Tuberculosis Genetic Epidemiology: A Latin American Perspective


Abstract
1. Introduction
2. Discussion
2.1. The Epidemiology of Tuberculosis in Latin America
2.2. The Genomic Sequencing of Mycobacterium tuberculosis
2.3. The Arrival of Tuberculosis in Latin America
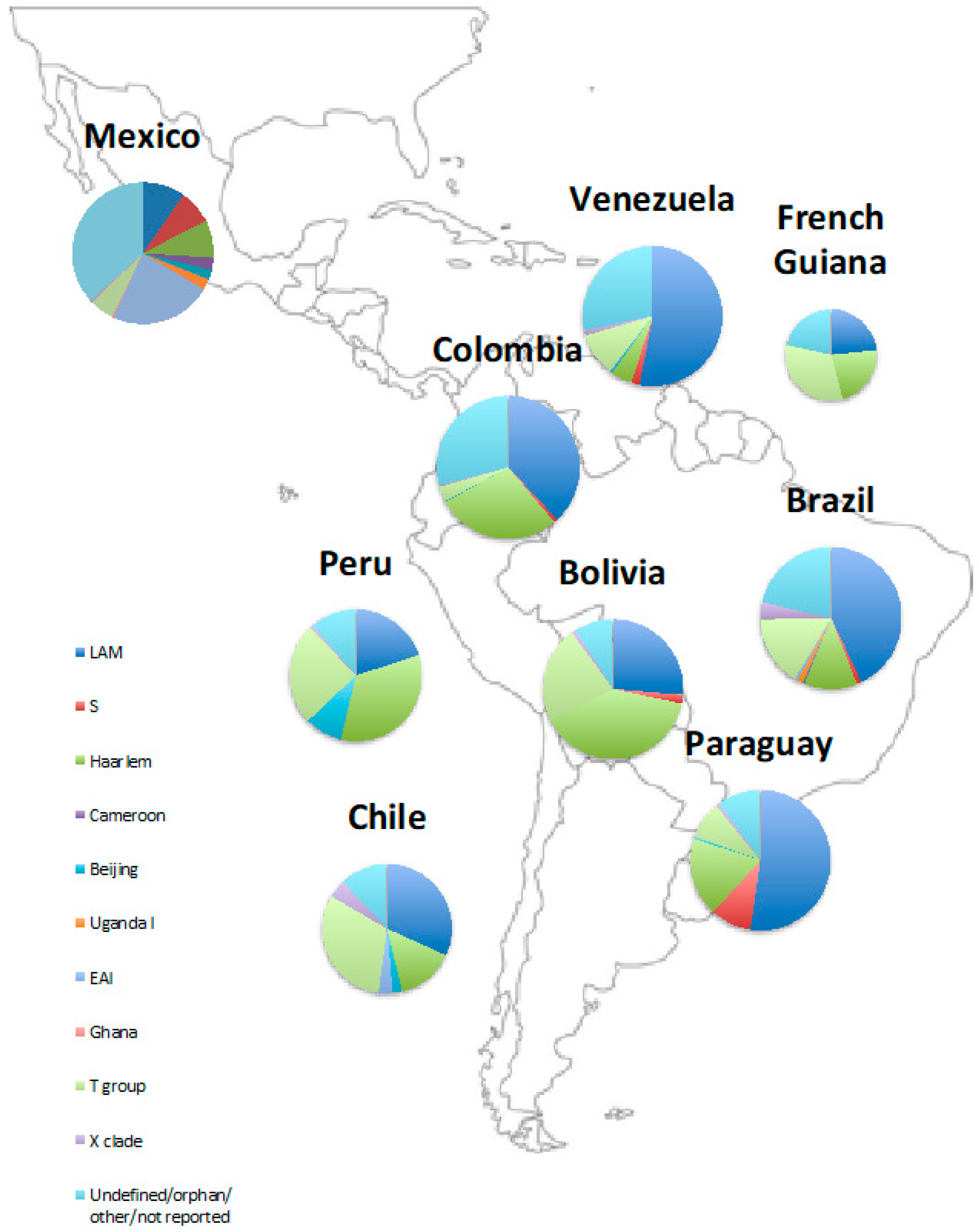
2.4. The Phylogenetics of Latin American Tuberculosis
2.5. Genetics of Mycobacterium tuberculosis Disease
2.6. Genetic Determinants of Disease Phenotype
2.7. Ability to Cause Active and Cavitating Disease
2.8. Ability to Transmit
2.9. Phylogenetic Associations with Acquired Drug Resistance
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2018; WHO: Geneva, Switzerland, 2018; Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 10 October 2018).
- Diacon, A.H.; Donald, P.R.; Pym, A.; Grobusch, M.; Patientia, R.F.; Mahanyele, R.; Bantubani, N.; Narasimooloo, R.; De Marez, T.; van Heeswijk, R.; et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: Long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob. Agents Chemother. 2012, 56, 3271–3276. [Google Scholar] [CrossRef] [PubMed]
- Gler, M.T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J.L.; Vargas-Vasquez, D.E.; Gao, M.; Awad, M.; Park, S.-K.; Shim, T.S.; et al. Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 2012, 366, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.J.R.; Ma, S.; Sherman, D.R.; Baliga, N.S. Network analysis identifies Rv0324 and Rv0880 as regulators of bedaquiline tolerance in Mycobacterium tuberculosis. Nat. Microbiol. 2016, 1, 16078. [Google Scholar] [CrossRef]
- Eldholm, V.; Norheim, G.; von der Lippe, B.; Kinander, W.; Dahle, U.R.; Caugant, D.A.; Mannsåker, T.; Mengshoel, A.T.; Dyrhol-Riise, A.M.; Balloux, F. Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biol. 2014, 15, 490. [Google Scholar] [CrossRef] [PubMed]
- The CRyPTIC consortium and the 100,000 Genomes Project. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N. Engl. J. Med. 2018, 379, 1403–1415. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.C.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef]
- Pan American Health Organisation. Tuberculosis in the Americas 2018; Pan American Health Organisation: Washington, DC, USA, 2018; Available online: www.paho.org (accessed on 10 October 2018).
