It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
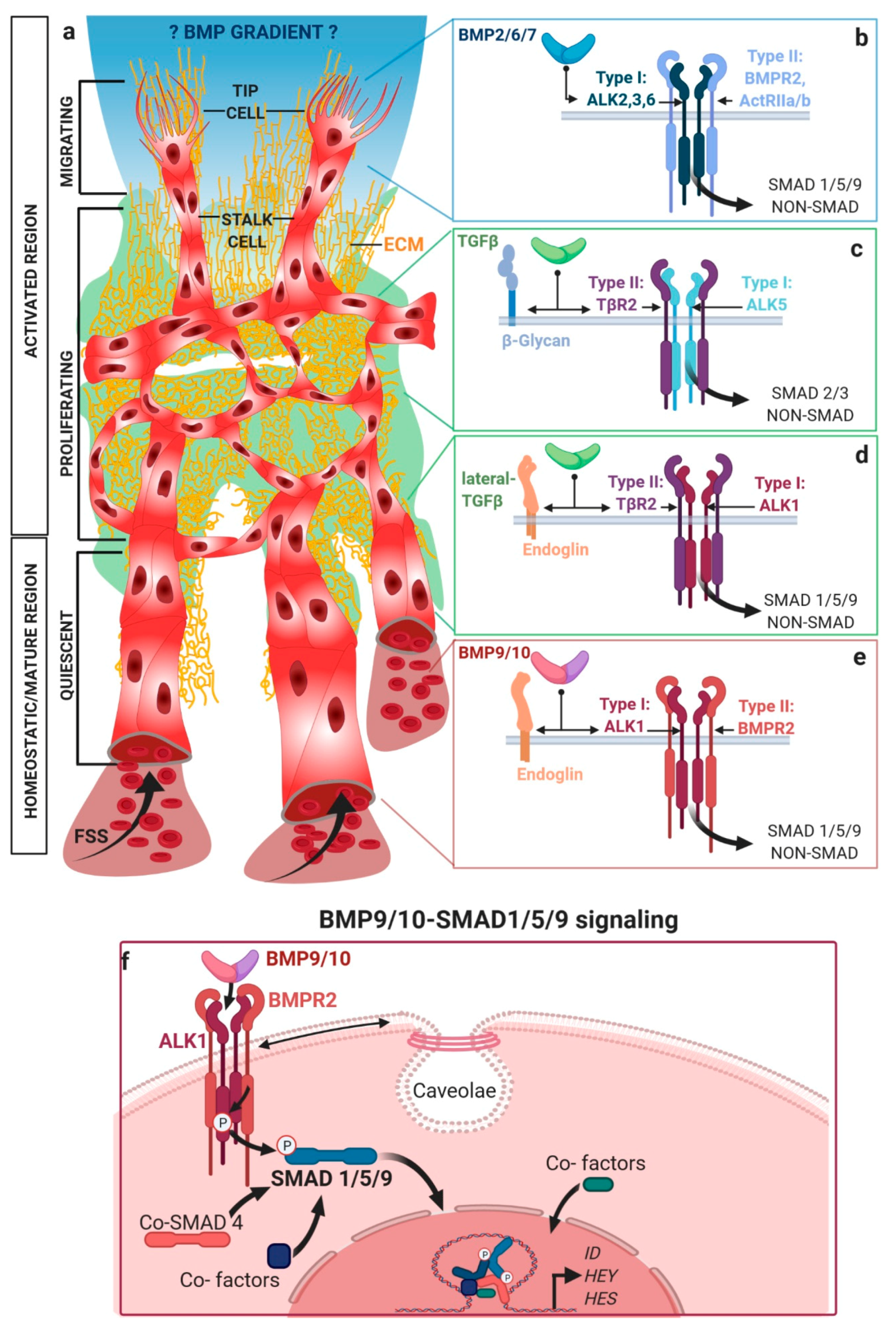
2. Activating Versus Homeostatic TGFβ/BMP Signaling in Endothelial Cells
2.1. Mechanical Regulation of Endocytosis Suggests Modulation of TGFβ/BMP-SMAD Signaling
2.2. TGFβ/BMP-SMAD Signaling Crosstalk to Mechanobiology at Distinct Subcellular Compartments
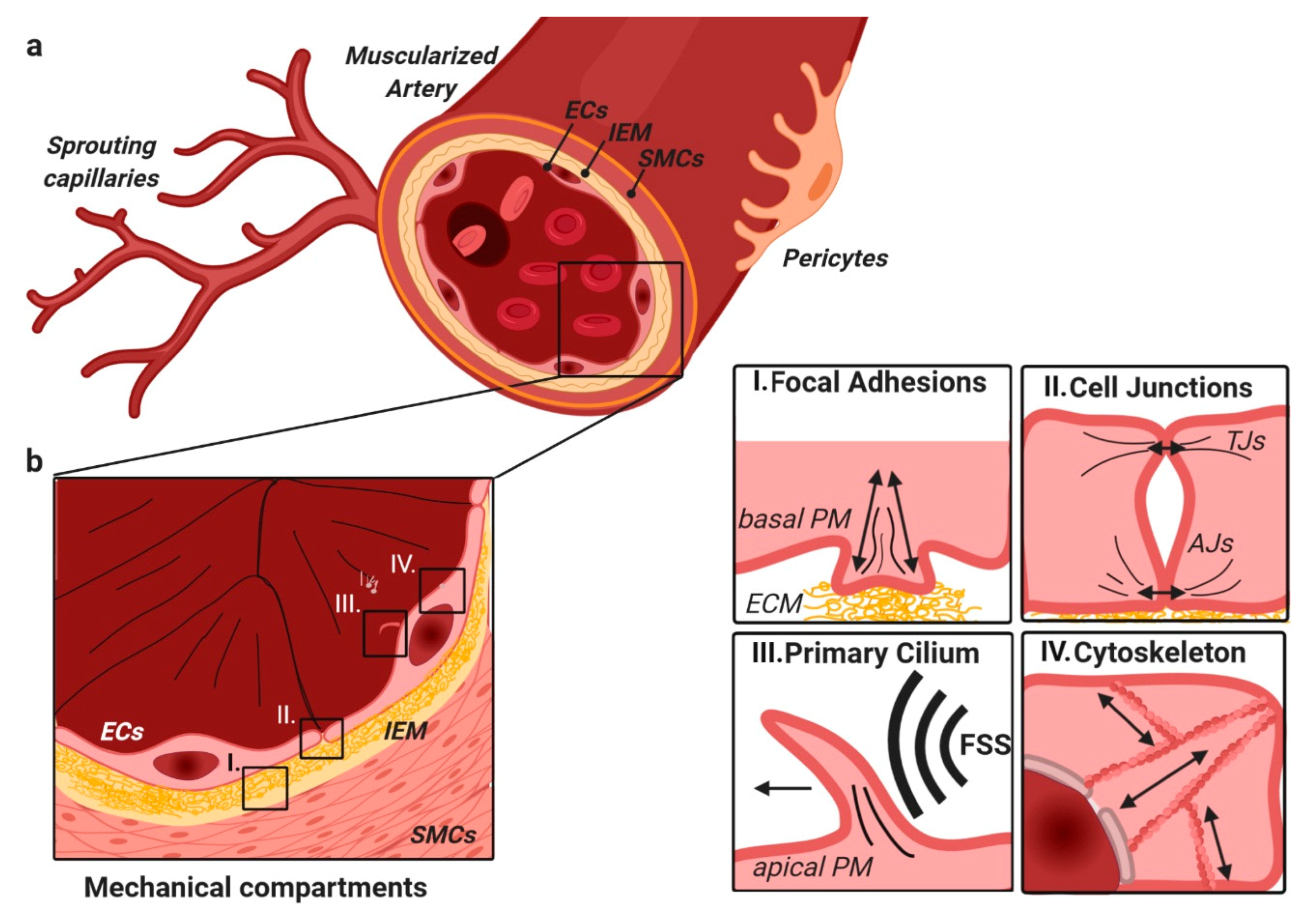
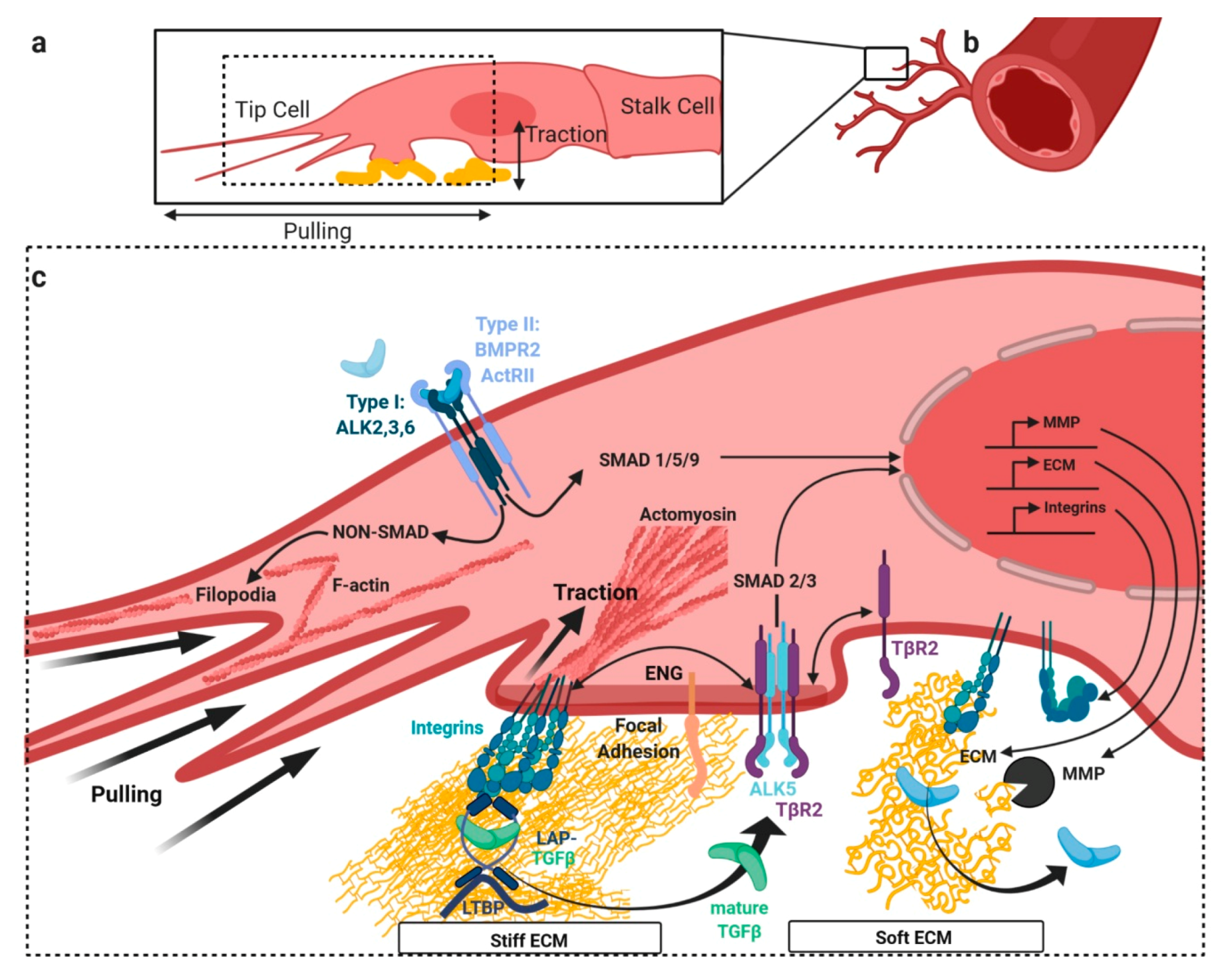
3. TGFβ/BMP Signaling Crosstalk at Focal Adhesions with Extracellular Matrix and Stiffness
3.1. Release of Latent TGFβ from Extracellular Depots by Mechanical Forces
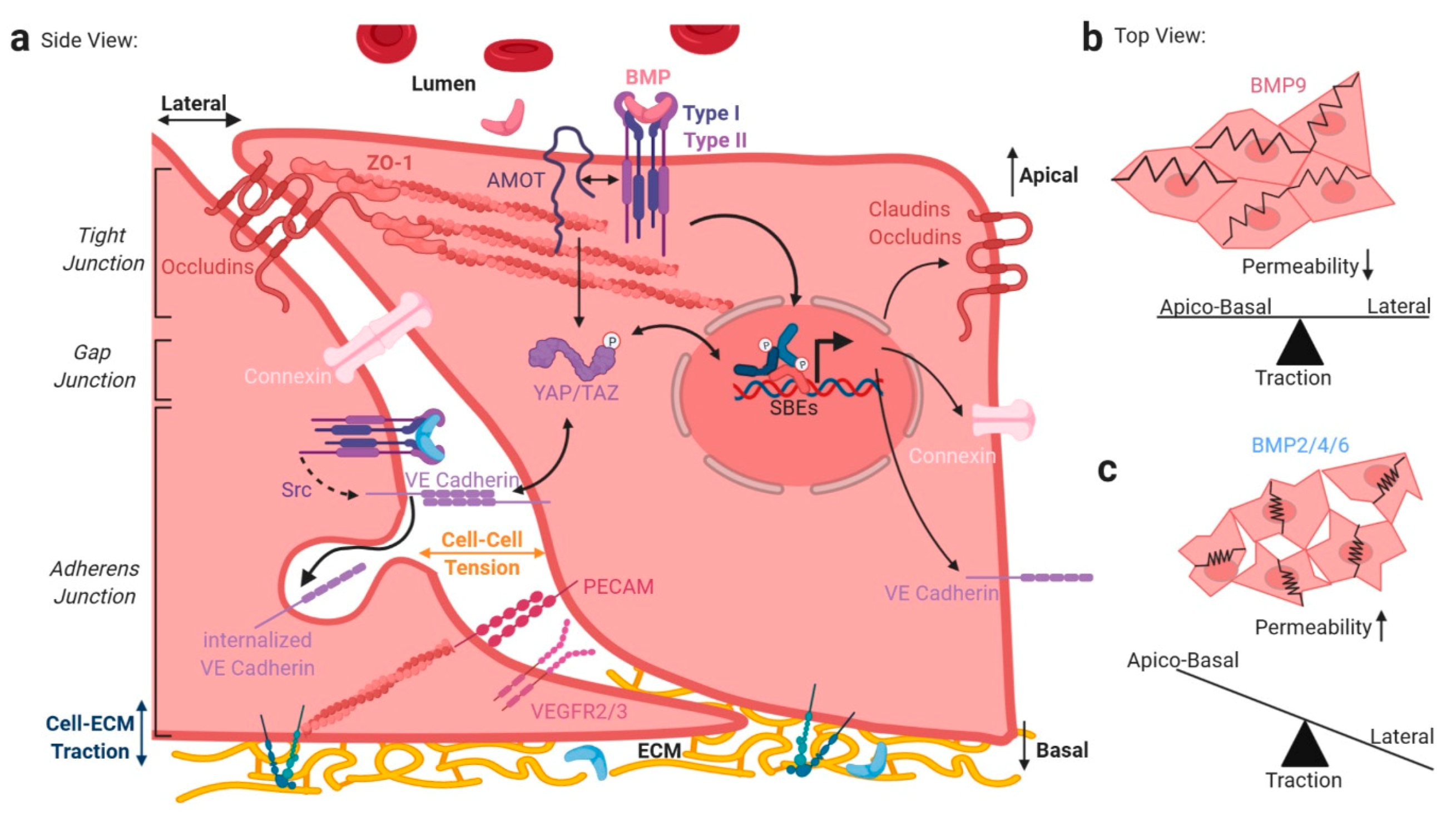
4. BMP/TGFβ Signaling and Integration of Basolateral Forces
5. Shear-Stress-Induced TGFβ/BMP Signaling at the Primary Cilium
The Endothelial Glycocalyx
6. Integrating Forces into Nuclear TGFβ/BMP Signaling
6.1. Forces Acting at the Nuclear Envelope
6.2. Forces Acting on the Chromosomal Architecture
6.3. Forces and SMAD Transcriptional Co-Factors
7. Perspective and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, D.S.J.; Schmierer, B.; Hill, C.S. Tgf-β family ligands exhibit distinct signalling dynamics that are driven by receptor localisation. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Conley, B.A.; Koleva, R.; Smith, J.D.; Kacer, D.; Zhang, D.; Bernabéu, C.; Vary, C.P. Endoglin controls cell migration and composition of focal adhesions: Function of the cytosolic domain. J. Biol. Chem. 2004, 279, 27440–27449. [Google Scholar] [CrossRef]
- López-Casillas, F.; Payne, H.M.; Andres, J.L.; Massagué, J. Betaglycan can act as a dual modulator of tgf-beta access to signaling receptors: Mapping of ligand binding and gag attachment sites. J. Cell Biol. 1994, 124, 557–568. [Google Scholar] [CrossRef]
- Brunner, P.; Hastar, N.; Kaehler, C.; Burdzinski, W.; Jatzlau, J.; Knaus, P. Amot130 drives bmp-smad signaling at the apical membrane in polarized cells. Mol. Biol. Cell 2020, 31, 118–130. [Google Scholar] [CrossRef]
- Miranda, M.Z.; Bialik, J.F.; Speight, P.; Dan, Q.; Yeung, T.; Szászi, K.; Pedersen, S.F.; Kapus, A. Tgf-β1 regulates the expression and transcriptional activity of taz protein via a smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017, 292, 14902–14920. [Google Scholar] [CrossRef] [PubMed]
- García de Vinuesa, A.; Abdelilah-Seyfried, S.; Knaus, P.; Zwijsen, A.; Bailly, S. Bmp signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 2016, 27, 65–79. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Feige, J.J.; Bailly, S. Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev. 2009, 20, 203–212. [Google Scholar] [CrossRef]
- Beets, K.; Huylebroeck, D.; Moya, I.M.; Umans, L.; Zwijsen, A. Robustness in angiogenesis: Notch and bmp shaping waves. Trends Genet. 2013, 29, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef]
- Cai, J.; Pardali, E.; Sánchez-Duffhues, G.; ten Dijke, P. Bmp signaling in vascular diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef]
- Cunha, S.I.; Magnusson, P.U.; Dejana, E.; Lampugnani, M.G. Deregulated tgf-β/bmp signaling in vascular malformations. Circ. Res. 2017, 121, 981–999. [Google Scholar] [CrossRef] [PubMed]
- MacCarrick, G.; Black, J.H., 3rd; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C., 3rd. Loeys-dietz syndrome: A primer for diagnosis and management. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 576–587. [Google Scholar]
- Yang, X.; Liaw, L.; Prudovsky, I.; Brooks, P.C.; Vary, C.; Oxburgh, L.; Friesel, R. Fibroblast growth factor signaling in the vasculature. Curr. Atheroscler. Rep. 2015, 17, 509. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Chong, D.C.; Ola, R.; Dunworth, W.P.; Meadows, S.; Ka, J.; Kaartinen, V.M.; Qyang, Y.; Cleaver, O.; Bautch, V.L.; et al. Alk2/acvr1 and alk3/bmpr1a provide essential function for bone morphogenetic protein-induced retinal angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 657–663. [Google Scholar] [CrossRef]
- Benn, A.; Hiepen, C.; Osterland, M.; Schutte, C.; Zwijsen, A.; Knaus, P. Role of bone morphogenetic proteins in sprouting angiogenesis: Differential bmp receptor-dependent signaling pathways balance stalk vs. Tip cell competence. FASEB J. 2017, 31, 4720–4733. [Google Scholar] [CrossRef]
- Yamashita, H.; Shimizu, A.; Kato, M.; Nishitoh, H.; Ichijo, H.; Hanyu, A.; Morita, I.; Kimura, M.; Makishima, F.; Miyazono, K. Growth/differentiation factor-5 induces angiogenesis in vivo. Exp. Cell Res. 1997, 235, 218–226. [Google Scholar] [CrossRef]
- Glienke, J.; Schmitt, A.O.; Pilarsky, C.; Hinzmann, B.; Weiss, B.; Rosenthal, A.; Thierauch, K.H. Differential gene expression by endothelial cells in distinct angiogenic states. Eur. J. Biochem. 2000, 267, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Miralles, I.; Schisler, J.C.; Patterson, C. New insights into bone morphogenetic protein signaling: Focus on angiogenesis. Curr. Opin. Hematol. 2009, 16, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A team effort coordinated by notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Silberberg, Y.R.; Abeyaratne, R.; Kamm, R.D.; Asada, H.H. Computational modeling of three-dimensional ecm-rigidity sensing to guide directed cell migration. Proc. Natl. Acad. Sci. USA 2018, 115, E390–E399. [Google Scholar] [CrossRef] [PubMed]
- Jacquemet, G.; Stubb, A.; Saup, R.; Miihkinen, M.; Kremneva, E.; Hamidi, H.; Ivaska, J. Filopodome mapping identifies p130cas as a mechanosensitive regulator of filopodia stability. Curr. Biol. CB 2019, 29, 202–216.e207. [Google Scholar] [CrossRef] [PubMed]
- Albuschies, J.; Vogel, V. The role of filopodia in the recognition of nanotopographies. Sci. Rep. 2013, 3, 1658. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. Vegf guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Phng, L.K.; Stanchi, F.; Gerhardt, H. Filopodia are dispensable for endothelial tip cell guidance. Development (Camb. Engl.) 2013, 140, 4031–4040. [Google Scholar] [CrossRef]
- Mouillesseaux, K.P.; Wiley, D.S.; Saunders, L.M.; Wylie, L.A.; Kushner, E.J.; Chong, D.C.; Citrin, K.M.; Barber, A.T.; Park, Y.; Kim, J.D.; et al. Notch regulates bmp responsiveness and lateral branching in vessel networks via smad6. Nat. Commun. 2016, 7, 13247. [Google Scholar] [CrossRef]
- Benn, A.; Bredow, C.; Casanova, I.; Vukičević, S.; Knaus, P. Ve-cadherin facilitates bmp-induced endothelial cell permeability and signaling. J. Cell Sci. 2016, 129, 206–218. [Google Scholar] [CrossRef]
- Pi, X.; Ren, R.; Kelley, R.; Zhang, C.; Moser, M.; Bohil, A.B.; Divito, M.; Cheney, R.E.; Patterson, C. Sequential roles for myosin-x in bmp6-dependent filopodial extension, migration, and activation of bmp receptors. J. Cell Biol. 2007, 179, 1569–1582. [Google Scholar] [CrossRef]
- Ren, R.; Charles, P.C.; Zhang, C.; Wu, Y.; Wang, H.; Patterson, C. Gene expression profiles identify a role for cyclooxygenase 2-dependent prostanoid generation in bmp6-induced angiogenic responses. Blood 2007, 109, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Valdimarsdottir, G.; Goumans, M.J.; Rosendahl, A.; Brugman, M.; Itoh, S.; Lebrin, F.; Sideras, P.; ten Dijke, P. Stimulation of id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 2002, 106, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Heinke, J.; Wehofsits, L.; Zhou, Q.; Zoeller, C.; Baar, K.M.; Helbing, T.; Laib, A.; Augustin, H.; Bode, C.; Patterson, C.; et al. Bmper is an endothelial cell regulator and controls bone morphogenetic protein-4-dependent angiogenesis. Circ. Res. 2008, 103, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Gangopahyay, A.; Oran, M.; Bauer, E.M.; Wertz, J.W.; Comhair, S.A.; Erzurum, S.C.; Bauer, P.M. Bone morphogenetic protein receptor ii is a novel mediator of endothelial nitric-oxide synthase activation. J. Biol. Chem. 2011, 286, 33134–33140. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, T.; Bataille, F.; Spruss, T.; Eissner, G.; Bosserhoff, A.K. Functional implication of bmp4 expression on angiogenesis in malignant melanoma. Oncogene 2007, 26, 4158–4170. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, I.; Yoshino, O.; Osuga, Y.; Shi, J.; Harada, M.; Koga, K.; Hirota, Y.; Hirata, T.; Fujii, T.; Saito, S.; et al. Bone morphogenetic protein 7 increased vascular endothelial growth factor (vegf)-a expression in human granulosa cells and vegf receptor expression in endothelial cells. Reprod. Sci. (Thousand OaksCalif.) 2014, 21, 477–482. [Google Scholar] [CrossRef]
