The Complete Mitogenome of Pyrrhocoris tibialis (Hemiptera: Pyrrhocoridae) and Phylogenetic Implications

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, and Sequencing
2.2. Annotation and Sequence Analysis
2.3. Phylogeneticsand Analyses of Sequence Heterogeneity
3. Results and Discussion
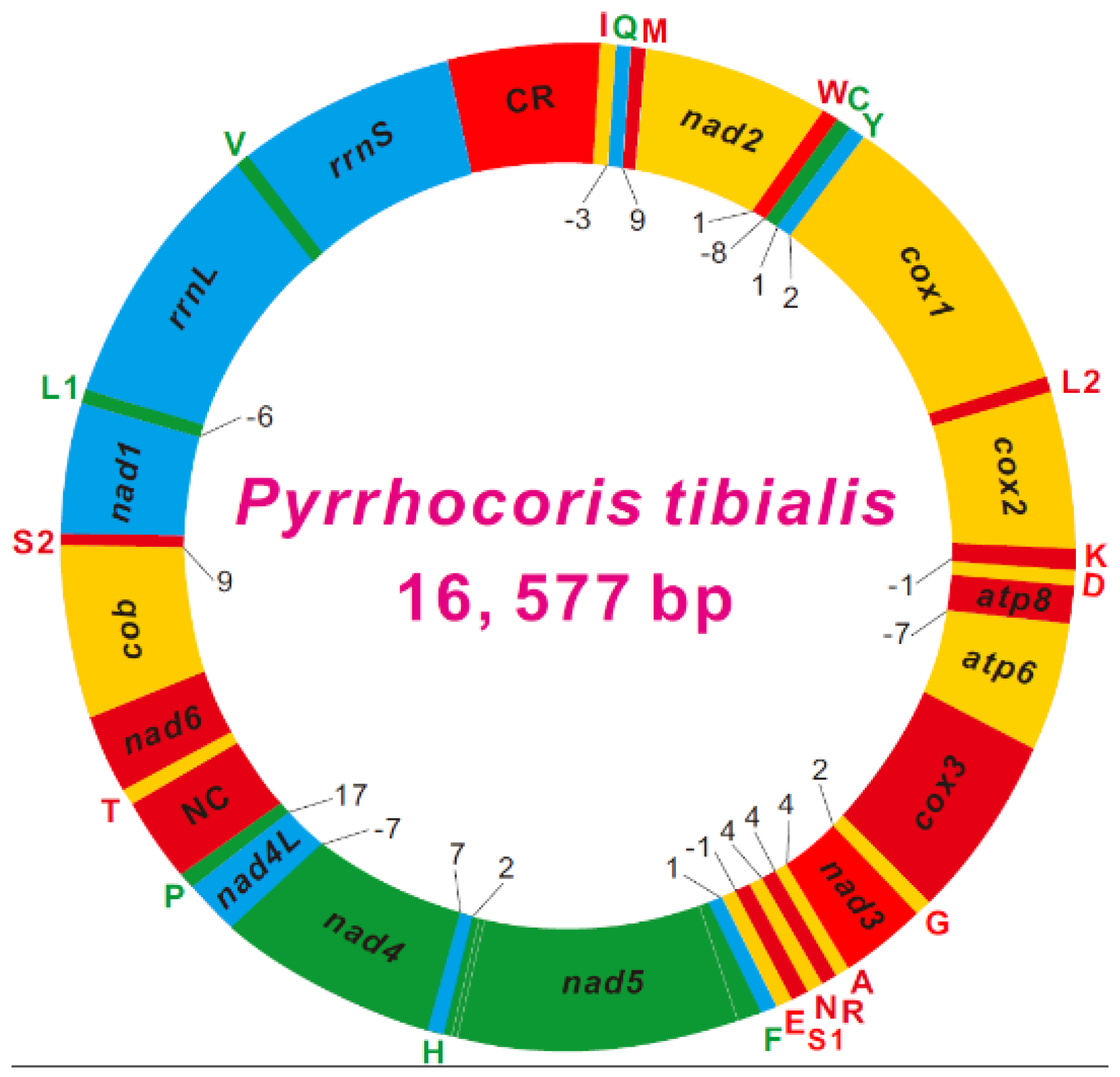
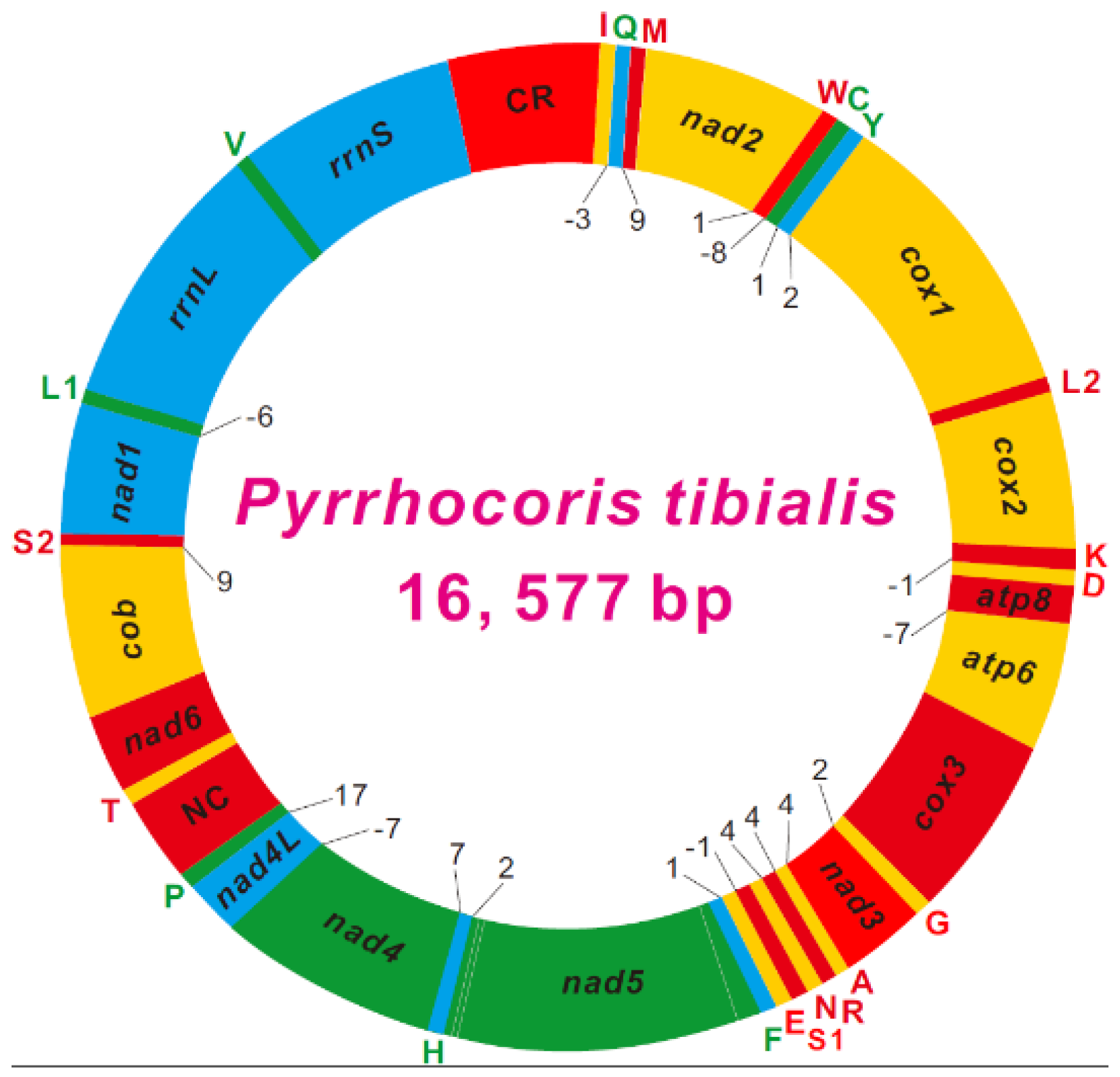
3.1. Genome Organization
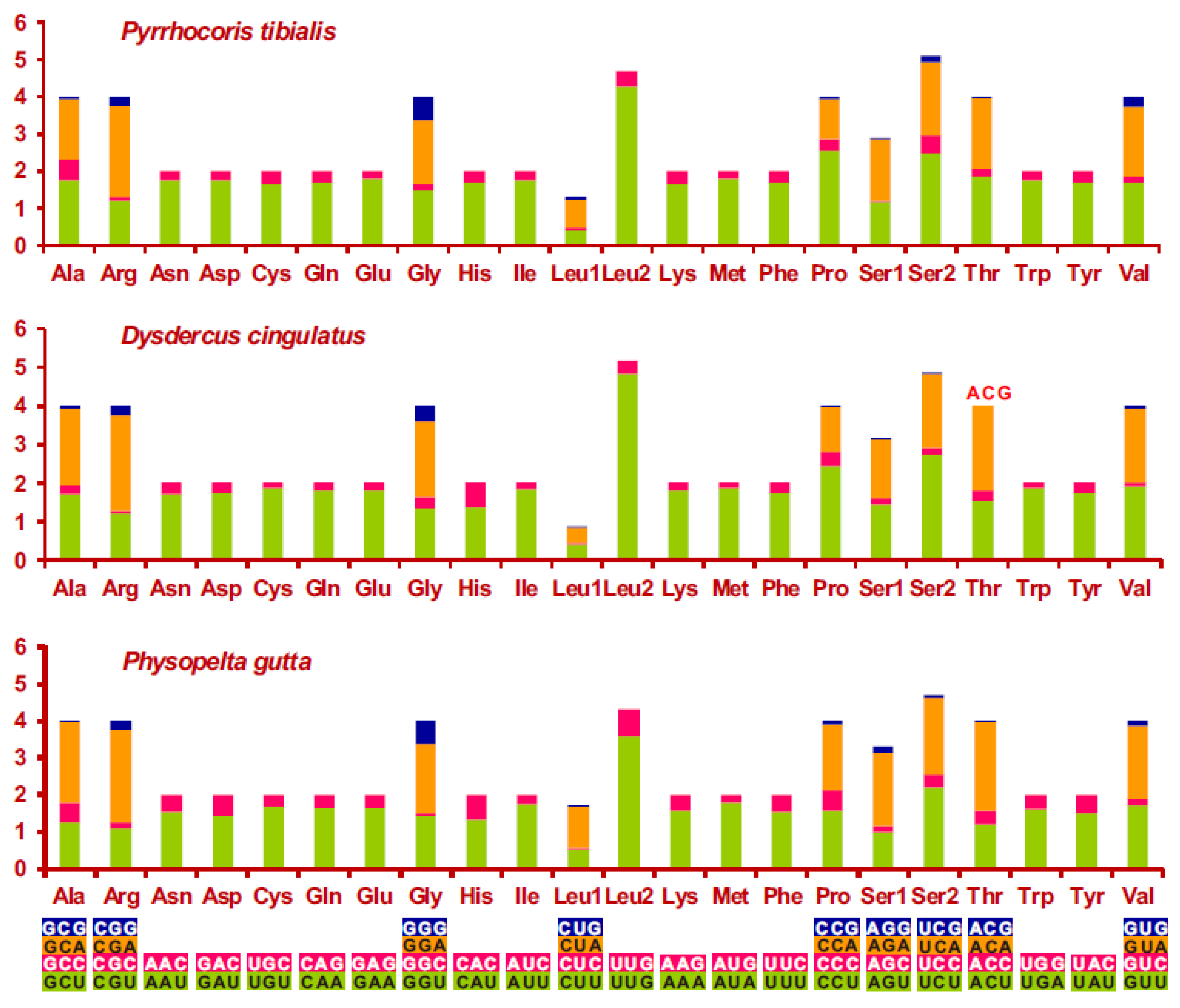
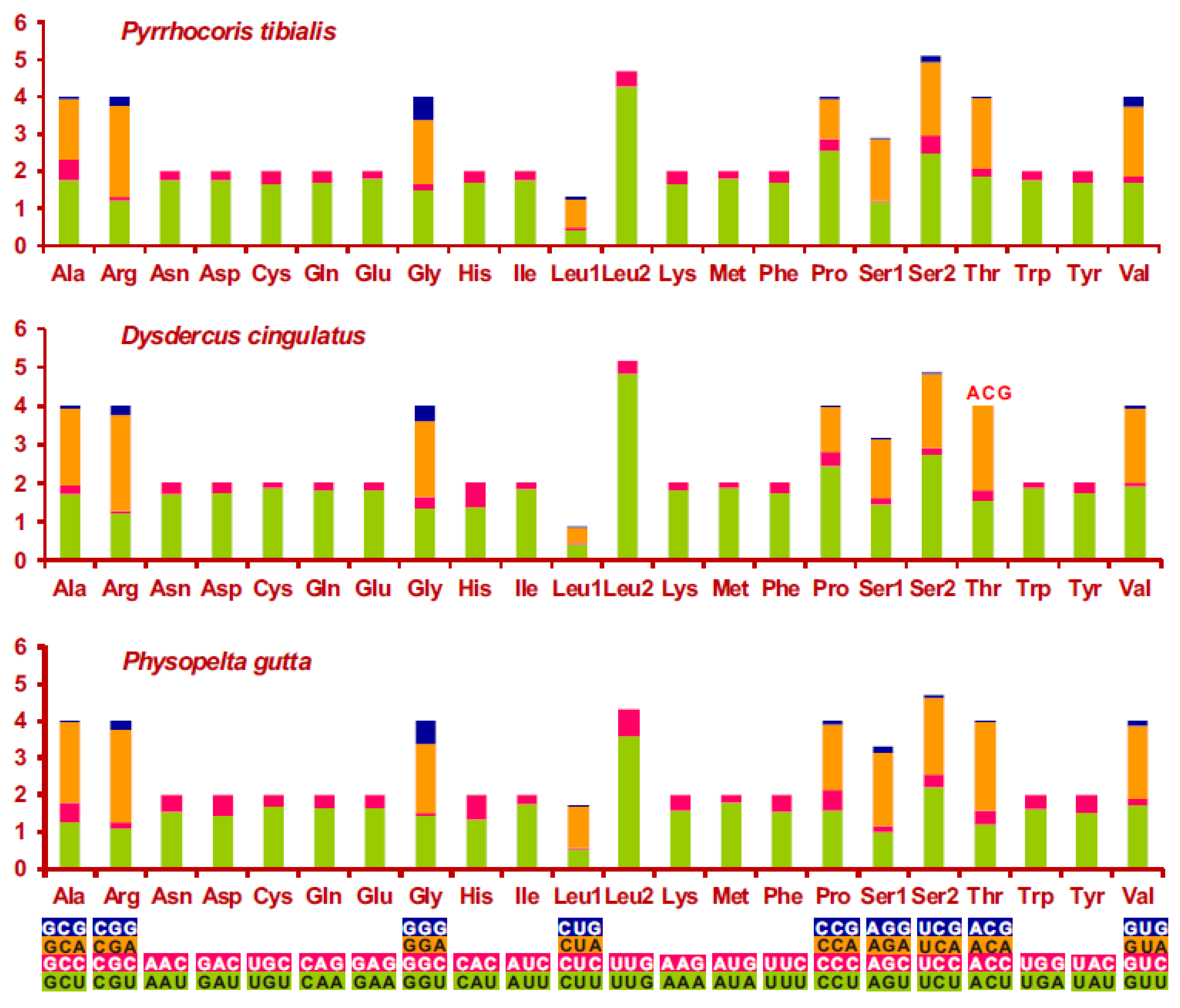
3.2. Nucleotide Composition and Codon Usage
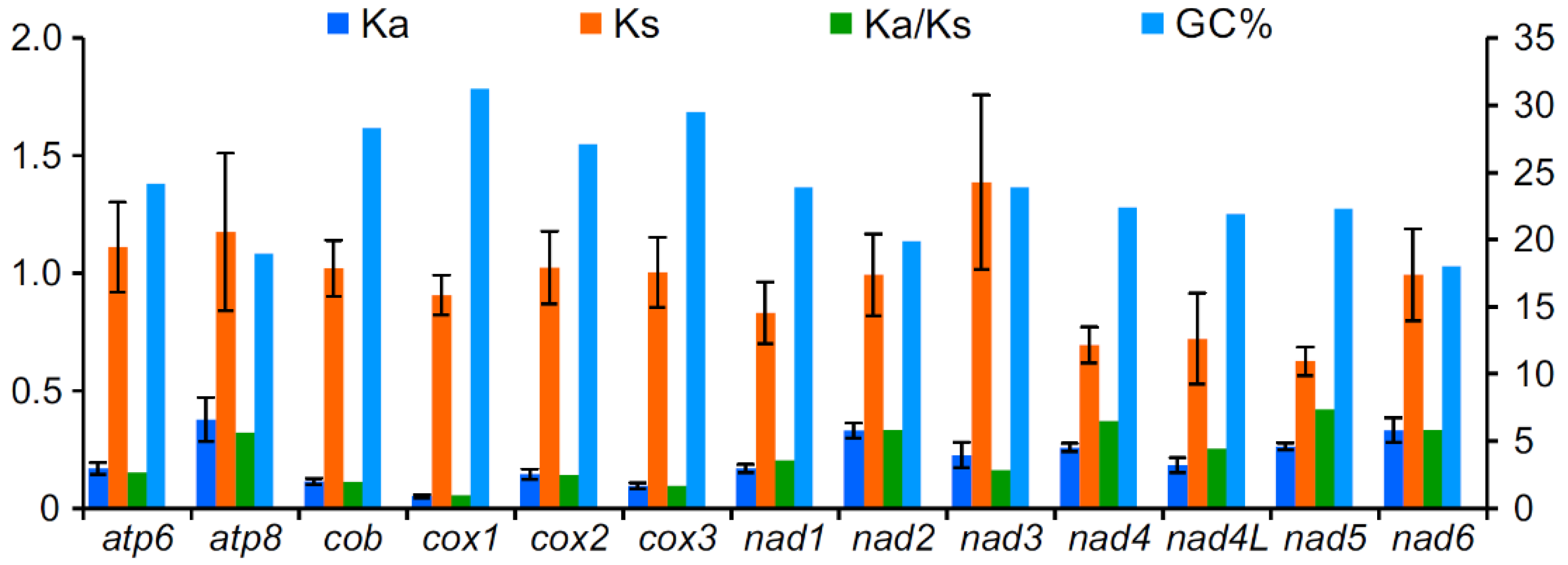
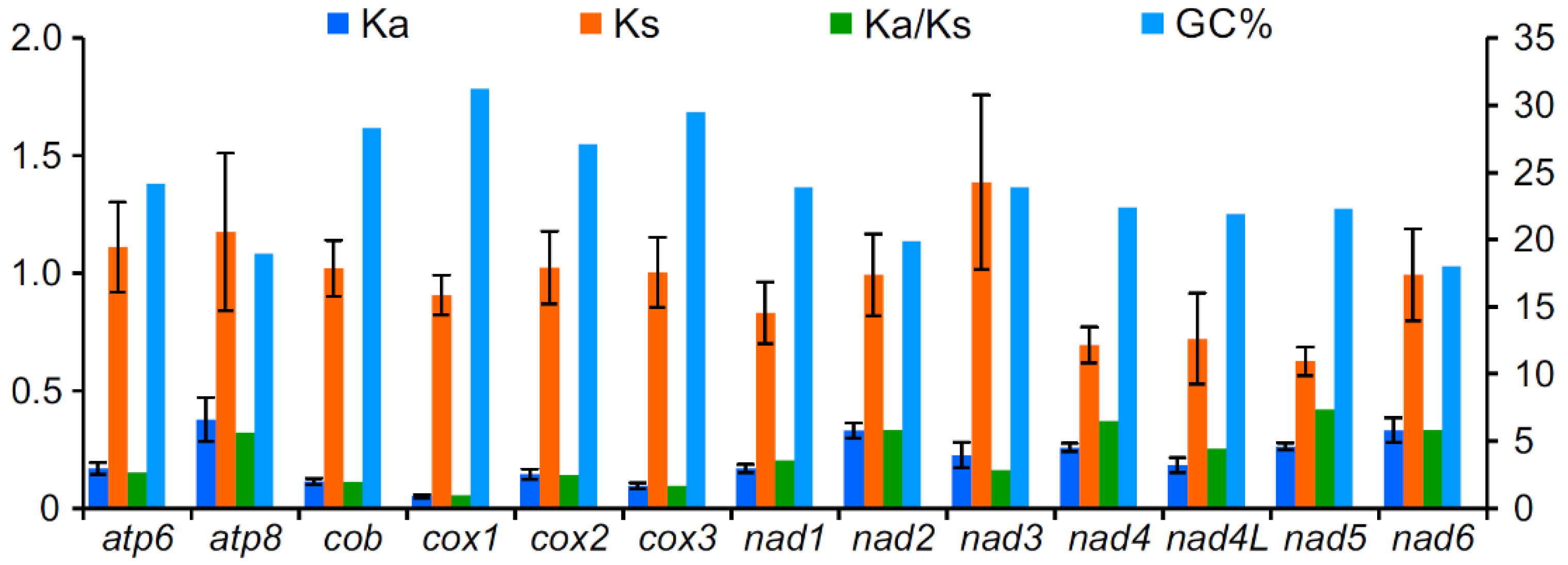
3.3. Protein-Coding Genes
3.4. Ribosomal and Transfer RNAs
3.5. Non-Coding Regions
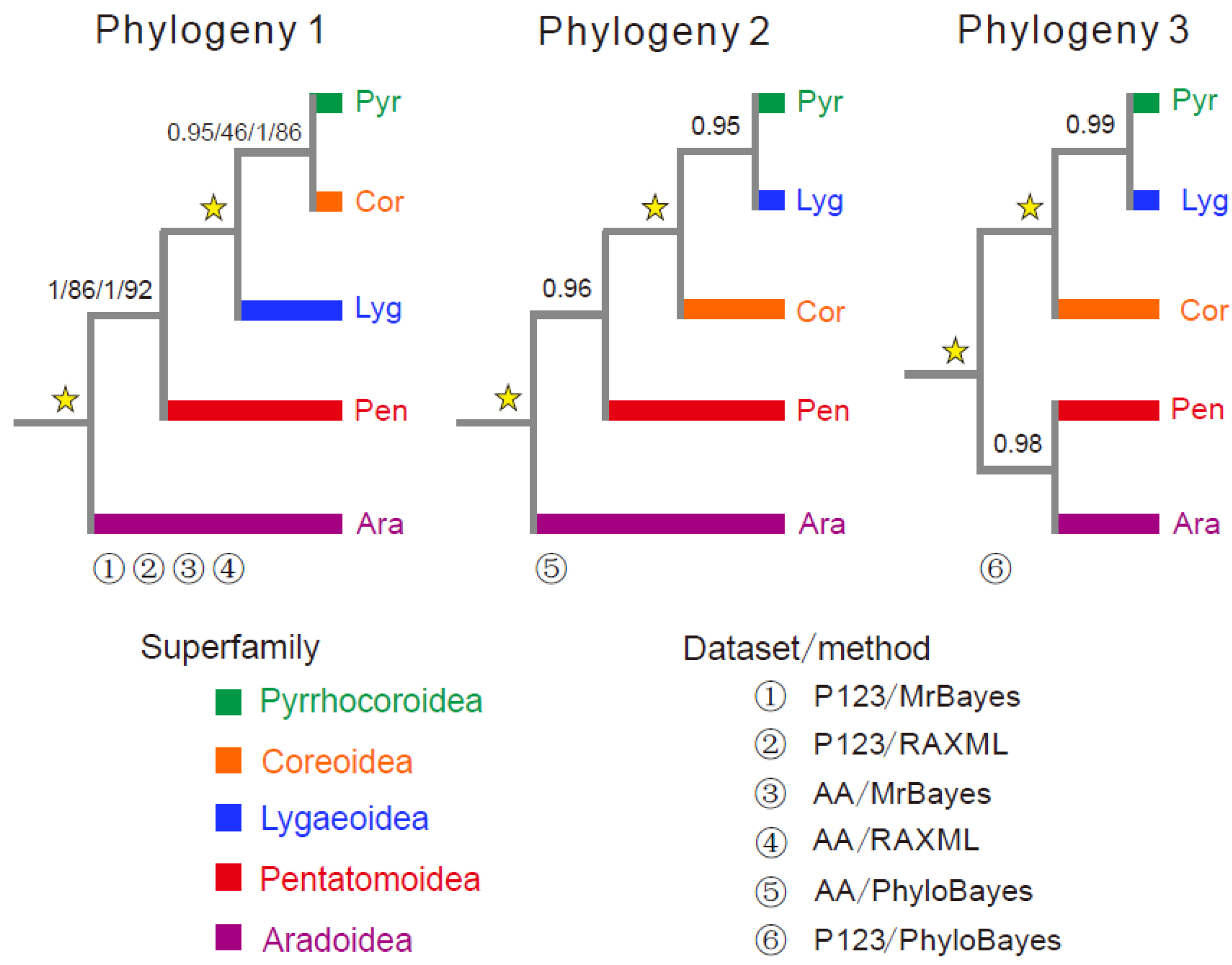
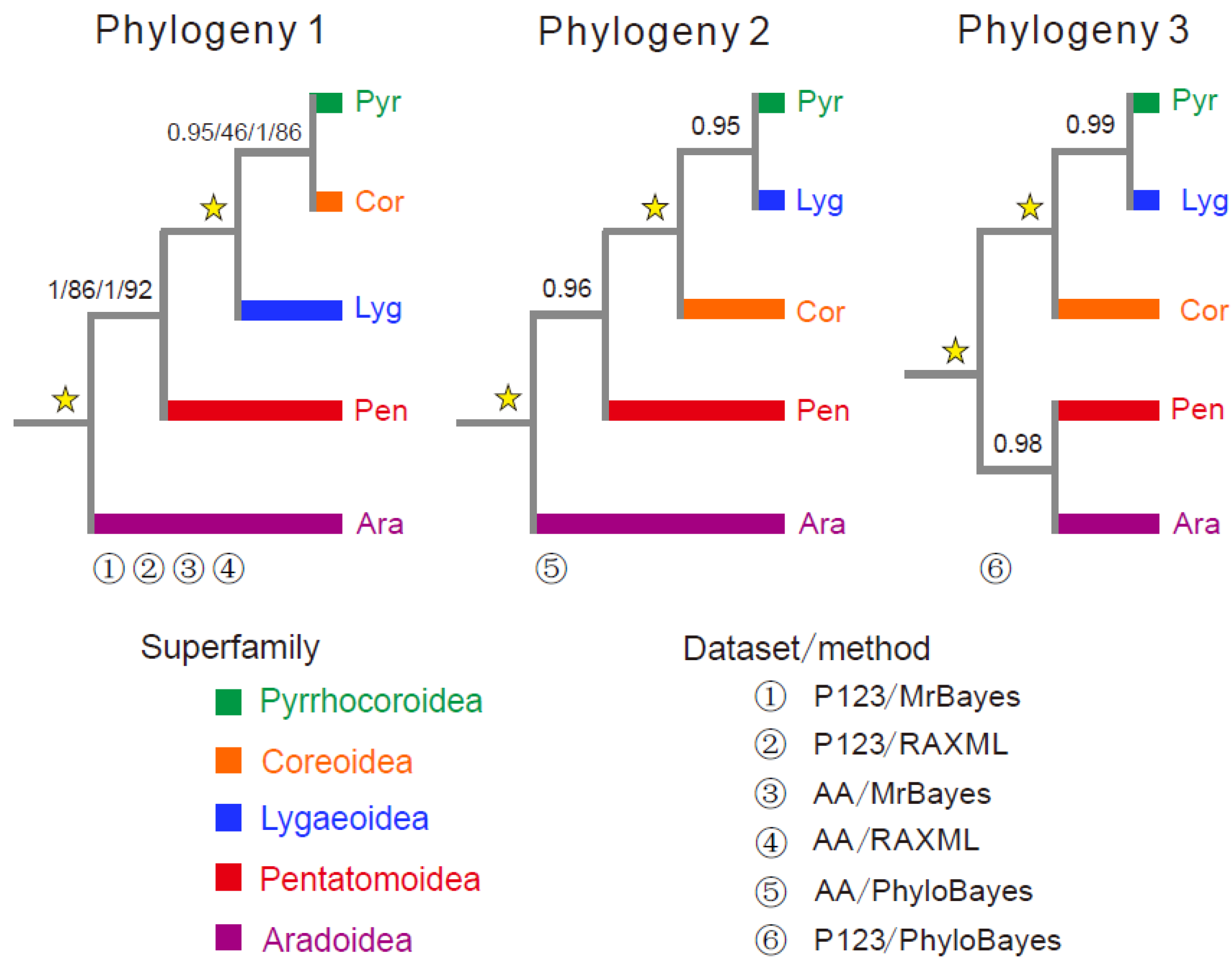
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Weirauch, C.; Schuh, R. Systematics and evolution of heteroptera: 25 years of progress. Annu. Rev. Entomol. 2011, 56, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Ming, L.; Dong, P.; Ying, C.; Bu, W. Comparative and phylogenomic studies on the mitochondrial genomes of pentatomomorpha (insecta: Hemiptera: Heteroptera). BMC Genom. 