- Matteelli, A.; Rendon, A.; Tiberi, S.; Al-Abri, S.; Voniatis, C.; Carvalho, A.C.C.; Centis, R.; D’Ambrosio, L.; Visca, D.; Spanevello, A.; et al. Tuberculosis elimination: Where are we now? Eur. Respir. Rev. 2018, 27, 180035. [Google Scholar] [CrossRef]
- Rendon, A.; Fuentes, Z.; Torres-Duque, C.A.; Granado, M.D.; Victoria, J.; Duarte, R.; Migliori, G.B. Roadmap for tuberculosis elimination in Latin American and Caribbean countries: A strategic alliance. Eur. Respir. J. 2016, 48, 1282–1287. [Google Scholar] [CrossRef]
- Muñoz-Torrico, M.; Caminero-Luna, J.; Migliori, G.B.; D’Ambrosio, L.; Carrillo-Alduenda, J.L.; Villareal-Velarde, H.; Torres-Cruz, A.; Flores-Vergara, H.; Martínez-Mendoza, D.; García-Sancho, C.; et al. Diabetes is associated with severe adverse events in multidrug-resistant tuberculosis. Arch. Bronconeumol. 2017, 53, 245–250. [Google Scholar] [CrossRef]
- Muñoz-Torrico, M.; Caminero Luna, J.; Migliori, G.B.; D’Ambrosio, L.; Carrillo-Alduenda, J.L.; Villareal-Velarde, H.; Torres-Cruz, A.; Flores-Ergara, H.; Martínez-Mendoza, D.; García-Sancho, C.; et al. Comparison of bacteriological conversion and treatment outcomes among MDR-TB patients with and without diabetes in Mexico: Preliminary data. Rev. Port. Pneumol. 2017, 23, 27–30. [Google Scholar] [CrossRef]
- Warren, J.L.; Grandjean, L.; Moore, D.A.J.; Lithgow, A.; Coronel, J.; Sheen, P.; Zelner, J.L.; Andrews, J.R.; Cohen, T. Investigating spillover of multidrug-resistant tuberculosis from a prison: A spatial and molecular epidemiological analysis. BMC Med. 2018, 16, 122. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M. Biological and epidemiological consequences of MTBC diversity. Adv. Exp. Med. Biol. 2017, 1019, 95–116. [Google Scholar] [CrossRef]
- Gagneux, S. Genetic Diversity in Mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 2013. [Google Scholar] [CrossRef]
- Gagneux, S.; Small, P.M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis. 2007, 7, 328–337. [Google Scholar] [CrossRef]
- Driscoll, J.R. Spoligotyping for molecular epidemiology of the Mycobacterium tuberculosis complex. In Methods in Molecular Biology (Clifton, N.J.); Springer: Berlin, Germany, 2009; Volume 551, pp. 117–128. [Google Scholar]
- Bifani, P.; Kurepina, N.; Mathema, B.; Wang, X.-M.; Kreiswirth, B. Genotyping of Mycobacterium tuberculosis clinical isolates using IS6110-based restriction fragment length polymorphism analysis. In Methods in Molecular Biology (Clifton, N.J.); Springer: Berlin, Germany, 2009; Volume 551, pp. 173–188. [Google Scholar]
- Brites, D.; Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 2015, 264, 6–24. [Google Scholar] [CrossRef]
- Blower, S.M.; McLean, A.R.; Porco, T.C.; Small, P.M.; Hopewell, P.C.; Sanchez, M.A.; Moss, A.R. The intrinsic transmission dynamics of tuberculosis epidemics. Nat. Med. 1995, 1, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C. Historia Antigua de Mexico: El Horizonte Posclásico y Algunos Aspectos Intelectuales de las Culturas Mesoamericanas; Consejo Nacional para la Cultura y las Artes: Austin, TX, USA, 1995; pp. 355–381. [Google Scholar]
- Mackowiak, P.A.; Blos, V.T.; Aguilar, M.; Buikstra, J.E. On the origin of American tuberculosis. Clin. Infect. Dis. 2005, 41, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Fagan, B.M. World Prehistory; Taylor and Francis: Abingdon, UK, 2016; ISBN 131 734 2445. [Google Scholar]
- Grandjean, L.; Iwamoto, T.; Lithgow, A.; Gilman, R.H.; Arikawa, K.; Nakanishi, N.; Martin, L.; Castillo, E.; Alarcon, V.; Coronel, J.; et al. The association between Mycobacterium tuberculosis genotype and drug resistance in Peru. PLoS ONE 2015, 10, e0126271. [Google Scholar] [CrossRef]
- Bos, K.I.; Harkins, K.M.; Herbig, A.; Coscolla, M.; Weber, N.; Comas, I.; Forrest, S.A.; Bryant, J.M.; Harris, S.R.; Schuenemann, V.J.; et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 2014, 514, 494. [Google Scholar] [CrossRef]
- Brynildsrud, O.B.; Pepperell, C.S.; Suffys, P.; Grandjean, L.; Monteserin, J.; Debech, N.; Bohlin, J.; Alfsnes, K.; Pettersson, J.O.-H.; Kirkeleite, I.; et al. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci. Adv. 2018, 4, eaat5869. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; McNerney, R.; Guerra-Assunção, J.A.; Glynn, J.R.; Perdigão, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014, 5, 4812. [Google Scholar] [CrossRef] [PubMed]
- Barletta, F.; Otero, L.; Collantes, J.; Asto, B.; de Jong, B.C.; Seas, C.; Rigouts, L. Genetic variability of Mycobacterium tuberculosis complex in patients with no known risk factors for MDR-TB in the North-Eastern part of Lima, Peru. BMC Infect. Dis. 2013, 13, 397. [Google Scholar] [CrossRef] [PubMed]
- Sheen, P.; Couvin, D.; Grandjean, L.; Zimic, M.; Dominguez, M.; Luna, G.; Gilman, R.H.; Rastogi, N.; Moore, D.A.J. Genetic diversity of Mycobacterium tuberculosis in Peru and exploration of phylogenetic associations with drug resistance. PLoS ONE 2013, 8, e65873. [Google Scholar] [CrossRef]