- Chen, W.C.; Chung, C.H.; Lu, Y.C.; Wu, M.H.; Chou, P.H.; Yen, J.Y.; Lai, Y.W.; Wang, G.S.; Liu, S.C.; Cheng, J.K.; et al. Bmp-2 induces angiogenesis by provoking integrin alpha6 expression in human endothelial progenitor cells. Biochem. Pharmacol. 2018, 150, 256–266. [Google Scholar] [CrossRef]
- Finkenzeller, G.; Hager, S.; Stark, G.B. Effects of bone morphogenetic protein 2 on human umbilical vein endothelial cells. Microvasc. Res. 2012, 84, 81–85. [Google Scholar] [CrossRef]
- Langenfeld, E.M.; Langenfeld, J. Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol. Cancer Res. Mcr 2004, 2, 141–149. [Google Scholar]
- Raida, M.; Clement, J.H.; Leek, R.D.; Ameri, K.; Bicknell, R.; Niederwieser, D.; Harris, A.L. Bone morphogenetic protein 2 (bmp-2) and induction of tumor angiogenesis. J. Cancer Res. Clin. Oncol. 2005, 131, 741–750. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ohga, N.; Morishita, Y.; Hida, K.; Miyazono, K.; Watabe, T. Bmp-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J. Cell Sci. 2010, 123, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Heinke, J.; Vargas, A.; Winnik, S.; Krauss, T.; Bode, C.; Patterson, C.; Moser, M. Erk signaling is a central regulator for bmp-4 dependent capillary sprouting. Cardiovasc. Res. 2007, 76, 390–399. [Google Scholar] [CrossRef]
- Kane, R.; Godson, C.; O’Brien, C. Chordin-like 1, a bone morphogenetic protein-4 antagonist, is upregulated by hypoxia in human retinal pericytes and plays a role in regulating angiogenesis. Mol. Vis. 2008, 14, 1138–1148. [Google Scholar] [PubMed]
- Tate, C.M.; Mc Entire, J.; Pallini, R.; Vakana, E.; Wyss, L.; Blosser, W.; Ricci-Vitiani, L.; D’Alessandris, Q.G.; Morgante, L.; Giannetti, S.; et al. A bmp7 variant inhibits tumor angiogenesis in vitro and in vivo through direct modulation of endothelial cell biology. PLoS ONE 2015, 10, e0125697. [Google Scholar] [CrossRef]
- Del Toro, R.; Prahst, C.; Mathivet, T.; Siegfried, G.; Kaminker, J.S.; Larrivee, B.; Breant, C.; Duarte, A.; Takakura, N.; Fukamizu, A.; et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood 2010, 116, 4025–4033. [Google Scholar] [CrossRef] [PubMed]
- Bier, E.; De Robertis, E.M. Embryo development. Bmp gradients: A paradigm for morphogen-mediated developmental patterning. Science 2015, 348, aaa5838. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, Y.; Fukuhara, S.; Ando, K.; Matsuda, M.; Mochizuki, N. Cdc42 mediates bmp-induced sprouting angiogenesis through fmnl3-driven assembly of endothelial filopodia in zebrafish. Dev. Cell 2015, 32, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Benn, A.; Alonso, F.; Mangelschots, J.; Génot, E.; Lox, M.; Zwijsen, A. Bmp-smad1/5 signaling regulates retinal vascular development. Biomolecules 2020, 10, 488. [Google Scholar] [CrossRef]
- Mallet, C.; Vittet, D.; Feige, J.J.; Bailly, S. Tgfbeta1 induces vasculogenesis and inhibits angiogenic sprouting in an embryonic stem cell differentiation model: Respective contribution of alk1 and alk5. Stem Cells 2006, 24, 2420–2427. [Google Scholar] [CrossRef]
- Ito, C.; Akimoto, T.; Ioka, T.; Kobayashi, T.; Kusano, E. Tgf-beta inhibits vascular sprouting through tgf-beta type i receptor in the mouse embryonic aorta. Tohoku J. Exp. Med. 2009, 218, 63–71. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct tgf-beta type i receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. Tgf-beta family signaling in tumor suppression and cancer progression. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.; Nornes, S.; Payne, S.; Wallace, M.D.; Fritzsche, M.; Louphrasitthiphol, P.; Wilkinson, R.N.; Chouliaras, K.M.; Liu, K.; Plant, K.; et al. Venous identity requires bmp signalling through alk3. Nat. Commun. 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Mallet, C.; Keramidas, M.; Lamande, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Rostama, B.; Turner, J.E.; Seavey, G.T.; Norton, C.R.; Gridley, T.; Vary, C.P.; Liaw, L. Dll4/notch1 and bmp9 interdependent signaling induces human endothelial cell quiescence via p27kip1 and thrombospondin-1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- Sengle, G.; Charbonneau, N.L.; Ono, R.N.; Sasaki, T.; Alvarez, J.; Keene, D.R.; Bächinger, H.P.; Sakai, L.Y. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J. Biol. Chem. 2008, 283, 13874–13888. [Google Scholar] [CrossRef]
- Kienast, Y.; Jucknischke, U.; Scheiblich, S.; Thier, M.; de Wouters, M.; Haas, A.; Lehmann, C.; Brand, V.; Bernicke, D.; Honold, K.; et al. Rapid activation of bone morphogenic protein 9 by receptor-mediated displacement of pro-domains. J. Biol. Chem. 2016, 291, 3395–3410. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of bmp9 and bmp10 as functional activators of the orphan activin receptor-like kinase 1 (alk1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. Bmp-9 signals via alk1 and inhibits bfgf-induced endothelial cell proliferation and vegf-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef]
- Townson, S.A.; Martinez-Hackert, E.; Greppi, C.; Lowden, P.; Sako, D.; Liu, J.; Ucran, J.A.; Liharska, K.; Underwood, K.W.; Seehra, J.; et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (alk1) signaling complex. J. Biol. Chem. 2012, 287, 27313–27325. [Google Scholar] [CrossRef]
- Ostrowski, M.A.; Huang, N.F.; Walker, T.W.; Verwijlen, T.; Poplawski, C.; Khoo, A.S.; Cooke, J.P.; Fuller, G.G.; Dunn, A.R. Microvascular endothelial cells migrate upstream and align against the shear stress field created by impinging flow. Biophys. J. 2014, 106, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Baker, B.M.; Chen, C.S.; Schwartz, M.A. Endothelial cell sensing of flow direction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, R. Endothelial Cell Culture; Roy Bicknell: Cambridge, UK, 1996; p. 152. [Google Scholar]
- Luo, J.; Tang, M.; Huang, J.; He, B.C.; Gao, J.L.; Chen, L.; Zuo, G.W.; Zhang, W.; Luo, Q.; Shi, Q.; et al. Tgfbeta/bmp type i receptors alk1 and alk2 are essential for bmp9-induced osteogenic signaling in mesenchymal stem cells. J. Biol. Chem. 2010, 285, 29588–29598. [Google Scholar] [CrossRef] [PubMed]
- Laux, D.W.; Young, S.; Donovan, J.P.; Mansfield, C.J.; Upton, P.D.; Roman, B.L. Circulating bmp10 acts through endothelial alk1 to mediate flow-dependent arterial quiescence. Development (Camb. Engl.) 2013, 140, 3403–3412. [Google Scholar] [CrossRef] [PubMed]
- Corti, P.; Young, S.; Chen, C.Y.; Patrick, M.J.; Rochon, E.R.; Pekkan, K.; Roman, B.L. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development (Camb. Engl.) 2011, 138, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Audiger, C.; Popovic, N.; Akla, N.; Lanthier, K.; Legault-Navarrete, I.; Melichar, H.; Costantino, S.; Lesage, S.; Larrivée, B. Bmp9 signaling promotes the normalization of tumor blood vessels. Oncogene 2020, 39, 2996–3014. [Google Scholar] [CrossRef]
- Young, K.; Conley, B.; Romero, D.; Tweedie, E.; O’Neill, C.; Pinz, I.; Brogan, L.; Lindner, V.; Liaw, L.; Vary, C.P. Bmp9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood 2012, 120, 4263–4273. [Google Scholar] [CrossRef]
- Capasso, T.L.; Li, B.; Volek, H.J.; Khalid, W.; Rochon, E.R.; Anbalagan, A.; Herdman, C.; Yost, H.J.; Villanueva, F.S.; Kim, K.; et al. Bmp10-mediated alk1 signaling is continuously required for vascular development and maintenance. Angiogenesis 2020, 23, 203–220. [Google Scholar] [CrossRef]