2008, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.J. Phylogenetic analysis of family groups within the infraorder pentatomomorpha (hemiptera: Heteroptera), with emphasis on the lygaeoidea. Ann. Entomol. Soc. Am. 1997, 90, 275–301. [Google Scholar] [CrossRef]
- Xie, Q.; Bu, W.; Zheng, L. The bayesian phylogenetic analysis of the 18S rRNA sequences from the main lineages of trichophora (insecta: Heteroptera: Pentatomomorpha). Mol. Phylogenet. Evol. 2005, 34, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Deng, R.Q.; Wang, J.W.; Chen, Z.Y.; Jia, F.L.; Wang, X.Z. A preliminary phylogeny of the pentatomomorpha (hemiptera: Heteroptera) based on nuclear 18S rDNA and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2005, 37, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Xie, Q.; Li, M.; Gao, C.; Cui, Y.; Xi, L.; Bu, W. Phylogeny of pentatomomorphan bugs (hemiptera-heteroptera: Pentatomomorpha) based on six HOX gene fragments. Zootaxa 2011, 2888, 57–68. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. The complete mitochondrial genome of corizus tetraspilus (hemiptera: Rhopalidae) and phylogenetic analysis of pentatomomorpha. PLoS ONE 2015, 10, e0129003. [Google Scholar] [CrossRef]