- Eldholm, V.; Monteserin, J.; Rieux, A.; Lopez, B.; Sobkowiak, B.; Ritacco, V.; Balloux, F. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat. Commun. 2015, 6, 7119. [Google Scholar] [CrossRef]
- Flores-Treviño, S.; Morfín-Otero, R.; Rodríguez-Noriega, E.; González-Díaz, E.; Pérez-Gómez, H.R.; Bocanegra-García, V.; Vera-Cabrera, L.; Garza-González, E. Genetic diversity of Mycobacterium tuberculosis from Guadalajara, Mexico and identification of a rare multidrug resistant Beijing genotype. PLoS ONE 2015, 10, e0118095. [Google Scholar] [CrossRef]
- Bocanegra-García, V.; Garza-González, E.; Cruz-Pulido, W.L.; Guevara-Molina, Y.L.; Cantú-Ramírez, R.; González, G.M.; Rivera, G.; Palma-Nicolas, J.P. Molecular assessment, drug-resistant profile, and spacer oligonucleotide typing (spoligotyping) of Mycobacterium tuberculosis strains from Tamaulipas, México. J. Clin. Lab. Anal. 2014, 28, 97–103. [Google Scholar] [CrossRef]
- Soares, R.O.; de Macedo, M.B.; von Groll, A.; da Silva, P.E. Mycobacterium tuberculosis belonging to family LAM and sublineage RD(Rio): Common strains in Southern Brazil for over 10 years. Braz. J. Microbiol. 2013, 44, 1251–1255. [Google Scholar] [CrossRef]
- Von Groll, A.; Martin, A.; Felix, C.; Prata, P.F.; Honscha, G.; Portaels, F.; Vandame, P.; da Silva, P.E.; Palomino, J.C. Fitness study of the RDRio lineage and Latin American-Mediterranean family of Mycobacterium tuberculosis in the city of Rio Grande, Brazil. FEMS Immunol. Med. Microbiol. 2010, 58, 119–127. [Google Scholar] [CrossRef]
- Dalla Costa, E.R.; Lazzarini, L.C.O.; Perizzolo, P.F.; Diaz, C.A.; Spies, F.S.; Costa, L.L.; Ribeiro, A.W.; Barroco, C.; Schuh, S.J.; da Silva Pereira, M.A.; et al. Mycobacterium tuberculosis of the RDRio genotype is the predominant cause of tuberculosis and associated with multidrug resistance in Porto Alegre City, South Brazil. J. Clin. Microbiol. 2013, 51, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Vasconcellos, S.E.; Acosta, C.C.; Gomes, L.L.; Conceicao, E.C.; Lima, K.V.; de Araujo, M.I.; Leite Mde, L.; Tannure, F.; Caldas, P.C.; Gomes, H.M.; et al. Strain classification of Mycobacterium tuberculosis isolates in Brazil based on genotypes obtained by spoligotyping, mycobacterial interspersed repetitive unit typing and the presence of large sequence and single nucleotide polymorphism. PLoS ONE 2014, 9, e107747. [Google Scholar] [CrossRef] [PubMed]
- Wiens, K.E.; Woyczynski, L.P.; Ledesma, J.R.; Ross, J.M.; Zenteno-Cuevas, R.; Goodridge, A.; Ullah, I.; Mathema, B.; Djoba Siawaya, J.F.; Biehl, M.H.; et al. Global variation in bacterial strains that cause tuberculosis disease: A systematic review and meta-analysis. BMC Med. 2018, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G.; et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 2013, 45, 1176–1182. [Google Scholar] [CrossRef]
- Hershberg, R.; Lipatov, M.; Small, P.M.; Sheffer, H.; Niemann, S.; Homolka, S.; Roach, J.C.; Kremer, K.; Petrov, D.A.; Feldman, M.W.; et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008, 6, e311. [Google Scholar] [CrossRef]
- Stucki, D.; Brites, D.; Jeljeli, L.; Coscolla, M.; Liu, Q.; Trauner, A.; Fenner, L.; Rutaihwa, L.; Borrell, S.; Luo, T.; et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 2016, 48, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Nava-Aguilera, E.; López-Vidal, Y.; Harris, E.; Morales-Pérez, A.; Mitchell, S.; Flores-Moreno, M.; Villegas-Arrizón, A.; Legorreta-Soberanis, J.; Ledogar, R.; Andersson, N. Clustering of Mycobacterium tuberculosis cases in Acapulco: Spoligotyping and risk factors. Clin. Dev. Immunol. 2011, 2011, 408375. [Google Scholar] [CrossRef]
- Bergad, L. The Comparative Histories of Slavery in Brazil, Cuba, and the United States; Cambridge University Press: Cambridge, UK, 2007; ISBN 978 051 180 3970. [Google Scholar]
- Lazzarini, L.C.; Huard, R.C.; Boechat, N.L.; Gomes, H.M.; Oelemann, M.C.; Kurepina, N.; Shashkina, E.; Mello, F.C.; Gibson, A.L.; Virginio, M.J.; et al. Discovery of a novel Mycobacterium tuberculosis lineage that is a major cause of tuberculosis in Rio de Janeiro, Brazil. J. Clin. Microbiol. 2007, 45, 3891–3902. [Google Scholar] [CrossRef]
- Homburger, J.R.; Moreno-Estrada, A.; Gignoux, C.R.; Nelson, D.; Sanchez, E.; Ortiz-Tello, P.; Pons-Estel, B.A.; Acevedo-Vasquez, E.; Miranda, P.; Langefeld, C.D.; et al. Genomic insights into the ancestry and demographic history of South America. PLoS Genet. 2015, 11, e1005602. [Google Scholar] [CrossRef] [PubMed]
- CIA South America. Argentina—The World Factbook; Central Intelligence Agency: McLean, VA, USA, 2019. Available online: https://www.cia.gov/library/publications/the-world-factbook/geos/ar.html#People (accessed on 10 October 2018).