- Richter, A.; Alexdottir, M.S.; Magnus, S.H.; Richter, T.R.; Morikawa, M.; Zwijsen, A.; Valdimarsdottir, G. Egfl7 mediates bmp9-induced sprouting angiogenesis of endothelial cells derived from human embryonic stem cells. Stem Cell Rep. 2019, 12, 1250–1259. [Google Scholar] [CrossRef]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. Taz controls smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional control by the smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. Yap/taz are mechanoregulators of tgf-β-smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. JASN 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Koinuma, D.; Tsutsumi, S.; Vasilaki, E.; Kanki, Y.; Heldin, C.H.; Aburatani, H.; Miyazono, K. Chip-seq reveals cell type-specific binding patterns of bmp-specific smads and a novel binding motif. Nucleic Acids Res. 2011, 39, 8712–8727. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (bmp) and activin type ii receptors balance bmp9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef]
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Bidart, M.; Feige, J.J.; Bailly, S. Bmp9 and bmp10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef]
- Lamouille, S.; Mallet, C.; Feige, J.J.; Bailly, S. Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood 2002, 100, 4495–4501. [Google Scholar] [CrossRef]
- Cheifetz, S.; Bellón, T.; Calés, C.; Vera, S.; Bernabeu, C.; Massagué, J.; Letarte, M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar]
- Barbara, N.P.; Wrana, J.L.; Letarte, M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J. Biol. Chem. 1999, 274, 584–594. [Google Scholar] [CrossRef]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural basis of the human endoglin-bmp9 interaction: Insights into bmp signaling and hht1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef]
- Blanco, F.J.; Santibanez, J.F.; Guerrero-Esteo, M.; Langa, C.; Vary, C.P.; Bernabeu, C. Interaction and functional interplay between endoglin and alk-1, two components of the endothelial transforming growth factor-beta receptor complex. J. Cell. Physiol. 2005, 204, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Huang, J.J.; Golzio, C.; Gao, X.; Hector-Greene, M.; Katsanis, N.; Blobe, G.C. Endoglin interacts with vegfr2 to promote angiogenesis. FASEB J. 2018, 32, 2934–2949. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Larrivée, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Chaouat, A.; Coulet, F.; Favre, C.; Simonneau, G.; Weitzenblum, E.; Soubrier, F.; Humbert, M. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004, 59, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Iyer, K.V.; Piscitello-Gómez, R.; Paijmans, J.; Jülicher, F.; Eaton, S. Epithelial viscoelasticity is regulated by mechanosensitive e-cadherin turnover. Curr. Biol. CB 2019, 29, 578–591. [Google Scholar] [CrossRef]
- Yoshida, A.; Sakai, N.; Uekusa, Y.; Imaoka, Y.; Itagaki, Y.; Suzuki, Y.; Yoshimura, S.H. Morphological changes of plasma membrane and protein assembly during clathrin-mediated endocytosis. PLoS Biol. 2018, 16, e2004786. [Google Scholar] [CrossRef]
- Wu, X.S.; Elias, S.; Liu, H.; Heureaux, J.; Wen, P.J.; Liu, A.P.; Kozlov, M.M.; Wu, L.G. Membrane tension inhibits rapid and slow endocytosis in secretory cells. Biophys. J. 2017, 113, 2406–2414. [Google Scholar] [CrossRef]
- Ehrlich, M. Endocytosis and trafficking of bmp receptors: Regulatory mechanisms for fine-tuning the signaling response in different cellular contexts. Cytokine Growth Factor Rev. 2016, 27, 35–42. [Google Scholar] [CrossRef]
- Zuo, W.; Chen, Y.G. Specific activation of mitogen-activated protein kinase by transforming growth factor-beta receptors in lipid rafts is required for epithelial cell plasticity. Mol. Biol. Cell 2009, 20, 1020–1029. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate tgf-beta receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef]
- Hayes, S.; Chawla, A.; Corvera, S. Tgf beta receptor internalization into eea1-enriched early endosomes: Role in signaling to smad2. J. Cell Biol. 2002, 158, 1239–1249. [Google Scholar] [CrossRef]
- Hartung, A.; Bitton-Worms, K.; Rechtman, M.M.; Wenzel, V.; Boergermann, J.H.; Hassel, S.; Henis, Y.I.; Knaus, P. Different routes of bone morphogenic protein (bmp) receptor endocytosis influence bmp signaling. Mol. Cell. Biol. 2006, 26, 7791–7805. [Google Scholar] [CrossRef] [PubMed]
- Bonor, J.; Adams, E.L.; Bragdon, B.; Moseychuk, O.; Czymmek, K.J.; Nohe, A. Initiation of bmp2 signaling in domains on the plasma membrane. J. Cell. Physiol. 2012, 227, 2880–2888. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, S.; Bragdon, B.; Moseychuk, O.; Bonor, J.; Dhurjati, P.; Nohe, A. Caveolae regulate smad signaling as verified by novel imaging and system biology approaches. J. Cell. Physiol. 2013, 228, 1060–1069. [Google Scholar] [CrossRef]
- He, Z.; Zhang, W.; Mao, S.; Li, N.; Li, H.; Lin, J.M. Shear stress-enhanced internalization of cell membrane proteins indicated by a hairpin-type DNA probe. Anal. Chem. 2018, 90, 5540–5545. [Google Scholar] [CrossRef] [PubMed]
- Urbano, R.L.; Furia, C.; Basehore, S.; Clyne, A.M. Stiff substrates increase inflammation-induced endothelial monolayer tension and permeability. Biophys. J. 2017, 113, 645–655. [Google Scholar] [CrossRef]
- Albinsson, S.; Nordström, I.; Swärd, K.; Hellstrand, P. Differential dependence of stretch and shear stress signaling on caveolin-1 in the vascular wall. Am. J. Physiol. Cell Physiol. 2008, 294, C271–C279. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Blanco, F.J.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; del Pozo, M.A.; Bernabeu, C. Caveolin-1 interacts and cooperates with the transforming growth factor-beta type i receptor alk1 in endothelial caveolae. Cardiovasc. Res. 2008, 77, 791–799. [Google Scholar] [CrossRef]
- Schwartz, E.A.; Reaven, E.; Topper, J.N.; Tsao, P.S. Transforming growth factor-beta receptors localize to caveolae and regulate endothelial nitric oxide synthase in normal human endothelial cells. Biochem. J. 2005, 390, 199–206. [Google Scholar] [CrossRef]
- Nohe, A.; Keating, E.; Underhill, T.M.; Knaus, P.; Petersen, N.O. Dynamics and interaction of caveolin-1 isoforms with bmp-receptors. J. Cell Sci. 2005, 118, 643–650. [Google Scholar] [CrossRef]
- Mathew, R. Pathogenesis of pulmonary hypertension: A case for caveolin-1 and cell membrane integrity. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H15–H25. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies alk-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Park, H.; Go, Y.M.; St John, P.L.; Maland, M.C.; Lisanti, M.P.; Abrahamson, D.R.; Jo, H. Plasma membrane cholesterol is a key molecule in shear stress-dependent activation of extracellular signal-regulated kinase. J. Biol. Chem. 1998, 273, 32304–32311. [Google Scholar] [CrossRef]
- Boyd, N.L.; Park, H.; Yi, H.; Boo, Y.C.; Sorescu, G.P.; Sykes, M.; Jo, H. Chronic shear induces caveolae formation and alters erk and akt responses in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1113–H1122. [Google Scholar] [CrossRef]
- Sun, X.; Fu, Y.; Gu, M.; Zhang, L.; Li, D.; Li, H.; Chien, S.; Shyy, J.Y.; Zhu, Y. Activation of integrin α5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2016, 113, 769–774. [Google Scholar] [CrossRef]
- Radel, C.; Rizzo, V. Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of csk to mediate actin reorganization. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H936–H945. [Google Scholar] [CrossRef][Green Version]
- Radel, C.; Carlile-Klusacek, M.; Rizzo, V. Participation of caveolae in beta1 integrin-mediated mechanotransduction. Biochem. Biophys. Res. Commun. 2007, 358, 626–631. [Google Scholar] [CrossRef][Green Version]
- Shi, F.; Sottile, J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef]