- Li, T.; Yang, J.; Li, Y.; Cui, Y.; Xie, Q.; Bu, W.; Hillis, D.M. A mitochondrial genome of rhyparochromidae (hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Xie, Q.; Tian, X.; Li, T.; Zhang, H.; Bu, W. Reanalysis of the phylogenetic relationships of the pentatomomorpha (hemiptera: Heteroptera) based on ribosomal, HOX and mitochondrial genes. Entomotaxonomia 2016, 38, 81–91. [Google Scholar]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. System. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: Retrospect and prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef]
- Wang, I.J. Recognizing the temporal distinctions between landscape genetics and phylogeography. Mol. Ecol. 2010, 19, 2605–2608. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.H.; Li, T.; Dang, K.; Bu, W.J. Compositional and mutational rate heterogeneity in mitochondrial genomes and its effect on the phylogenetic inferences of cimicomorpha (hemiptera: Heteroptera). BMC Genom. 2018, 19, 264. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B. Taxon sampling and the accuracy of large phylogenies. Syst. Biol. 1998, 47, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-L.; Zhang, G.-L.; Yuan, M.-L.; Dong, Z.-X.; Li, H.-W.; Guo, J.; Wang, F.; Deng, X.-Y.; Chen, J.-Y.; Lin, L.-B. A phylogenomic framework and divergence history of cephalochordata amphioxus. Front. Physiol. 2018, 9, 1833. [Google Scholar] [CrossRef]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef]
- Regier, J.C.; Shultz, J.W.; Zwick, A.; Hussey, A.; Ball, B.; Wetzer, R.; Martin, J.W.; Cunningham, C.W. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 2010, 463, 1079. [Google Scholar] [CrossRef]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.-J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef]
- Rota-Stabelli, O.; Lartillot, N.; Philippe, H.; Pisani, D. Serine codon-usage bias in deep phylogenomics: Pancrustacean relationships as a case study. Syst. Biol. 2012, 62, 121–133. [Google Scholar] [CrossRef]
- Schaefer, C.W.; Ahmad, I. Cotton Stainers and Their Relatives (Pyrrhocoroidea: Pyrrhocoridae and Largidae); CRC Press: Boca Raton, FL, USA, 2000; pp. 271–307. [Google Scholar]
- Yuan, M.-L.; Zhang, Q.-L.; Zhang, L.; Jia, C.-L.; Li, X.-P.; Yang, X.-Z.; Feng, R.-Q. Mitochondrial phylogeny, divergence history and high-altitude adaptation of grassland caterpillars (lepidoptera: Lymantriinae: Gynaephora) inhabiting the tibetan plateau. Mol. Phylogenet. Evol. 2018, 122, 116–124. [Google Scholar] [CrossRef]