- Balcells, M.E.; Garcia, P.; Meza, P.; Pena, C.; Cifuentes, M.; Couvin, D.; Rastogi, N. A first insight on the population structure of Mycobacterium tuberculosis complex as studied by spoligotyping and MIRU-VNTRs in Santiago, Chile. PLoS ONE 2015, 10, e0118007. [Google Scholar] [CrossRef]
- Tuite, A.R.; Thomas-Bachli, A.; Acosta, H.; Bhatia, D.; Huber, C.; Petrasek, K.; Watts, A.; Yong, J.H.E.; Bogoch, I.I.; Khan, K. Infectious disease implications of large-scale migration of Venezuelan nationals. J. Travel. Med. 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- Flores-Lopez, C.A.; Zenteno-Cuevas, R.; Laniado-Laborin, R.; Reynaud, Y.; Garcia-Ortiz, R.A.; Gonzalez, Y.M.J.A.; Rivera, S.; Vazquez-Chacon, C.A.; Vaughan, G.; Martinez-Guarneros, J.A.; et al. Molecular epidemiology of Mycobacterium tuberculosis in Baja California, Mexico: A result of human migration? Infect. Genet. Evol. 2017, 55, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, I.; Jimenez, Y.; Hernandez, J.; Zozio, T.; Murcia, M.I.; Rastogi, N. A first insight on the population structure of Mycobacterium tuberculosis complex as studied by spoligotyping and MIRU-VNTRs in Bogota, Colombia. Infect. Genet. Evol. 2012, 12, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Cardoso Oelemann, M.; Gomes, H.M.; Willery, E.; Possuelo, L.; Batista Lima, K.V.; Allix-Beguec, C.; Locht, C.; Goguet de la Salmoniere, Y.O.; Gutierrez, M.C.; Suffys, P.; et al. The forest behind the tree: Phylogenetic exploration of a dominant Mycobacterium tuberculosis strain lineage from a high tuberculosis burden country. PLoS ONE 2011, 6, e18256. [Google Scholar] [CrossRef] [PubMed]
- Noguti, E.N.; Leite, C.Q.; Malaspina, A.C.; Santos, A.C.; Hirata, R.D.; Hirata, M.H.; Mamizuka, E.M.; Cardoso, R.F. Genotyping of Mycobacterium tuberculosis isolates from a low-endemic setting in northwestern state of Parana in Southern Brazil. Mem. Inst. Oswaldo Cruz 2010, 105, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Dantas, N.G.; Suffys, P.N.; Carvalho Wda, S.; Gomes, H.M.; de Almeida, I.N.; de Assis, L.J.; Augusto, C.J.; Gomgnimbou, M.K.; Refregier, G.; Sola, C.; et al. Genetic diversity and molecular epidemiology of multidrug-resistant Mycobacterium tuberculosis in Minas Gerais State, Brazil. BMC Infect. Dis. 2015, 15, 306. [Google Scholar] [CrossRef]
- Malaspina, A.C.; Cavalcanti, H.R.; Leite, C.Q.; Machado, S.M.; Viana, B.H.; Silva, R.M.; Hage, E.F.; Figueiredo, W.M.; Marques, E.; Ferrazoli, L.; et al. Usefulness of Mycobacterium tuberculosis molecular typing in a tuberculosis low-endemic agro-industrial setting of Brazil. Jpn. J. Infect. Dis. 2008, 61, 231–233. [Google Scholar]
- Huber, F.D.; Sanchez, A.; Gomes, H.M.; Vasconcellos, S.; Massari, V.; Barreto, A.; Cesconi, V.; de Almeida Machado, S.M.; Gomgnimbou, M.K.; Sola, C.; et al. Insights into the population structure of Mycobacterium tuberculosis using spoligotyping and RDRio in a southeastern Brazilian prison unit. Infect. Genet. Evol. 2014, 26, 194–202. [Google Scholar] [CrossRef]
- Meza, P.; Balcells, M.E.; Miranda, C.; Cifuentes, M.; Wozniak, A.; Garcia, P. Presence of Bejing genotype among Mycobacterium tuberculosis strains in two centres of the Region Metropolitana of Chile. Rev. Chil. Infectol. 2014, 31, 21–27. [Google Scholar] [CrossRef]
- Taype, C.A.; Agapito, J.C.; Accinelli, R.A.; Espinoza, J.R.; Godreuil, S.; Goodman, S.J.; Banuls, A.L.; Shaw, M.A. Genetic diversity, population structure and drug resistance of Mycobacterium tuberculosis in Peru. Infect. Genet. Evol. 2012, 12, 577–585. [Google Scholar] [CrossRef]
- Caceres, O.; Rastogi, N.; Bartra, C.; Couvin, D.; Galarza, M.; Asencios, L.; Mendoza-Ticona, A. Characterization of the genetic diversity of extensively-drug resistant Mycobacterium tuberculosis clinical isolates from pulmonary tuberculosis patients in Peru. PLoS ONE 2014, 9, e112789. [Google Scholar] [CrossRef]
- Realpe, T.; Correa, N.; Rozo, J.C.; Ferro, B.E.; Gomez, V.; Zapata, E.; Ribon, W.; Puerto, G.; Castro, C.; Nieto, L.M.; et al. Population structure among Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients in Colombia. PLoS ONE 2014, 9, e93848. [Google Scholar] [CrossRef] [PubMed]
- Guernier, V.; Sola, C.; Brudey, K.; Guegan, J.F.; Rastogi, N. Use of cluster-graphs from spoligotyping data to study genotype similarities and a comparison of three indices to quantify recent tuberculosis transmission among culture positive cases in French Guiana during a eight year period. BMC Infect. Dis. 2008, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Monteserin, J.; Camacho, M.; Barrera, L.; Palomino, J.C.; Ritacco, V.; Martin, A. Genotypes of Mycobacterium tuberculosis in patients at risk of drug resistance in Bolivia. Infect. Genet. Evol. 2013, 17, 195–201. [Google Scholar] [CrossRef]
- Candia, N.; Lopez, B.; Zozio, T.; Carrivale, M.; Diaz, C.; Russomando, G.; de Romero, N.J.; Jara, J.C.; Barrera, L.; Rastogi, N.; et al. First insight into Mycobacterium tuberculosis genetic diversity in Paraguay. BMC Microbiol. 2007, 7, 75. [Google Scholar] [CrossRef]
- Abadia, E.; Sequera, M.; Ortega, D.; Mendez, M.V.; Escalona, A.; Da Mata, O.; Izarra, E.; Rojas, Y.; Jaspe, R.; Motiwala, A.S.; et al. Mycobacterium tuberculosis ecology in Venezuela: Epidemiologic correlates of common spoligotypes and a large clonal cluster defined by MIRU-VNTR-24. BMC Infect. Dis. 2009, 9, 122. [Google Scholar] [CrossRef]