- Zhou, J.; Lee, P.L.; Lee, C.I.; Wei, S.Y.; Lim, S.H.; Lin, T.E.; Chien, S.; Chiu, J.J. Bmp receptor-integrin interaction mediates responses of vascular endothelial smad1/5 and proliferation to disturbed flow. J. Thromb. Haemost. JTH 2013, 11, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, M.; Ando, J.; Yamamoto, K.; Fujita, T.; Ying, Y.; Anderson, R.G. Sites of ca(2+) wave initiation move with caveolae to the trailing edge of migrating cells. J. Cell Sci. 2002, 115, 475–484. [Google Scholar] [PubMed]
- Basagiannis, D.; Zografou, S.; Murphy, C.; Fotsis, T.; Morbidelli, L.; Ziche, M.; Bleck, C.; Mercer, J.; Christoforidis, S. Vegf induces signalling and angiogenesis by directing vegfr2 internalisation through macropinocytosis. J. Cell Sci. 2016, 129, 4091–4104. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross talk among tgf-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef]
- Zhou, J.; Lee, P.L.; Tsai, C.S.; Lee, C.I.; Yang, T.L.; Chuang, H.S.; Lin, W.W.; Lin, T.E.; Lim, S.H.; Wei, S.Y.; et al. Force-specific activation of smad1/5 regulates vascular endothelial cell cycle progression in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2012, 109, 7770–7775. [Google Scholar] [CrossRef]
- Jin, Z.G.; Ueba, H.; Tanimoto, T.; Lungu, A.O.; Frame, M.D.; Berk, B.C. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ. Res. 2003, 93, 354–363. [Google Scholar] [CrossRef]
- Peacock, H.M.; Tabibian, A.; Criem, N.; Caolo, V.; Hamard, L.; Deryckere, A.; Haefliger, J.A.; Kwak, B.R.; Zwijsen, A.; Jones, E.A.V. Impaired smad1/5 mechanotransduction and cx37 (connexin37) expression enable pathological vessel enlargement and shunting. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e87–e104. [Google Scholar] [CrossRef]
- Santos-Oliveira, P.; Correia, A.; Rodrigues, T.; Ribeiro-Rodrigues, T.M.; Matafome, P.; Rodríguez-Manzaneque, J.C.; Seiça, R.; Girão, H.; Travasso, R.D. The force at the tip--modelling tension and proliferation in sprouting angiogenesis. PLoS Comput. Biol. 2015, 11, e1004436. [Google Scholar] [CrossRef]
- Bibi, H.; Armoni, M.; Ohali, M.; Pollak, S. tuberculosis in early childhood. Harefuah 1996, 131, 166–215. (In Hebrew) [Google Scholar]
- Fantin, A.; Lampropoulou, A.; Gestri, G.; Raimondi, C.; Senatore, V.; Zachary, I.; Ruhrberg, C. Nrp1 regulates cdc42 activation to promote filopodia formation in endothelial tip cells. Cell Rep. 2015, 11, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Ruhrberg, C.; Abramsson, A.; Fujisawa, H.; Shima, D.; Betsholtz, C. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev. Dyn. 2004, 231, 503–509. [Google Scholar] [CrossRef]
- Lee-Hoeflich, S.T.; Causing, C.G.; Podkowa, M.; Zhao, X.; Wrana, J.L.; Attisano, L. Activation of limk1 by binding to the bmp receptor, bmprii, regulates bmp-dependent dendritogenesis. EMBO J. 2004, 23, 4792–4801. [Google Scholar] [CrossRef]
- Hiepen, C.; Benn, A.; Denkis, A.; Lukonin, I.; Weise, C.; Boergermann, J.H.; Knaus, P. Bmp2-induced chemotaxis requires pi3k p55γ/p110α-dependent phosphatidylinositol (3,4,5)-triphosphate production and ll5β recruitment at the cytocortex. BMC Biol. 2014, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Sieminski, A.L.; Hebbel, R.P.; Gooch, K.J. The relative magnitudes of endothelial force generation and matrix stiffness modulate capillary morphogenesis in vitro. Exp. Cell Res. 2004, 297, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Mason, B.N.; Starchenko, A.; Williams, R.M.; Bonassar, L.J.; Reinhart-King, C.A. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater. 2013, 9, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef]
- Wong, L.; Kumar, A.; Gabela-Zuniga, B.; Chua, J.; Singh, G.; Happe, C.L.; Engler, A.J.; Fan, Y.; McCloskey, K.E. Substrate stiffness directs diverging vascular fates. Acta Biomater. 2019, 96, 321–329. [Google Scholar] [CrossRef]
- Xue, C.; Zhang, T.; Xie, X.; Zhang, Q.; Zhang, S.; Zhu, B.; Lin, Y.; Cai, X. Substrate stiffness regulates arterial-venous differentiation of endothelial progenitor cells via the ras/mek pathway. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 1799–1808. [Google Scholar] [CrossRef]
- Frye, M.; Taddei, A.; Dierkes, C.; Martinez-Corral, I.; Fielden, M.; Ortsäter, H.; Kazenwadel, J.; Calado, D.P.; Ostergaard, P.; Salminen, M.; et al. Matrix stiffness controls lymphatic vessel formation through regulation of a gata2-dependent transcriptional program. Nat. Commun. 2018, 9, 1511. [Google Scholar] [CrossRef]
- Kniazeva, E.; Putnam, A.J. Endothelial cell traction and ecm density influence both capillary morphogenesis and maintenance in 3-d. Am. J. Physiol. Cell Physiol. 2009, 297, C179–C187. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Liu, S.L.; Byfield, F.J.; Janmey, P.A.; Assoian, R.K. Measuring the stiffness of ex vivo mouse aortas using atomic force microscopy. J. Vis. Exp. Jove 2016, 116. [Google Scholar] [CrossRef]
- Spronck, B.; Humphrey, J. Arterial stiffness: Different metrics, different meanings. J. Biomech. Eng. 2019, 141, 0910041–09100412. [Google Scholar]
- Woodrum, D.A.; Romano, A.J.; Lerman, A.; Pandya, U.H.; Brosh, D.; Rossman, P.J.; Lerman, L.O.; Ehman, R.L. Vascular wall elasticity measurement by magnetic resonance imaging. Magn. Reson. Med. 2006, 56, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Bastounis, E.E.; Yeh, Y.T.; Theriot, J.A. Subendothelial stiffness alters endothelial cell traction force generation while exerting a minimal effect on the transcriptome. Sci. Rep. 2019, 9, 18209. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, J.; Huynh, J.; Williams, R.M.; Reinhart-King, C.A. Indentation measurements of the subendothelial matrix in bovine carotid arteries. J. Biomech. 2011, 44, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Nishimura, N.; Rana, K.; Peloquin, J.M.; Califano, J.P.; Montague, C.R.; King, M.R.; Schaffer, C.B.; Reinhart-King, C.A. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. 2011, 3, 112ra122. [Google Scholar] [CrossRef]
- De Beaufort, H.W.L.; Ferrara, A.; Conti, M.; Moll, F.L.; van Herwaarden, J.A.; Figueroa, C.A.; Bismuth, J.; Auricchio, F.; Trimarchi, S. Comparative analysis of porcine and human thoracic aortic stiffness. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2018, 55, 560–566. [Google Scholar] [CrossRef]
- Kothapalli, D.; Liu, S.L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by apoe and apoe-hdl linked to suppression of ecm gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef]
- Teng, Z.; Zhang, Y.; Huang, Y.; Feng, J.; Yuan, J.; Lu, Q.; Sutcliffe, M.P.F.; Brown, A.J.; Jing, Z.; Gillard, J.H. Material properties of components in human carotid atherosclerotic plaques: A uniaxial extension study. Acta Biomater. 2014, 10, 5055–5063. [Google Scholar] [CrossRef] [PubMed]
- Zhong, A.; Mirzaei, Z.; Simmons, C.A. The roles of matrix stiffness and ß-catenin signaling in endothelial-to-mesenchymal transition of aortic valve endothelial cells. Cardiovasc. Eng. Technol. 2018, 9, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chen, W.L.; Sider, K.L.; Yip, C.Y.; Simmons, C.A. Β-catenin mediates mechanically regulated, transforming growth factor-β1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, H.; Wang, L.M.; Ren, K.F.; Martins, M.C.; Barbosa, M.A.; Ji, J. Effect of polyelectrolyte film stiffness on endothelial cells during endothelial-to-mesenchymal transition. Biomacromolecules 2015, 16, 3584–3593. [Google Scholar] [CrossRef] [PubMed]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef]
- Vaeyens, M.M.; Jorge-Peñas, A.; Barrasa-Fano, J.; Steuwe, C.; Heck, T.; Carmeliet, P.; Roeffaers, M.; Van Oosterwyck, H. Matrix deformations around angiogenic sprouts correlate to sprout dynamics and suggest pulling activity. Angiogenesis 2020, 23, 315–324. [Google Scholar] [CrossRef]
- LaValley, D.J.; Zanotelli, M.R.; Bordeleau, F.; Wang, W.; Schwager, S.C.; Reinhart-King, C.A. Matrix stiffness enhances vegfr-2 internalization, signaling, and proliferation in endothelial cells. Converg. Sci. Phys. Oncol. 2017, 3, 044001. [Google Scholar] [CrossRef]
