≥50,000 bp) | 355 | 301 | 280 |
| Largest scaffold (bp) | 7,830,761 | 10,389,188 | 12,760,714 |
| Total scaffold length (bp) | 375,813,451 | 328,208,105 | 359,075,955 |
| GC (%) | 35.33 | 35.5 | 35.56 |
| N50 (bp) | 1,628,260 | 1,737,254 | 2,326,860 |
| N75 (bp) | 449,524 | 1,019,983 | 1,198,934 |
| L50 (bp) | 66 | 57 | 44 |
| L75 (bp) | 172 | 120 | 98 |
| # N’s per 100 kb | 15,724 | 2919 | 5453 |
| Types | Tni-FNL | Bombyx mori | S. frugiperda |
|---|---|---|---|
| Total length (bp) | 359,075,955 | 481,803,763 | 358,050,723 |
| Total scaffolds | 280 | 43,462 | 37,243 |
| Ungapped length (bp) | 339,494,557 | 431,707,935 | 332,569,779 |
| Scaffold N50 (bp) | 2,326,860 | 4,008,358 | 53,779 |
| Total contigs | 2,043 | 88,672 | >49,244 |
| Contig N50 (bp) | 893,993 | 15,508 | 7,851 |
| Largest Scaffold (bp) | 12,760,714 | 14,496,184 | 641,448 |
| Largest Contig (bp) | 6,104,547 | 139,031 | 234,570 |
| GC (%) | 35.56 | 37.70 | 32.97 |
| Gap size (bp) | 19,581,398 | 50,095,828 | 25,480,944 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talsania, K.; Mehta, M.; Raley, C.; Kriga, Y.; Gowda, S.; Grose, C.; Drew, M.; Roberts, V.; Cheng, K.T.; Burkett, S.; et al. Genome Assembly and Annotation of the Trichoplusia ni Tni-FNL Insect Cell Line Enabled by Long-Read Technologies. Genes 2019, 10, 79. https://doi.org/10.3390/genes10020079
Talsania K, Mehta M, Raley C, Kriga Y, Gowda S, Grose C, Drew M, Roberts V, Cheng KT, Burkett S, et al. Genome Assembly and Annotation of the Trichoplusia ni Tni-FNL Insect Cell Line Enabled by Long-Read Technologies. Genes. 2019; 10(2):79. https://doi.org/10.3390/genes10020079
Chicago/Turabian StyleTalsania, Keyur, Monika Mehta, Castle Raley, Yuliya Kriga, Sujatha Gowda, Carissa Grose, Matthew Drew, Veronica Roberts, Kwong Tai Cheng, Sandra Burkett, and et al. 2019. "Genome Assembly and Annotation of the Trichoplusia ni Tni-FNL Insect Cell Line Enabled by Long-Read Technologies" Genes 10, no. 2: 79. https://doi.org/10.3390/genes10020079
APA StyleTalsania, K., Mehta, M., Raley, C., Kriga, Y., Gowda, S., Grose, C., Drew, M., Roberts, V., Cheng, K. T., Burkett, S., Oeser, S., Stephens, R., Soppet, D., Chen, X., Kumar, P., German, O., Smirnova, T., Hautman, C., Shetty, J., ... Esposito, D. (2019). Genome Assembly and Annotation of the Trichoplusia ni Tni-FNL Insect Cell Line Enabled by Long-Read Technologies. Genes, 10(2), 79. https://doi.org/10.3390/genes10020079