- Grandjean, L. Investigating the pathogen genomic determinants of tuberculosis transmission. Am. J. Respir. Crit. Care Med. 2017, 195, 1418–1420. [Google Scholar] [CrossRef]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E.; Freedman, V.H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757. [Google Scholar] [CrossRef]
- Ribeiro, S.C.M.; Gomes, L.L.; Amaral, E.P.; Andrade, M.R.M.; Almeida, F.M.; Rezende, A.L.; Lanes, V.R.; Carvalho, E.C.Q.; Suffys, P.N.; Mokrousov, I.; et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J. Clin. Microbiol. 2014, 52, 2615–2624. [Google Scholar] [CrossRef]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef]
- Brites, D.; Gagneux, S. Old and new selective pressures on Mycobacterium tuberculosis. Infect. Genet. Evol. 2012, 12, 678–685. [Google Scholar] [CrossRef]
- Rodrigo, T.; Caylà, J.A.; García de Olalla, P.; Galdós-Tangüis, H.; Jansà, J.M.; Miranda, P.; Brugal, T. Characteristics of tuberculosis patients who generate secondary cases. Int. J. Tuberc. Lung Dis. 1997, 1, 352–357. [Google Scholar]
- Forrellad, M.A.; Klepp, L.I.; Gioffré, A.; Sabio y García, J.; Morbidoni, H.R.; de la Paz Santangelo, M.; Cataldi, A.A.; Bigi, F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef] [PubMed]
- Smith, I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 2003, 16, 463–496. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, D.A.; Wallace, J.G.; Bhatia, A.L.; Selkon, J.B.; Subbaiah, T.V.; Lancaster, M.C. A comparison of the virulence in guinea-pigs of South Indian and British tubercle bacilli. Tubercle 1960, 41, 1–22. [Google Scholar] [CrossRef]
- Bhatia, A.L.; Jacob, C.V.; Hitze, K.L.; Ramachandran, K.; Selkon, J.B. A comparison of the virulence in the guinea-pig of tubercle bacilli from thai and british patients. Bull. World Health Organ. 1963, 29, 483–490. [Google Scholar] [PubMed]
- Valway, S.E.; Sanchez, M.P.; Shinnick, T.F.; Orme, I.; Agerton, T.; Hoy, D.; Jones, J.S.; Westmoreland, H.; Onorato, I.M. An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N. Engl. J. Med. 1998, 338, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Small, P.M.; Hopewell, P.C.; Singh, S.P.; Paz, A.; Parsonnet, J.; Ruston, D.C.; Schecter, G.F.; Daley, C.L.; Schoolnik, G.K. The epidemiology of tuberculosis in San Francisco. A population-based study using conventional and molecular methods. N. Engl. J. Med. 1994, 330, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gong, J.; Yang, Z.; Samten, B.; Cave, M.D.; Barnes, P.F. Enhanced capacity of a widespread strain of Mycobacterium tuberculosis to grow in human macrophages. J. Infect. Dis. 1999, 179, 1213–1217. [Google Scholar] [CrossRef]
- Rajakumar, K.; Shafi, J.; Smith, R.J.; Stabler, R.A.; Andrew, P.W.; Modha, D.; Bryant, G.; Monk, P.; Hinds, J.; Butcher, P.D.; et al. Use of genome level-informed PCR as a new investigational approach for analysis of outbreak-associated Mycobacterium tuberculosis isolates. J. Clin. Microbiol. 2004, 42, 1890–1896. [Google Scholar] [CrossRef]
- Gao, Q.; Kripke, K.E.; Saldanha, A.J.; Yan, W.; Holmes, S.; Small, P.M. Gene expression diversity among Mycobacterium tuberculosis clinical isolates. Microbiology 2005, 151, 5–14. [Google Scholar] [CrossRef]
- Homolka, S.; Meyer, C.G.; Hillemann, D.; Owusu-Dabo, E.; Adjei, O.; Horstmann, R.D.; Browne, E.N.L.; Chinbuah, A.; Osei, I.; Gyapong, J.; et al. Unequal distribution of resistance-conferring mutations among Mycobacterium tuberculosis and Mycobacterium africanum strains from Ghana. Int. J. Med. Microbiol. 2010, 300, 489–495. [Google Scholar] [CrossRef]
- Manca, C.; Tsenova, L.; Barry, C.E.; Bergtold, A.; Freeman, S.; Haslett, P.A.; Musser, J.M.; Freedman, V.H.; Kaplan, G. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J. Immunol. 1999, 162, 6740–6746. [Google Scholar] [PubMed]
- Newton, S.M.; Smith, R.J.; Wilkinson, K.A.; Nicol, M.P.; Garton, N.J.; Staples, K.J.; Stewart, G.R.; Wain, J.R.; Martineau, A.R.; Fandrich, S.; et al. A deletion defining a common Asian lineage of Mycobacterium tuberculosis associates with immune subversion. Proc. Natl. Acad. Sci. USA 2006, 103, 15594–15598. [Google Scholar] [CrossRef]
- Reed, M.B.; Domenech, P.; Manca, C.; Su, H.; Barczak, A.K.; Kreiswirth, B.N.; Kaplan, G.; Barry, C.E. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 2004, 431, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Alland, D.; Eisen, J.A.; Carpenter, L.; White, O.; Peterson, J.; DeBoy, R.; Dodson, R.; Gwinn, M.; Haft, D.; et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J. Bacteriol. 2002, 184, 5479–5490. [Google Scholar] [CrossRef] [PubMed]
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K.; Ernst, J.D.; Gagneux, S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 2010, 42, 498–503. [Google Scholar] [CrossRef]
- Deitsch, K.W.; Lukehart, S.A.; Stringer, J.R. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 2009, 7, 493–503. [Google Scholar] [CrossRef]
- Li, Q.; Whalen, C.C.; Albert, J.M.; Larkin, R.; Zukowski, L.; Cave, M.D.; Silver, R.F. Differences in rate and variability of intracellular growth of a panel of Mycobacterium tuberculosis clinical isolates within a human monocyte model. Infect. Immun. 2002, 70, 6489–6493. [Google Scholar] [CrossRef]
- Manca, C.; Reed, M.B.; Freeman, S.; Mathema, B.; Kreiswirth, B.; Barry, C.E.; Kaplan, G. Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infect. Immun. 2004, 72, 5511–5514. [Google Scholar] [CrossRef]