- Rys, J.P.; DuFort, C.C.; Monteiro, D.A.; Baird, M.A.; Oses-Prieto, J.A.; Chand, S.; Burlingame, A.L.; Davidson, M.W.; Alliston, T.N. Discrete spatial organization of tgfβ receptors couples receptor multimerization and signaling to cellular tension. eLife 2015, 4, e09300. [Google Scholar] [CrossRef]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into bmp receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Isogai, Z.; Ono, R.N.; Ushiro, S.; Keene, D.R.; Chen, Y.; Mazzieri, R.; Charbonneau, N.L.; Reinhardt, D.P.; Rifkin, D.B.; Sakai, L.Y. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 2003, 278, 2750–2757. [Google Scholar] [CrossRef]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of tgfbeta1. J. Cell Biol. 2007, 176, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, J.; Taipale, J.; Keski-Oja, J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein ltbp-1. EMBO J. 1996, 15, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-cys repeats of tgf-beta binding proteins, ltbps, creates a hydrophobic interaction surface for binding of small latent tgf-beta. Mol. Biol. Cell 2000, 11, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Hirani, R.; Hanssen, E.; Gibson, M.A. Ltbp-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with ltbp-1 for binding to this microfibrillar protein. Matrix Biol. J. Int. Soc. Matrix Biol. 2007, 26, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005, 24, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Mu, Z.; Dabovic, B.; Jurukovski, V.; Yu, D.; Sung, J.; Xiong, X.; Munger, J.S. Absence of integrin-mediated tgfbeta1 activation in vivo recapitulates the phenotype of tgfbeta1-null mice. J. Cell Biol. 2007, 176, 787–793. [Google Scholar] [CrossRef]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M.; Costell, M.; Alman, B.A.; Genot, E.; Hinz, B. Prestress in the extracellular matrix sensitizes latent tgf-β1 for activation. J. Cell Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of tgf-β. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent tgf-β1 complex. Curr. Biol. CB 2011, 21, 2046–2054. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent tgf-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Qin, Y.; Garrison, B.S.; Ma, W.; Wang, R.; Jiang, A.; Li, J.; Mistry, M.; Bronson, R.T.; Santoro, D.; Franco, C.; et al. A milieu molecule for tgf-β required for microglia function in the nervous system. Cell 2018, 174, 156–171.e116. [Google Scholar] [CrossRef] [PubMed]
- Roca-Cusachs, P.; Iskratsch, T.; Sheetz, M.P. Finding the weakest link: Exploring integrin-mediated mechanical molecular pathways. J. Cell Sci. 2012, 125, 3025–3038. [Google Scholar] [CrossRef] [PubMed]
- De Laporte, L.; Rice, J.J.; Tortelli, F.; Hubbell, J.A. Tenascin c promiscuously binds growth factors via its fifth fibronectin type iii-like domain. PLoS ONE 2013, 8, e62076. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, R.; Hoffmann, E.; Sebald, W. Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur. J. Biochem. 1996, 237, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, J.; Ishihara, A.; Fukunaga, K.; Sasaki, K.; White, M.J.V.; Briquez, P.S.; Hubbell, J.A. Laminin heparin-binding peptides bind to several growth factors and enhance diabetic wound healing. Nat. Commun. 2018, 9, 2163. [Google Scholar] [CrossRef] [PubMed]
- Von Offenberg Sweeney, N.; Cummins, P.M.; Birney, Y.A.; Cullen, J.P.; Redmond, E.M.; Cahill, P.A. Cyclic strain-mediated regulation of endothelial matrix metalloproteinase-2 expression and activity. Cardiovasc. Res. 2004, 63, 625–634. [Google Scholar] [CrossRef]
- Wang, B.W.; Chang, H.; Lin, S.; Kuan, P.; Shyu, K.G. Induction of matrix metalloproteinases-14 and -2 by cyclical mechanical stretch is mediated by tumor necrosis factor-alpha in cultured human umbilical vein endothelial cells. Cardiovasc. Res. 2003, 59, 460–469. [Google Scholar] [CrossRef]
- Von Offenberg Sweeney, N.; Cummins, P.M.; Cotter, E.J.; Fitzpatrick, P.A.; Birney, Y.A.; Redmond, E.M.; Cahill, P.A. Cyclic strain-mediated regulation of vascular endothelial cell migration and tube formation. Biochem. Biophys. Res. Commun. 2005, 329, 573–582. [Google Scholar] [CrossRef]
- Enenstein, J.; Waleh, N.S.; Kramer, R.H. Basic fgf and tgf-beta differentially modulate integrin expression of human microvascular endothelial cells. Exp. Cell Res. 1992, 203, 499–503. [Google Scholar] [CrossRef]
- Pepper, M.S. Transforming growth factor-beta: Vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997, 8, 21–43. [Google Scholar] [CrossRef]
- Baranska, P.; Jerczynska, H.; Pawlowska, Z.; Koziolkiewicz, W.; Cierniewski, C.S. Expression of integrins and adhesive properties of human endothelial cell line ea.Hy 926. Cancer Genom. Proteom. 2005, 2, 265–269. [Google Scholar]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. Bmpr2 acts as a gatekeeper to protect endothelial cells from increased tgfβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef]
- Tian, H.; Mythreye, K.; Golzio, C.; Katsanis, N.; Blobe, G.C. Endoglin mediates fibronectin/α5β1 integrin and tgf-β pathway crosstalk in endothelial cells. EMBO J. 2012, 31, 3885–3900. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Tweedie, E.; Conley, B.; Ames, J.; FitzSimons, M.; Brooks, P.; Liaw, L.; Vary, C.P. Bmp9 crosstalk with the hippo pathway regulates endothelial cell matricellular and chemokine responses. PLoS ONE 2015, 10, e0122892. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Burg, N.; Yoshinaga, K.; Janczak, C.A.; Rifkin, D.B.; Coller, B.S. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood 2008, 112, 3650–3660. [Google Scholar] [CrossRef]
- Baker, A.B.; Ettenson, D.S.; Jonas, M.; Nugent, M.A.; Iozzo, R.V.; Edelman, E.R. Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine tgf-beta signaling pathway. Circ. Res. 2008, 103, 289–297. [Google Scholar] [CrossRef]
- Kouzbari, K.; Hossan, M.R.; Arrizabalaga, J.H.; Varshney, R.; Simmons, A.D.; Gostynska, S.; Nollert, M.U.; Ahamed, J. Oscillatory shear potentiates latent tgf-β1 activation more than steady shear as demonstrated by a novel force generator. Sci. Rep. 2019, 9, 6065. [Google Scholar] [CrossRef]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for tgfbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes snail-mediated endothelial-mesenchymal transition through convergence of smad-dependent and smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef]
- Maruthamuthu, V.; Sabass, B.; Schwarz, U.S.; Gardel, M.L. Cell-ecm traction force modulates endogenous tension at cell-cell contacts. Proc. Natl. Acad. Sci. USA 2011, 108, 4708–4713. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G. Endothelial cell-to-cell junctions: Adhesion and signaling in physiology and pathology. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Rüffer, C.; Strey, A.; Janning, A.; Kim, K.S.; Gerke, V. Cell-cell junctions of dermal microvascular endothelial cells contain tight and adherens junction proteins in spatial proximity. Biochemistry 2004, 43, 5360–5369. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu Leclerc, V.; Roy, O.; Santerre, K.; Proulx, S. Tgf-β1 promotes cell barrier function upon maturation of corneal endothelial cells. Sci. Rep. 2018, 8, 4438. [Google Scholar] [CrossRef]
- Dohgu, S.; Yamauchi, A.; Takata, F.; Naito, M.; Tsuruo, T.; Higuchi, S.; Sawada, Y.; Kataoka, Y. Transforming growth factor-beta1 upregulates the tight junction and p-glycoprotein of brain microvascular endothelial cells. Cell. Mol. Neurobiol. 2004, 24, 491–497. [Google Scholar] [CrossRef]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. Tgf-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef]
- Gkatzis, K.; Thalgott, J.; Dos-Santos-Luis, D.; Martin, S.; Lamandé, N.; Carette, M.F.; Disch, F.; Snijder, R.J.; Westermann, C.J.; Mager, J.J.; et al. Interaction between alk1 signaling and connexin40 in the development of arteriovenous malformations. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 707–717. [Google Scholar] [CrossRef]