- Tamura, K.; Filipski, A.; Peterson, D.; Stecher, G.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the Cipres Science Gateway for Inference of Large Phylogenetic Trees; Gateway Computing Environments Workshop (GCE): New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Kück, P.; Meid, S.A.; Groβ, C.; Wägele, J.W.; Misof, B. AliGROOVE – visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of Log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Strimmer, K.; Rambaut, A. Inferring confidence sets of possibly misspecified gene trees. Proc. Biol. Sci. 2002, 269, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zheng, Y.; Murphy, R.W.; Zeng, X. Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genom. 2016, 17, 965. [Google Scholar] [CrossRef] [PubMed]
- San, M.; Gower, D.; Zardoya, R.; Wilkinson, M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol. Biol. Evol. 2006, 23, 227–234. [Google Scholar]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef]
- Wei, S.-J.; Li, Q.; van Achterberg, K.; Chen, X.-X. Two mitochondrial genomes from the families Bethylidae and Mutillidae: Independent rearrangement of protein-coding genes and higher-level phylogeny of the Hymenoptera. Mol. Phylogenet. Evol. 2014, 77, 1–10. [Google Scholar] [CrossRef]
- Timmermans, M.J.T.N.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-level sampling of mitochondrial genomes in coleoptera: Compositional heterogeneity and phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of holometabola with mitochondrial genome data and bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, C.J.; Wang, W.; Goffinet, B. Mitochondrial phylogenomics of early land plants: Mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst. Biol. 2014, 63, 862–878. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. Comparative mitogenomic analysis of the superfamily pentatomoidea (insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genom. 2015, 16, 460. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A + T% | AT-Skew | GC-Skew | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PT | DC | PG | PT | DC | PG | PT | DC | PG | |
| Whole genome | 75.91 | 77.69 | 74.51 | 0.101 | 0.135 | 0.206 | −0.181 | −0.220 | −0.207 |