- Tsenova, L.; Ellison, E.; Harbacheuski, R.; Moreira, A.L.; Kurepina, N.; Reed, M.B.; Mathema, B.; Barry, C.E.; Kaplan, G. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J. Infect. Dis. 2005, 192, 98–106. [Google Scholar] [CrossRef]
- Chacón-Salinas, R.; Serafín-López, J.; Ramos-Payán, R.; Méndez-Aragón, P.; Hernández-Pando, R.; Van Soolingen, D.; Flores-Romo, L.; Estrada-Parra, S.; Estrada-García, I. Differential pattern of cytokine expression by macrophages infected in vitro with different Mycobacterium tuberculosis genotypes. Clin. Exp. Immunol. 2005, 140, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Ramírez, L.M.; Estrada-García, I.; López-Marín, L.M.; Segura-Salinas, E.; Méndez-Aragón, P.; Van Soolingen, D.; Torres-González, R.; Chacón-Salinas, R.; Estrada-Parra, S.; Maldonado-Bernal, C.; et al. Mycobacterium tuberculosis lipids regulate cytokines, TLR-2/4 and MHC class II expression in human macrophages. Tuberculosis 2008, 88, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Subbian, S.; Bandyopadhyay, N.; Tsenova, L.; O’Brien, P.; Khetani, V.; Kushner, N.L.; Peixoto, B.; Soteropoulos, P.; Bader, J.S.; Karakousis, P.C.; et al. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun. Signal. 2013, 11, 60. [Google Scholar] [CrossRef]
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 2005, 25, 694–701. [Google Scholar] [CrossRef]
- Comas, I.; Gagneux, S. A role for systems epidemiology in tuberculosis research. Trends Microbiol. 2011, 19, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Reiling, N.; Homolka, S.; Walter, K.; Brandenburg, J.; Niwinski, L.; Ernst, M.; Herzmann, C.; Lange, C.; Diel, R.; Ehlers, S.; et al. Clade-specific virulence patterns of Mycobacterium tuberculosis complex strains in human primary macrophages and aerogenically infected mice. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Portevin, D.; Gagneux, S.; Comas, I.; Young, D. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. 2011, 7, e1001307. [Google Scholar] [CrossRef]
- De Jong, B.C.; Hill, P.C.; Aiken, A.; Awine, T.; Antonio, M.; Adetifa, I.M.; Jackson-Sillah, D.J.; Fox, A.; DeRiemer, K.; Gagneux, S.; et al. Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis Lineage in The Gambia. J. Infect. Dis. 2008, 198, 1037–1043. [Google Scholar] [CrossRef]
- Meyer, C.G.; Scarisbrick, G.; Niemann, S.; Browne, E.N.L.; Chinbuah, M.A.; Gyapong, J.; Osei, I.; Owusu-Dabo, E.; Kubica, T.; Rüsch-Gerdes, S.; et al. Pulmonary tuberculosis: Virulence of Mycobacterium africanum and relevance in HIV co-infection. Tuberculosis 2008, 88, 482–489. [Google Scholar] [CrossRef]
- Via, L.E.; Weiner, D.M.; Schimel, D.; Lin, P.L.; Dayao, E.; Tankersley, S.L.; Cai, Y.; Coleman, M.T.; Tomko, J.; Paripati, P.; et al. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus). Infect. Immun. 2013, 81, 2909–2919. [Google Scholar] [CrossRef]
- Peres, R.L.; Vinhas, S.A.; Ribeiro, F.K.C.; Palaci, M.; do Prado, T.N.; Reis-Santos, B.; Zandonade, E.; Suffys, P.N.; Golub, J.E.; Riley, L.W.; et al. Risk factors associated with cluster size of Mycobacterium tuberculosis (MTB) of different RFLP lineages in Brazil. BMC Infect. Dis. 2018, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, S.A.; Gibson, A.L.; Huard, R.C.; Kurepina, N.; Bang, H.; Lazzarini, L.C.O.; Chiu, Y.; Li, J.; Ahuja, S.; Driscoll, J.; et al. Distinct clinical and epidemiological features of tuberculosis in New York City caused by the RDRio Mycobacterium tuberculosis sublineage. Infect. Genet. Evol. 2012, 12, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, L.C.O.; Spindola, S.M.; Bang, H.; Gibson, A.L.; Weisenberg, S.; da Silva Carvalho, W.; Augusto, C.J.; Huard, R.C.; Kritski, A.L.; Ho, J.L. RDRio Mycobacterium tuberculosis infection is associated with a higher frequency of cavitary pulmonary disease. J. Clin. Microbiol. 2008, 46, 2175–2183. [Google Scholar] [CrossRef]
- Barbosa, C. De B.; Lazzarini, L.C.O.; Elias, A.R.; Leung, J.A.M.; Ribeiro, S.B.; da Silva, M.G.; Duarte, R.S.; Suffys, P.; Gomes, H.M.; Kritski, A.L.; et al. Tuberculosis caused by RD Rio Mycobacterium tuberculosis is not associated with differential clinical features. Int. J. Tuberc. Lung Dis. 2012, 16, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Cave, M.D.; Zhang, L.; Foxman, B.; Marrs, C.F.; Bates, J.H.; Yang, Z.H. Population-based study of deletions in five different genomic regions of Mycobacterium tuberculosis and possible clinical relevance of the deletions. J. Clin. Microbiol. 2006, 44, 3940–3946. [Google Scholar] [CrossRef]
- Kong, Y.; Cave, M.D.; Zhang, L.; Foxman, B.; Marrs, C.F.; Bates, J.H.; Yang, Z.H. Association between Mycobacterium tuberculosis Beijing/W lineage strain infection and extrathoracic tuberculosis: Insights from epidemiologic and clinical characterization of the three principal genetic groups of M. tuberculosis clinical isolates. J. Clin. Microbiol. 2007, 45, 409–414. [Google Scholar] [CrossRef]
- Pareek, M.; Evans, J.; Innes, J.; Smith, G.; Hingley-Wilson, S.; Lougheed, K.E.; Sridhar, S.; Dedicoat, M.; Hawkey, P.; Lalvani, A. Ethnicity and mycobacterial lineage as determinants of tuberculosis disease phenotype. Thorax 2013, 68, 221–229. [Google Scholar] [CrossRef]