- Seebach, J.; Dieterich, P.; Luo, F.; Schillers, H.; Vestweber, D.; Oberleithner, H.; Galla, H.J.; Schnittler, H.J. Endothelial barrier function under laminar fluid shear stress. Lab. Investig. A J. Tech. Methods Pathol. 2000, 80, 1819–1831. [Google Scholar] [CrossRef]
- Helbing, T.; Arnold, L.; Wiltgen, G.; Hirschbihl, E.; Gabelmann, V.; Hornstein, A.; Esser, J.S.; Diehl, P.; Grundmann, S.; Busch, H.J.; et al. Endothelial bmp4 regulates leukocyte diapedesis and promotes inflammation. Inflammation 2017, 40, 1862–1874. [Google Scholar] [CrossRef]
- Hussein, K.A.; Choksi, K.; Akeel, S.; Ahmad, S.; Megyerdi, S.; El-Sherbiny, M.; Nawaz, M.; Abu El-Asrar, A.; Al-Shabrawey, M. Bone morphogenetic protein 2: A potential new player in the pathogenesis of diabetic retinopathy. Exp. Eye Res. 2014, 125, 79–88. [Google Scholar] [CrossRef]
- Helbing, T.; Wiltgen, G.; Hornstein, A.; Brauers, E.Z.; Arnold, L.; Bauer, A.; Esser, J.S.; Diehl, P.; Grundmann, S.; Fink, K.; et al. Bone morphogenetic protein-modulator bmper regulates endothelial barrier function. Inflammation 2017, 40, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, L.; Yang, X.; Tong, Z.; Southwood, M.; Caruso, P.; Upton, P.D.; Yang, P.; Bocobo, G.A.; Nikolic, I.; et al. Circulating bmp9 protects the pulmonary endothelium during inflammation-induced lung injury in mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Akla, N.; Viallard, C.; Popovic, N.; Lora Gil, C.; Sapieha, P.; Larrivée, B. Bmp9 (bone morphogenetic protein-9)/alk1 (activin-like kinase receptor type i) signaling prevents hyperglycemia-induced vascular permeability. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1821–1836. [Google Scholar] [CrossRef] [PubMed]
- Abu Taha, A.; Taha, M.; Seebach, J.; Schnittler, H.J. Arp2/3-mediated junction-associated lamellipodia control ve-cadherin-based cell junction dynamics and maintain monolayer integrity. Mol. Biol. Cell 2014, 25, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Ehling, M.; März, S.; Seebach, J.; Tarbashevich, K.; Sixta, T.; Pitulescu, M.E.; Werner, A.C.; Flach, B.; Montanez, E.; et al. Polarized actin and ve-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat. Commun. 2017, 8, 2210. [Google Scholar] [CrossRef]
- Cao, J.; Schnittler, H. Putting ve-cadherin into jail for junction remodeling. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Coon, B.G.; Baeyens, N.; Han, J.; Budatha, M.; Ross, T.D.; Fang, J.S.; Yun, S.; Thomas, J.L.; Schwartz, M.A. Intramembrane binding of ve-cadherin to vegfr2 and vegfr3 assembles the endothelial mechanosensory complex. J. Cell Biol. 2015, 208, 975–986. [Google Scholar] [CrossRef]
- Rudini, N.; Felici, A.; Giampietro, C.; Lampugnani, M.; Corada, M.; Swirsding, K.; Garrè, M.; Liebner, S.; Letarte, M.; ten Dijke, P.; et al. Ve-cadherin is a critical endothelial regulator of tgf-beta signalling. EMBO J. 2008, 27, 993–1004. [Google Scholar] [CrossRef]
- Hirate, Y.; Sasaki, H. The role of angiomotin phosphorylation in the hippo pathway during preimplantation mouse development. Tissue Barriers 2014, 2, e28127. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel hippo pathway component that inhibits yap oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, C.; Disanza, A.; Bravi, L.; Barrios-Rodiles, M.; Corada, M.; Frittoli, E.; Savorani, C.; Lampugnani, M.G.; Boggetti, B.; Niessen, C.; et al. The actin-binding protein eps8 binds ve-cadherin and modulates yap localization and signaling. J. Cell Biol. 2015, 211, 1177–1192. [Google Scholar] [CrossRef]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, C.A.; et al. Yap and taz regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef]
- Young, Y.N.; Downs, M.; Jacobs, C.R. Dynamics of the primary cilium in shear flow. Biophys. J. 2012, 103, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.G.; Steed, E.; Ferreira, R.R.; Roth, S.; Ramspacher, C.; Boselli, F.; Charvin, G.; Liebling, M.; Wyart, C.; Schwab, Y.; et al. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep. 2014, 6, 799–808. [Google Scholar] [CrossRef]
- Schwartz, E.A.; Leonard, M.L.; Bizios, R.; Bowser, S.S. Analysis and modeling of the primary cilium bending response to fluid shear. Am. J. Physiol. 1997, 272, F132–F138. [Google Scholar] [CrossRef]
- Hoey, D.A.; Downs, M.E.; Jacobs, C.R. The mechanics of the primary cilium: An intricate structure with complex function. J. Biomech. 2012, 45, 17–26. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Gradilone, S.A.; LaRusso, N.F. Calcium signaling in cilia and ciliary-mediated intracellular calcium signaling: Are they independent or coordinated molecular events? Hepatology (Baltim. Md.) 2014, 60, 1783–1785. [Google Scholar] [CrossRef]
- Hartmannsgruber, V.; Heyken, W.T.; Kacik, M.; Kaistha, A.; Grgic, I.; Harteneck, C.; Liedtke, W.; Hoyer, J.; Köhler, R. Arterial response to shear stress critically depends on endothelial trpv4 expression. PLoS ONE 2007, 2, e827. [Google Scholar] [CrossRef]
- Corrigan, M.A.; Johnson, G.P.; Stavenschi, E.; Riffault, M.; Labour, M.N.; Hoey, D.A. Trpv4-mediates oscillatory fluid shear mechanotransduction in mesenchymal stem cells in part via the primary cilium. Sci. Rep. 2018, 8, 3824. [Google Scholar] [CrossRef] [PubMed]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. Trpv4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef]
- Sharma, S.; Goswami, R.; Zhang, D.X.; Rahaman, S.O. Trpv4 regulates matrix stiffness and tgfβ1-induced epithelial-mesenchymal transition. J. Cell. Mol. Med. 2019, 23, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Sreekumar, V.; Aspera-Werz, R.H.; Sajadian, S.O.; Wintermeyer, E.; Sandmann, G.H.; Bahrs, C.; Hengstler, J.G.; Godoy, P.; Nussler, A.K. Tgf-β(1) impairs mechanosensation of human osteoblasts via hdac6-mediated shortening and distortion of primary cilia. J. Mol. Med. (Berl. Ger.) 2017, 95, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Jung, J.K.; Im, S.S.; Lee, S.R.; Jang, B.C.; Park, K.M.; Kim, J.I. Deficiency of primary cilia in kidney epithelial cells induces epithelial to mesenchymal transition. Biochem. Biophys. Res. Commun. 2018, 496, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Ezura, Y.; Hayata, T.; Notomi, T.; Izu, Y.; Noda, M. Tgf-β suppresses ift88 expression in chondrocytic atdc5 cells. J. Cell. Physiol. 2015, 230, 2788–2795. [Google Scholar] [CrossRef]
- Egorova, A.D.; Khedoe, P.P.; Goumans, M.J.; Yoder, B.K.; Nauli, S.M.; ten Dijke, P.; Poelmann, R.E.; Hierck, B.P. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res. 2011, 108, 1093–1101. [Google Scholar] [CrossRef]
- Sánchez-Duffhues, G.; de Vinuesa, A.G.; Lindeman, J.H.; Mulder-Stapel, A.; DeRuiter, M.C.; Van Munsteren, C.; Goumans, M.J.; Hierck, B.P.; Ten Dijke, P. Slug is expressed in endothelial cells lacking primary cilia to promote cellular calcification. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 616–627. [Google Scholar] [CrossRef]
- Clement, C.A.; Ajbro, K.D.; Koefoed, K.; Vestergaard, M.L.; Veland, I.R.; Henriques de Jesus, M.P.; Pedersen, L.B.; Benmerah, A.; Andersen, C.Y.; Larsen, L.A.; et al. Tgf-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef]
- Vion, A.C.; Alt, S.; Klaus-Bergmann, A.; Szymborska, A.; Zheng, T.; Perovic, T.; Hammoutene, A.; Oliveira, M.B.; Bartels-Klein, E.; Hollfinger, I.; et al. Primary cilia sensitize endothelial cells to bmp and prevent excessive vascular regression. J. Cell Biol. 2018, 217, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Koefoed, K.; Skat-Rørdam, J.; Andersen, P.; Warzecha, C.B.; Pye, M.; Andersen, T.A.; Ajbro, K.D.; Bendsen, E.; Narimatsu, M.; Vilhardt, F.; et al. The e3 ubiquitin ligase smurf1 regulates cell-fate specification and outflow tract septation during mammalian heart development. Sci. Rep. 2018, 8, 9542. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.G.; Koch, D.L.; Paszek, M.J. Equilibrium modeling of the mechanics and structure of the cancer glycocalyx. Biophys. J. 2019, 116, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Gouverneur, M.; Berg, B.; Nieuwdorp, M.; Stroes, E.; Vink, H. Vasculoprotective properties of the endothelial glycocalyx: Effects of fluid shear stress. J. Intern. Med. 2006, 259, 393–400. [Google Scholar] [CrossRef]