| Protein-coding genes | 75.15 | 76.86 | 73.55 | −0.134 | −0.127 | −0.099 | 0.020 | 0.015 | −0.004 |
| 1st codon positions | 69.46 | 71.62 | 69.01 | −0.002 | −0.010 | 0.033 | 0.220 | 0.240 | 0.198 |
| 2nd codon positions | 67.88 | 68.09 | 67.61 | −0.401 | −0.408 | −0.419 | −0.106 | −0.088 | −0.100 |
| 3rd codon positions | 88.10 | 90.86 | 84.02 | −0.033 | −0.009 | 0.051 | −0.153 | −0.325 | −0.203 |
| tRNA genes | 76.69 | 78.44 | 77.46 | 0.020 | 0.035 | 0.028 | 0.142 | 0.162 | 0.138 |
| rrnL genes | 79.94 | 81.23 | 78.68 | −0.103 | −0.109 | −0.204 | 0.260 | 0.286 | 0.257 |
| rrnS genes | 77.91 | 79.42 | 75.35 | −0.115 | −0.122 | −0.175 | 0.297 | 0.256 | 0.323 |
| Control region | 73.46 | 79.22 | 75.45 | −0.061 | 0.176 | 0.101 | −0.219 | −0.411 | −0.309 |
| Dataset | Topology | logL | KH | SH | WKH | WSH | ELW | AU |
|---|---|---|---|---|---|---|---|---|
| PCG | Phylogeny 1: ((((Pyr, Cor), Lyg), Pen), Ara) | −176,055.8 | 0.671 | 1 | 0.671 | 0.808 | 0.557 | 0.657 |
| Phylogeny 2: ((((Pyr, Lyg), Cor), Pen), Ara) | −176,060.0 | 0.329 | 0.581 | 0.329 | 0.554 | 0.205 | 0.386 | |
| Phylogeny 3: (((Pyr, Lyg), Cor), (Pen, Ara)) | −176,063.8 | 0.296 | 0.363 | 0.296 | 0.384 | 0.236 | 0.362 | |