- Click, E.S.; Moonan, P.K.; Winston, C.A.; Cowan, L.S.; Oeltmann, J.E. Relationship between Mycobacterium tuberculosis phylogenetic lineage and clinical site of tuberculosis. Clin. Infect. Dis. 2012, 54, 211–219. [Google Scholar] [CrossRef]
- Caws, M.; Thwaites, G.; Dunstan, S.; Hawn, T.R.; Thi Ngoc Lan, N.; Thuong, N.T.T.; Stepniewska, K.; Huyen, M.N.T.; Bang, N.D.; Huu Loc, T.; et al. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 2008, 4, e1000034. [Google Scholar] [CrossRef]
- Firdessa, R.; Berg, S.; Hailu, E.; Schelling, E.; Gumi, B.; Erenso, G.; Gadisa, E.; Kiros, T.; Habtamu, M.; Hussein, J.; et al. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg. Infect. Dis. 2013, 19, 460–463. [Google Scholar] [CrossRef]
- Van Crevel, R.; Nelwan, R.H.; de Lenne, W.; Veeraragu, Y.; van der Zanden, A.G.; Amin, Z.; van der Meer, J.W.; van Soolingen, D. Mycobacterium tuberculosis Beijing genotype strains associated with febrile response to treatment. Emerg. Infect. Dis. 2001, 7, 880–883. [Google Scholar] [CrossRef]
- Borgdorff, M.W.; van Deutekom, H.; de Haas, P.E.; Kremer, K.; van Soolingen, D. Mycobacterium tuberculosis, Beijing genotype strains not associated with radiological presentation of pulmonary tuberculosis. Tuberculosis 2004, 84, 337–340. [Google Scholar] [CrossRef]
- Nicol, M.P.; Sola, C.; February, B.; Rastogi, N.; Steyn, L.; Wilkinson, R.J. Distribution of strain families of Mycobacterium tuberculosis causing pulmonary and extrapulmonary disease in hospitalized children in Cape Town, South Africa. J. Clin. Microbiol. 2005, 43, 5779–5781. [Google Scholar] [CrossRef] [PubMed]
- Wampande, E.M.; Mupere, E.; Debanne, S.M.; Asiimwe, B.B.; Nsereko, M.; Mayanja, H.; Eisenach, K.; Kaplan, G.; Boom, H.W.; Gagneux, S.; et al. Long-term dominance of Mycobacterium tuberculosis Uganda family in peri-urban Kampala-Uganda is not associated with cavitary disease. BMC Infect. Dis. 2013, 13, 484. [Google Scholar] [CrossRef]
- Van der Spuy, G.D.; Warren, R.M.; Richardson, M.; Beyers, N.; Behr, M.A.; van Helden, P.D. Use of genetic distance as a measure of ongoing transmission of Mycobacterium tuberculosis. J. Clin. Microbiol. 2003, 41, 5640–5644. [Google Scholar] [CrossRef]
- Borgdorff, M.W.; van Soolingen, D. The re-emergence of tuberculosis: What have we learnt from molecular epidemiology? Clin. Microbiol. Infect. 2013, 19, 889–901. [Google Scholar] [CrossRef]
- Buu, T.N.; van Soolingen, D.; Huyen, M.N.T.; Lan, N.T.N.; Quy, H.T.; Tiemersma, E.W.; Kremer, K.; Borgdorff, M.W.; Cobelens, F.G.J. Increased transmission of Mycobacterium tuberculosis Beijing genotype strains associated with resistance to streptomycin: A population-based study. PLoS ONE 2012, 7, e42323. [Google Scholar] [CrossRef]
- Yang, C.; Luo, T.; Sun, G.; Qiao, K.; Sun, G.; DeRiemer, K.; Mei, J.; Gao, Q. Mycobacterium tuberculosis Beijing strains favor transmission but not drug resistance in China. Clin. Infect. Dis. 2012, 55, 1179–1187. [Google Scholar] [CrossRef]
- Wirth, T.; Hildebrand, F.; Allix-Béguec, C.; Wölbeling, F.; Kubica, T.; Kremer, K.; van Soolingen, D.; Rüsch-Gerdes, S.; Locht, C.; Brisse, S.; et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 2008, 4, e1000160. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Chang, J.-R.; Huang, W.-F.; Hsu, S.-C.; Kuo, S.-C.; Sun, J.-R.; Dou, H.-Y. The pattern of cytokine production in vitro induced by ancient and modern Beijing Mycobacterium tuberculosis strains. PLoS ONE 2014, 9, e94296. [Google Scholar] [CrossRef]
- Duc Anh, D.; Borgdorff, M.W.; Van, L.N.; Lan, N.T.; van Gorkom, T.; Kremer, K.; van Soolingen, D. Mycobacterium tuberculosis Beijing genotype emerging in Vietnam. Emerg. Infect. Dis. 2000, 6, 302–305. [Google Scholar] [CrossRef]
- European concerted action on new generation genetic markers and techniques for the epidemiology and control of tuberculosis. Beijing/W genotype Mycobacterium tuberculosis and drug resistance. Emerg. Infect. Dis. 2006, 12, 736–743. [CrossRef]
- Hanekom, M.; van der Spuy, G.D.; Streicher, E.; Ndabambi, S.L.; McEvoy, C.R.E.; Kidd, M.; Beyers, N.; Victor, T.C.; van Helden, P.D.; Warren, R.M. A recently evolved sublineage of the Mycobacterium tuberculosis Beijing strain family is associated with an increased ability to spread and cause disease. J. Clin. Microbiol. 2007, 45, 1483–1490. [Google Scholar] [CrossRef]
- Cowley, D.; Govender, D.; February, B.; Wolfe, M.; Steyn, L.; Evans, J.; Wilkinson, R.J.; Nicol, M.P. Recent and rapid emergence of W-Beijing strains of Mycobacterium tuberculosis in Cape Town, South Africa. Clin. Infect. Dis. 2008, 47, 1252–1259. [Google Scholar] [CrossRef]
- Van der Spuy, G.D.; Kremer, K.; Ndabambi, S.L.; Beyers, N.; Dunbar, R.; Marais, B.J.; van Helden, P.D.; Warren, R.M. Changing Mycobacterium tuberculosis population highlights clade-specific pathogenic characteristics. Tuberculosis 2009, 89, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Tuite, A.R.; Guthrie, J.L.; Alexander, D.C.; Whelan, M.S.; Lee, B.; Lam, K.; Ma, J.; Fisman, D.N.; Jamieson, F.B. Epidemiological evaluation of spatiotemporal and genotypic clustering of Mycobacterium tuberculosis in Ontario, Canada. Int. J. Tuberc. Lung Dis. 2013, 17, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Lillebaek, T.; Andersen, Å.B.; Dirksen, A.; Glynn, J.R.; Kremer, K. Mycobacterium tuberculosis Beijing Genotype1. Emerg. Infect. Dis. 2003, 9, 1553–1557. [Google Scholar] [CrossRef] [PubMed]