- Van den Berg, B.M.; Spaan, J.A.; Rolf, T.M.; Vink, H. Atherogenic region and diet diminish glycocalyx dimension and increase intima-to-media ratios at murine carotid artery bifurcation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H915–H920. [Google Scholar] [CrossRef]
- Ueda, A.; Shimomura, M.; Ikeda, M.; Yamaguchi, R.; Tanishita, K. Effect of glycocalyx on shear-dependent albumin uptake in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2287–H2294. [Google Scholar] [CrossRef]
- Galie, P.A.; Nguyen, D.H.; Choi, C.K.; Cohen, D.M.; Janmey, P.A.; Chen, C.S. Fluid shear stress threshold regulates angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2014, 111, 7968–7973. [Google Scholar] [CrossRef]
- Uchida, C.; Haas, T.L. Endothelial cell timp-1 is upregulated by shear stress via sp-1 and the tgfβ1 signaling pathways. Biochem. Cell Biol. Biochim. Et Biol. Cell. 2014, 92, 77–83. [Google Scholar] [CrossRef]
- Negishi, M.; Lu, D.; Zhang, Y.Q.; Sawada, Y.; Sasaki, T.; Kayo, T.; Ando, J.; Izumi, T.; Kurabayashi, M.; Kojima, I.; et al. Upregulatory expression of furin and transforming growth factor-beta by fluid shear stress in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 785–790. [Google Scholar] [CrossRef]
- McCaffrey, T.A.; Falcone, D.J.; Du, B. Transforming growth factor-beta 1 is a heparin-binding protein: Identification of putative heparin-binding regions and isolation of heparins with varying affinity for tgf-beta 1. J. Cell. Physiol. 1992, 152, 430–440. [Google Scholar] [CrossRef]
- Rider, C.C.; Mulloy, B. Heparin, heparan sulphate and the tgf-β cytokine superfamily. Molecules 2017, 22, 713. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Gaynor, K.U.; Rowan, S.C.; Walsh, S.M.; Fabre, A.; Boylan, J.; Keane, M.P.; McLoughlin, P. Altered expression of bone morphogenetic protein accessory proteins in murine and human pulmonary fibrosis. Am. J. Pathol. 2016, 186, 600–615. [Google Scholar] [CrossRef] [PubMed]
- Paine-Saunders, S.; Viviano, B.L.; Economides, A.N.; Saunders, S. Heparan sulfate proteoglycans retain noggin at the cell surface: A potential mechanism for shaping bone morphogenetic protein gradients. J. Biol. Chem. 2002, 277, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Resnick, N.; Collins, T.; Atkinson, W.; Bonthron, D.T.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Platelet-derived growth factor b chain promoter contains a cis-acting fluid shear-stress-responsive element. Proc. Natl. Acad. Sci. USA 1993, 90, 4591–4595. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go with the flow”: How krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef]
- Tkachenko, E.; Gutierrez, E.; Saikin, S.K.; Fogelstrand, P.; Kim, C.; Groisman, A.; Ginsberg, M.H. The nucleus of endothelial cell as a sensor of blood flow direction. Biol. Open 2013, 2, 1007–1012. [Google Scholar] [CrossRef]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Coan, D.E.; Wechezak, A.R.; Viggers, R.F.; Sauvage, L.R. Effect of shear stress upon localization of the golgi apparatus and microtubule organizing center in isolated cultured endothelial cells. J. Cell Sci. 1993, 104, 1145–1153. [Google Scholar]
- Cucina, A.; Sterpetti, A.V.; Pupelis, G.; Fragale, A.; Lepidi, S.; Cavallaro, A.; Giustiniani, Q.; Santoro D’Angelo, L. Shear stress induces changes in the morphology and cytoskeleton organisation of arterial endothelial cells. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 1995, 9, 86–92. [Google Scholar] [CrossRef]
- Joukov, V.; De Nicolo, A. The Centrosome and the Primary Cilium: The Yin and Yang of a Hybrid Organelle. Cells 2019, 8, 701. [Google Scholar] [CrossRef]
- Lai, J.K.; Stainier, D.Y. Pushing yap into the nucleus with shear force. Dev. Cell 2017, 40, 517–518. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force triggers yap nuclear entry by regulating transport across nuclear pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Na, S.; Collin, O.; Chowdhury, F.; Tay, B.; Ouyang, M.; Wang, Y.; Wang, N. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc. Natl. Acad. Sci. USA 2008, 105, 6626–6631. [Google Scholar] [CrossRef]
- Isermann, P.; Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. Curr. Biol. CB 2013, 23, R1113–R1121. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Chen, C.S.; Ingber, D.E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA 1997, 94, 849–854. [Google Scholar] [CrossRef]
- Ramdas, N.M.; Shivashankar, G.V. Cytoskeletal control of nuclear morphology and chromatin organization. J. Mol. Biol. 2015, 427, 695–706. [Google Scholar] [CrossRef]
- Martin, M.; Veloso, A.; Wu, J.; Katrukha, E.A.; Akhmanova, A. Control of endothelial cell polarity and sprouting angiogenesis by non-centrosomal microtubules. eLife 2018, 7. [Google Scholar] [CrossRef]
- Kushner, E.J.; Ferro, L.S.; Yu, Z.; Bautch, V.L. Excess centrosomes perturb dynamic endothelial cell repolarization during blood vessel formation. Mol. Biol. Cell 2016, 27, 1911–1920. [Google Scholar] [CrossRef]
- Wu, J.; Misra, G.; Russell, R.J.; Ladd, A.J.; Lele, T.P.; Dickinson, R.B. Effects of dynein on microtubule mechanics and centrosome positioning. Mol. Biol. Cell 2011, 22, 4834–4841. [Google Scholar] [CrossRef]
- Kim, J.K.; Louhghalam, A.; Lee, G.; Schafer, B.W.; Wirtz, D.; Kim, D.H. Nuclear lamin a/c harnesses the perinuclear apical actin cables to protect nuclear morphology. Nat. Commun. 2017, 8, 2123. [Google Scholar] [CrossRef]
- Yoshigi, M.; Hoffman, L.M.; Jensen, C.C.; Yost, H.J.; Beckerle, M.C. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 2005, 171, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Shiu, J.Y.; Aires, L.; Lin, Z.; Vogel, V. Nanopillar force measurements reveal actin-cap-mediated yap mechanotransduction. Nat. Cell Biol. 2018, 20, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, E.C.; Foisner, R. Proteins that associate with lamins: Many faces, many functions. Exp. Cell Res. 2007, 313, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins a and c but not lamin b1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef]
- Ihalainen, T.O.; Aires, L.; Herzog, F.A.; Schwartlander, R.; Moeller, J.; Vogel, V. Differential basal-to-apical accessibility of lamin a/c epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat. Mater. 2015, 14, 1252–1261. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of a-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-a scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef]
- Qi, Y.X.; Jiang, J.; Jiang, X.H.; Wang, X.D.; Ji, S.Y.; Han, Y.; Long, D.K.; Shen, B.R.; Yan, Z.Q.; Chien, S.; et al. Pdgf-bb and tgf-{beta}1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1908–1913. [Google Scholar] [CrossRef]
- Jiang, Y.; Ji, J.Y. Expression of nuclear lamin proteins in endothelial cells is sensitive to cell passage and fluid shear stress. Cell. Mol. Bioeng. 2018, 11, 53–64. [Google Scholar] [CrossRef]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. Erk1/2 directly acts on ctgf/ccn2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin a/c gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef]
- Hata, A.; Lagna, G.; Massagué, J.; Hemmati-Brivanlou, A. Smad6 inhibits bmp/smad1 signaling by specifically competing with the smad4 tumor suppressor. Genes Dev. 1998, 12, 186–197. [Google Scholar] [CrossRef]