| Amino acid | Phylogeny 1: ((((Pyr, Cor), Lyg), Pen), Ara) | −89,145.0 | 0.853 | 1 | 0.853 | 0.929 | 0.845 | 0.915 |
| Phylogeny 2: ((((Pyr, Lyg), Cor), Pen), Ara) | −89,157.7 | 0.147 | 0.295 | 0.147 | 0.254 | 0.133 | 0.135 | |
| Phylogeny 3: (((Pyr, Lyg), Cor), (Pen, Ara)) | −89,177.4 | 0.032 | 0.039 | 0.032 | 0.058 | 0.022 | 0.040 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.-L.; Feng, R.-Q.; Li, M.; Guo, Z.-L.; Zhang, L.-J.; Luo, F.-Z.; Cao, Y.; Yuan, M.-L. The Complete Mitogenome of Pyrrhocoris tibialis (Hemiptera: Pyrrhocoridae) and Phylogenetic Implications. Genes 2019, 10, 820. https://doi.org/10.3390/genes10100820
Zhang Q-L, Feng R-Q, Li M, Guo Z-L, Zhang L-J, Luo F-Z, Cao Y, Yuan M-L. The Complete Mitogenome of Pyrrhocoris tibialis (Hemiptera: Pyrrhocoridae) and Phylogenetic Implications. Genes. 2019; 10(10):820. https://doi.org/10.3390/genes10100820
Chicago/Turabian StyleZhang, Qi-Lin, Run-Qiu Feng, Min Li, Zhong-Long Guo, Li-Jun Zhang, Fang-Zhen Luo, Ya Cao, and Ming-Long Yuan. 2019. "The Complete Mitogenome of Pyrrhocoris tibialis (Hemiptera: Pyrrhocoridae) and Phylogenetic Implications" Genes 10, no. 10: 820. https://doi.org/10.3390/genes10100820