- Albanna, A.S.; Reed, M.B.; Kotar, K.V.; Fallow, A.; McIntosh, F.A.; Behr, M.A.; Menzies, D. Reduced transmissibility of East African Indian strains of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e25075. [Google Scholar] [CrossRef] [PubMed]
- Marais, B.J.; Hesseling, A.C.; Schaaf, H.S.; Gie, R.P.; van Helden, P.D.; Warren, R.M. Mycobacterium tuberculosis transmission is not related to household genotype in a setting of high endemicity. J. Clin. Microbiol. 2009, 47, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Langlois-Klassen, D.; Senthilselvan, A.; Chui, L.; Kunimoto, D.; Saunders, L.D.; Menzies, D.; Long, R. Transmission of Mycobacterium tuberculosis Beijing Strains, Alberta, Canada, 1991–2007. Emerg. Infect. Dis. 2013, 19, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, G.; Caws, M.; Chau, T.T.H.; D’Sa, A.; Lan, N.T.N.; Huyen, M.N.T.; Gagneux, S.; Anh, P.T.H.; Tho, D.Q.; Torok, E.; et al. Relationship between Mycobacterium tuberculosis genotype and the clinical phenotype of pulmonary and meningeal tuberculosis. J. Clin. Microbiol. 2008, 46, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.B.; Shah, R.R.; Maeda, M.K.; Gagneux, S.; Murray, M.B.; Cohen, T.; Johnston, J.C.; Gardy, J.; Lipsitch, M.; Fortune, S.M. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 2013, 45, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Verver, S.; Warren, R.M.; Munch, Z.; Richardson, M.; van der Spuy, G.D.; Borgdorff, M.W.; Behr, M.A.; Beyers, N.; van Helden, P.D. Proportion of tuberculosis transmission that takes place in households in a high-incidence area. Lancet 2004, 363, 212–214. [Google Scholar] [CrossRef]


{kind=link}
| Country/Region | LAM (%) | S (%) | Haarlem (%) | Cameroon (%) | Beijing (%) | Uganda (%) | EAI (%) | Ghana (%) | T Group (%) | X Clade (%) | Undefined/Orphan/Other (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acapulco, Mexico [42] | - | 1.9 | 3 | - | - | - | 44.6 | - | 11.2 | 1.1 | 4.9 |
| Baja California, Mexico [49] | 19.3 | 13.6 | 14.3 | 5.7 | 4.3 | 5.0 | 2.1 | 0.7 | - | - | 35.0 |
| Bogota, Colombia [50] | 49.3 | 3.3 | 25.0 | - | 0.7 | - | - | - | 13.8 | 1.3 | 6.6 |
| Rio Grande, Brazil [35] | 54.0 | 16.0 | - | - | - | - | - | 22.0 | - | - | |
| Rio Grande, Brazil [34] | 34.0 | 4.2 | 12.8 | 2.1 | - | 10.6 | - | - | - | 14.9 | 2.1 |
| Rio de Janeiro, Brazil [37] | 43.6 | 18.3 | - | 0.5 | - | 2.3 | - | 34.9 | 0.5 | - | |
| Rio de Janeiro, Porte Alegre, and Belem, Brazil [51] | 66.2 | 2.5 | 9.7 | - | 0.5 | - | 3.0 | - | 2.0 | 5.2 | 10.1 |
| Parana, Brazil [52] | 26.9 | - | 17.2 | - | - | - | - | - | 11.8 | - | - |
| Minas Gerais state, Brazil [53] | 66.3 | 1.9 | 5.8 | - | - | - | - | - | 14.4 | 1.9 | 8.7 |
| Sao Paulo, Brazil [54] | 2.8 | - | 5.7 | - | - | - | - | - | 35.4 | 2.8 | 7.1 |
| Southeast Brazil (prison population) [55] | 50.0 | - | 11.5 | - | - | - | - | - | 8.7 | 5.7 | 9.2 |
| Metropolitan region, Chile [56] | 39.5 | - | 7.0 | - | 4.7 | - | - | - | 32.5 | 2.3 | - |
| Peru [57] | 23.8 | - | 23.8 | - | 9.3 | - | - | - | 22.3 | - | |
| Peru [58] | 16.2 | - | 43.7 | - | 9.1 | - | - | - | 27.5 | 1.4 | - |
| Santiago, Chile [47] | 23.9 | - | 22.1 | - | - | - | 7.1 | - | 29.9 | 6.7 | - |
| Medellin, Colombia [59] | 39.6 | - | 48.7 | - | - | - | - | - | - | - | 6.4 |
| Cali, Colombia [59] | 39.1 | - | 39.0 | - | - | - | - | - | - | - | 11 |
| Cauca State, Colombia [59] | 24.0 | - | - | - | - | - | - | - | - | - | 40 |
| French Guiana [60] | 23.3 | - | 22.6 | - | - | - | - | - | 32.6 | - | - |
| Bolivia [61] | 26.3 | 2.0 | 39.4 | - | - | - | - | - | 22.2 | 1.0 | 9.1 |
| Paraguay [62] | 52.3 | 9.5 | 18.2 | - | 0.5 | - | - | - | 8.6 | 0.9 | - |
| Venezuela [63] | 53.0 | 1.9 | 5.0 | - | 0.4 | - | 0.2 | - | 10.0 | 1.2 | - |
| Country | LAM (%) | S (%) | Haarlem (%) | Cameroon (%) | Beijing (%) | Uganda I (%) | EAI (%) | Ghana (%) | T Group (%) | X Clade (%) | Undefined/Orphan/Other (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mexico [42,49] | 9.65 | 7.75 | 8.65 | 2.85 | 2.15 | 2.5 | 23.35 | 0.35 | 5.6 | 0.55 | 19.95 |
| Brazil [34,35,37,51,52,53,54,55] | 42.99 | 1.09 | 12.12 | 0.27 | 0.12 | 1.33 | 0.66 | - | 16.15 | 3.87 | 4.64 |
| Peru [57,58] | 20 | - | 33.73 | - | 9.22 | - | - | - | 24.88 | 0.71 | - |
| Chile [47,56] | 31.7 | - | 14.55 | - | 2.35 | - | 3.55 | - | 31.2 | 4.5 | - |
| Colombia [50,59] | 38.01 | 0.82 | 28.18 | - | 0.16 | - | - | - | 3.46 | 0.33 | 1.64 |
| French Guiana [60] | 23.3 | - | 22.6 | - | - | - | - | - | 32.6 | - | - |
| Bolivia [61] | 26.3 | 2 | 39.4 | - | - | - | - | - | 22.2 | 1 | 9.1 |
| Paraguay [62] | 52.3 | 9.5 | 18.2 | - | 0.5 | - | - | - | 8.6 | 0.9 | - |
| Venezuela [63] | 53 | 1.9 | 5 | - | 0.4 | - | 0.2 | - | 10 | 1.2 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodman, M.; Haeusler, I.L.; Grandjean, L. Tuberculosis Genetic Epidemiology: A Latin American Perspective. Genes 2019, 10, 53. https://doi.org/10.3390/genes10010053
Woodman M, Haeusler IL, Grandjean L. Tuberculosis Genetic Epidemiology: A Latin American Perspective. Genes. 2019; 10(1):53. https://doi.org/10.3390/genes10010053
Chicago/Turabian StyleWoodman, Marc, Ilsa L. Haeusler, and Louis Grandjean. 2019. "Tuberculosis Genetic Epidemiology: A Latin American Perspective" Genes 10, no. 1: 53. https://doi.org/10.3390/genes10010053
APA StyleWoodman, M., Haeusler, I. L., & Grandjean, L. (2019). Tuberculosis Genetic Epidemiology: A Latin American Perspective. Genes, 10(1), 53. https://doi.org/10.3390/genes10010053