- Altraja, S.; Jaama, J.; Valk, E.; Altraja, A. Changes in the proteome of human bronchial epithelial cells following stimulation with leucotriene e4 and transforming growth factor-beta1. Respirology 2009, 14, 39–45. [Google Scholar] [CrossRef]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic instability is induced by persistent proliferation of cells undergoing epithelial-to-mesenchymal transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Estévez-Salmerón, L.D.; Stroschein, S.L.; Zhu, X.; He, J.; Zhou, S.; Luo, K. The integral inner nuclear membrane protein man1 physically interacts with the r-smad proteins to repress signaling by the transforming growth factor-{beta} superfamily of cytokines. J. Biol. Chem. 2005, 280, 15992–16001. [Google Scholar] [CrossRef]
- Lin, F.; Morrison, J.M.; Wu, W.; Worman, H.J. Man1, an integral protein of the inner nuclear membrane, binds smad2 and smad3 and antagonizes transforming growth factor-beta signaling. Hum. Mol. Genet. 2005, 14, 437–445. [Google Scholar] [CrossRef]
- Raju, G.P.; Dimova, N.; Klein, P.S.; Huang, H.C. Sane, a novel lem domain protein, regulates bone morphogenetic protein signaling through interaction with smad1. J. Biol. Chem. 2003, 278, 428–437. [Google Scholar] [CrossRef]
- Osada, S.; Ohmori, S.Y.; Taira, M. Xman1, an inner nuclear membrane protein, antagonizes bmp signaling by interacting with smad1 in xenopus embryos. Development (Camb. Engl.) 2003, 130, 1783–1794. [Google Scholar] [CrossRef]
- Bourgeois, B.; Gilquin, B.; Tellier-Lebègue, C.; Östlund, C.; Wu, W.; Pérez, J.; El Hage, P.; Lallemand, F.; Worman, H.J.; Zinn-Justin, S. Inhibition of tgf-β signaling at the nuclear envelope: Characterization of interactions between man1, smad2 and smad3, and ppm1a. Sci. Signal. 2013, 6, ra49. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef]
- Cain, N.E.; Jahed, Z.; Schoenhofen, A.; Valdez, V.A.; Elkin, B.; Hao, H.; Harris, N.J.; Herrera, L.A.; Woolums, B.M.; Mofrad, M.R.K.; et al. Conserved sun-kash interfaces mediate linc complex-dependent nuclear movement and positioning. Curr. Biol. CB 2018, 28, 3086–3097. [Google Scholar] [CrossRef]
- Han, Y.; Wang, L.; Yao, Q.P.; Zhang, P.; Liu, B.; Wang, G.L.; Shen, B.R.; Cheng, B.; Wang, Y.; Jiang, Z.L.; et al. Nuclear envelope proteins nesprin2 and lamina regulate proliferation and apoptosis of vascular endothelial cells in response to shear stress. Biochim. Biophys. Acta 2015, 1853, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.N.; Eckes, B.; Glöckner, G.; Groth, M.; Neumann, S.; Gloy, J.; Sellin, L.; Walz, G.; Schneider, M.; Karakesisoglou, I.; et al. The nuclear envelope protein nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus (AustinTex.) 2012, 3, 172–186. [Google Scholar] [CrossRef]
- Van Steensel, B.; Belmont, A.S. Lamina-associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Uhler, C.; Shivashankar, G.V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727. [Google Scholar] [CrossRef]
- Schlereth, K.; Weichenhan, D.; Bauer, T.; Heumann, T.; Giannakouri, E.; Lipka, D.; Jaeger, S.; Schlesner, M.; Aloy, P.; Eils, R.; et al. The transcriptomic and epigenetic map of vascular quiescence in the continuous lung endothelium. eLife 2018, 7. [Google Scholar] [CrossRef]
- Hayashi, M.; Nimura, K.; Kashiwagi, K.; Harada, T.; Takaoka, K.; Kato, H.; Tamai, K.; Kaneda, Y. Comparative roles of twist-1 and id1 in transcriptional regulation by bmp signaling. J. Cell Sci. 2007, 120, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Lassar, A.B. Smad-dependent recruitment of a histone deacetylase/sin3a complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of nkx3.2. Mol. Cell. Biol. 2003, 23, 8704–8717. [Google Scholar] [CrossRef]
- Wotton, D.; Lo, R.S.; Lee, S.; Massagué, J. A smad transcriptional corepressor. Cell 1999, 97, 29–39. [Google Scholar] [CrossRef]
- Frontelo, P.; Leader, J.E.; Yoo, N.; Potocki, A.C.; Crawford, M.; Kulik, M.; Lechleider, R.J. Suv39h histone methyltransferases interact with smads and cooperate in bmp-induced repression. Oncogene 2004, 23, 5242–5251. [Google Scholar] [CrossRef]
- Grannas, K.; Arngården, L.; Lönn, P.; Mazurkiewicz, M.; Blokzijl, A.; Zieba, A.; Söderberg, O. Crosstalk between hippo and tgfβ: Subcellular localization of yap/taz/smad complexes. J. Mol. Biol. 2015, 427, 3407–3415. [Google Scholar] [CrossRef]
- Alarcón, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear cdks drive smad transcriptional activation and turnover in bmp and tgf-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Wang, Y.; Zhang, P.; Chen, H.; Xu, Z.; Jiao, J.; Yuan, Z. Bmp2-smad signaling represses the proliferation of embryonic neural stem cells through yap. J. Neurosci. 2014, 34, 12039–12048. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. Tead mediates yap-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef]
- Bartholin, L.; Wessner, L.L.; Chirgwin, J.M.; Guise, T.A. The human cyr61 gene is a transcriptional target of transforming growth factor beta in cancer cells. Cancer Lett. 2007, 246, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, T.; Cheng, A.S.; Yu, J.; Kang, W.; To, K.F. The tead family and its oncogenic role in promoting tumorigenesis. Int. J. Mol. Sci. 2016, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. Tgf-β synergizes with defects in the hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef]
- Huang, Z.; Hu, J.; Pan, J.; Wang, Y.; Hu, G.; Zhou, J.; Mei, L.; Xiong, W.C. Yap stabilizes smad1 and promotes bmp2-induced neocortical astrocytic differentiation. Development (Camb. Engl.) 2016, 143, 2398–2409. [Google Scholar] [CrossRef]
- Mihira, H.; Suzuki, H.I.; Akatsu, Y.; Yoshimatsu, Y.; Igarashi, T.; Miyazono, K.; Watabe, T. Tgf-β-induced mesenchymal transition of ms-1 endothelial cells requires smad-dependent cooperative activation of rho signals and mrtf-a. J. Biochem. 2012, 151, 145–156. [Google Scholar] [CrossRef]
- Morita, T.; Mayanagi, T.; Sobue, K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007, 179, 1027–1042. [Google Scholar] [CrossRef]
- Wang, D.Z.; Li, S.; Hockemeyer, D.; Sutherland, L.; Wang, Z.; Schratt, G.; Richardson, J.A.; Nordheim, A.; Olson, E.N. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 14855–14860. [Google Scholar] [CrossRef]
- Iwasaki, K.; Hayashi, K.; Fujioka, T.; Sobue, K. Rho/rho-associated kinase signal regulates myogenic differentiation via myocardin-related transcription factor-a/smad-dependent transcription of the id3 gene. J. Biol. Chem. 2008, 283, 21230–21241. [Google Scholar] [CrossRef]
- Vartiainen, M.K.; Guettler, S.; Larijani, B.; Treisman, R. Nuclear actin regulates dynamic subcellular localization and activity of the srf cofactor mal. Science 2007, 316, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Weissbach, J.; Schikora, F.; Weber, A.; Kessels, M.; Posern, G. Myocardin-related transcription factor a activation by competition with wh2 domain proteins for actin binding. Mol. Cell. Biol. 2016, 36, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Finch-Edmondson, M.; Sudol, M. Framework to function: Mechanosensitive regulators of gene transcription. Cell. Mol. Biol. Lett. 2016, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. Yap/taz regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef]
- Speight, P.; Kofler, M.; Szászi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated mrtf and taz and tgfβ-regulated smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiepen, C.; Mendez, P.-L.; Knaus, P. It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology. Cells 2020, 9, 1965. https://doi.org/10.3390/cells9091965
Hiepen C, Mendez P-L, Knaus P. It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology. Cells. 2020; 9(9):1965. https://doi.org/10.3390/cells9091965
Chicago/Turabian StyleHiepen, Christian, Paul-Lennard Mendez, and Petra Knaus. 2020. "It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology" Cells 9, no. 9: 1965. https://doi.org/10.3390/cells9091965
APA StyleHiepen, C., Mendez, P.-L., & Knaus, P. (2020). It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology. Cells, 9(9), 1965. https://doi.org/10.3390/cells9091965