New Insights into the Role of Sphingolipid Metabolism in Melanoma

 ,
,
Abstract
1. Introduction
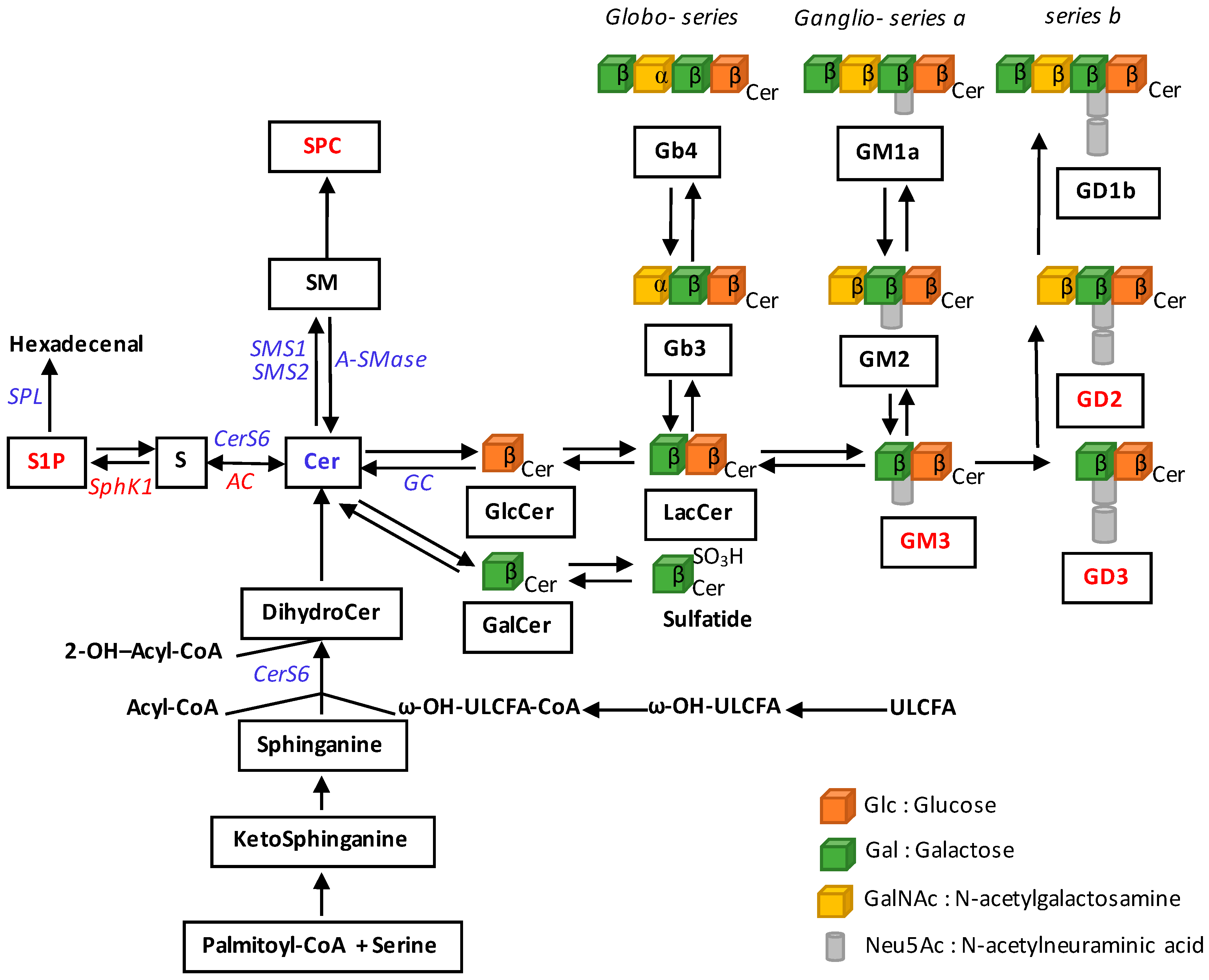
2. Alterations of Sphingolipid Metabolism in Melanoma
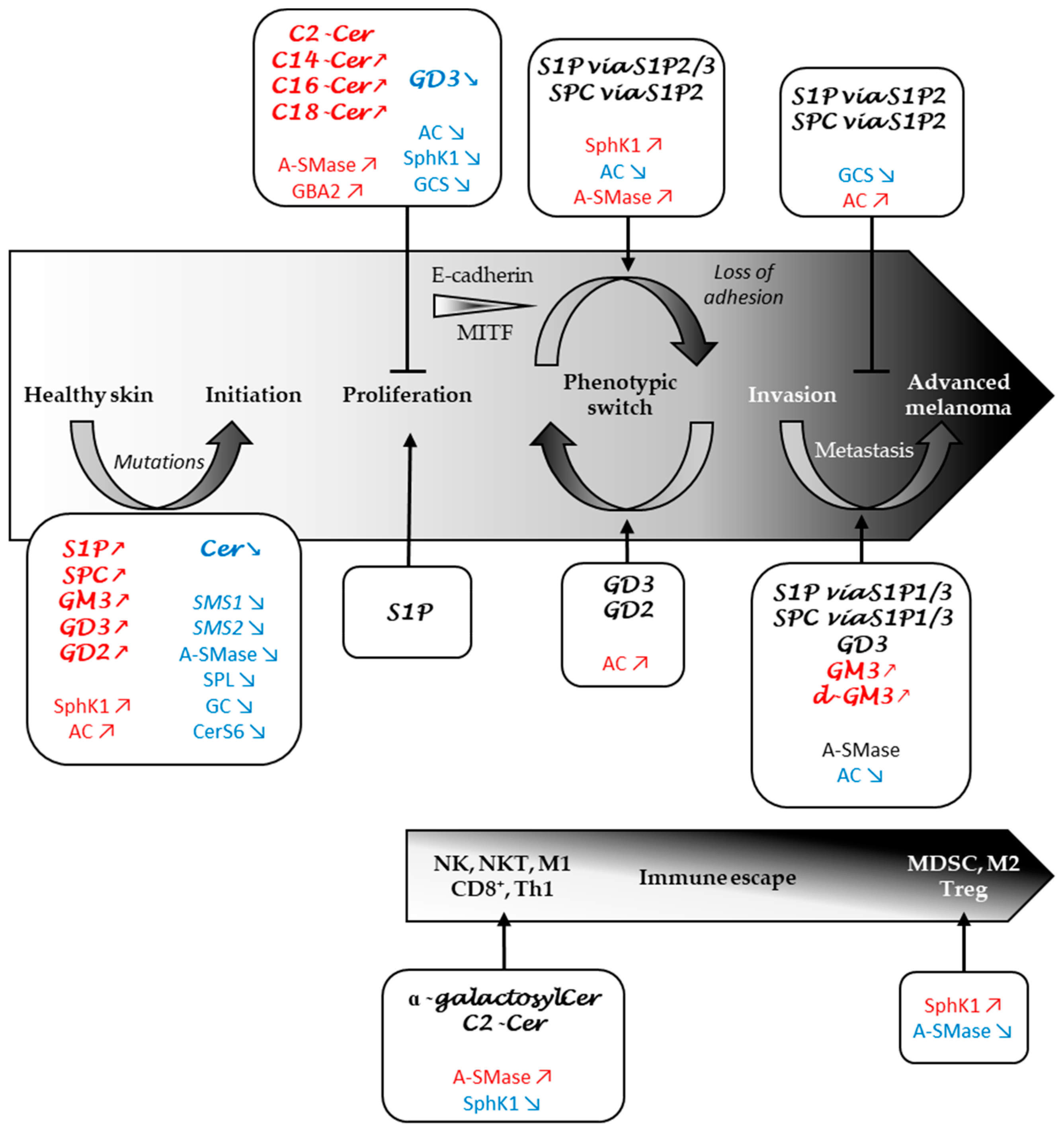
3. Role of the Sphingolipid Metabolism in Melanomagenesis
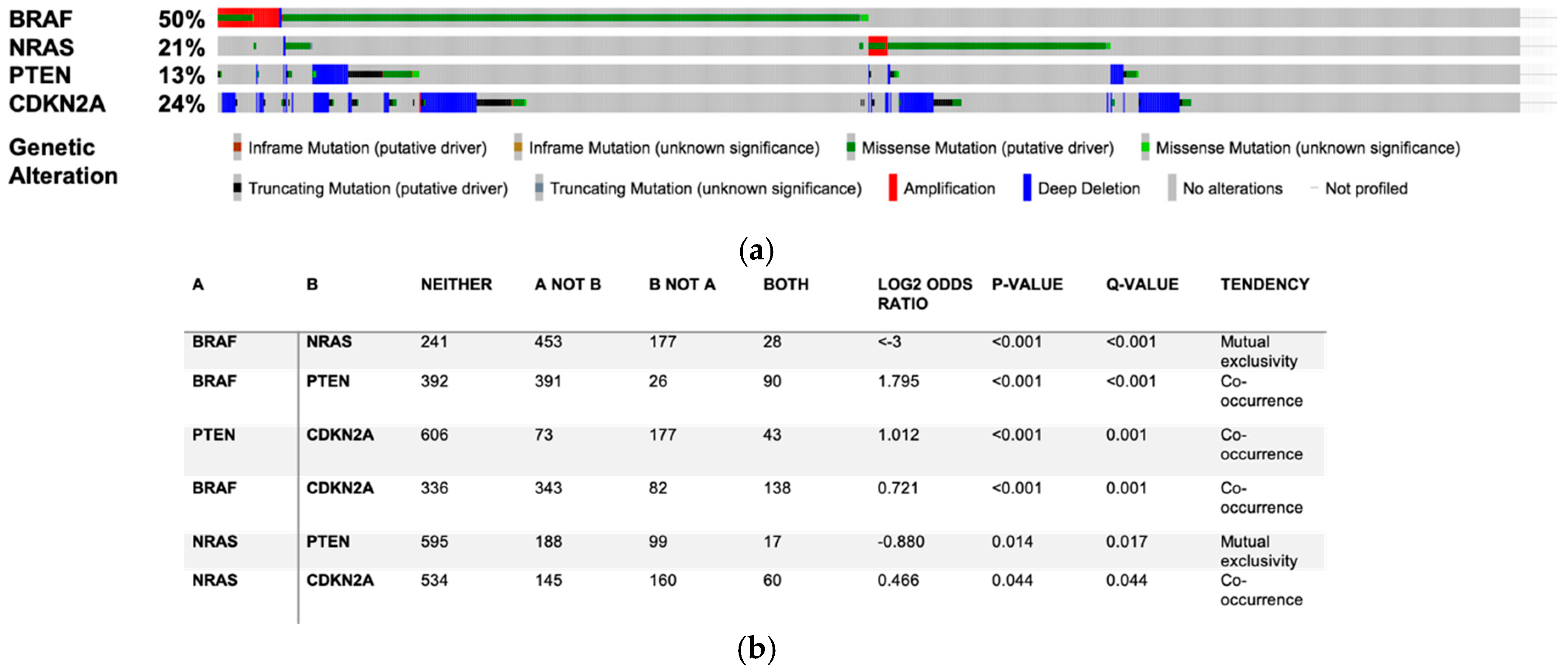
3.1. Do SL Metabolism Alterations Increase the Risk to Develop Melanoma?
3.2. Sphingolipid Metabolism Modulates Melanoma Cell Proliferation and Survival
4. Role of the Sphingolipid Metabolism in Melanoma Progression
4.1. SL Metabolism Regulates Melanoma Cell Adhesion
4.2. SL Metabolism as a Determinant of Melanoma Plasticity
4.3. SL Metabolism as a Major Regulator of Melanoma Aggressiveness
5. Role of SL Metabolism in the Immune Response to Melanoma
5.1. S1P in Lymphocyte Traffic and Differentiation
5.2. S1P Impairs the Immune Response in Melanoma
5.3. Ceramide and Its Derivatives in the Immune Response
5.4. Melanoma-Derived Exosomes Are Vectors of Immunosuppression
6. Potential Therapeutic Strategies for Melanoma Patients
7. Conclusions
Funding
Conflicts of Interest
References
- Gershenwald, J.E.; Guy, G.P. Stemming the Rising Incidence of Melanoma: Calling Prevention to Action. JNCI J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, W.; Gotlieb, V. The rapidly evolving therapies for advanced melanoma—Towards immunotherapy, molecular targeted therapy, and beyond. Crit. Rev. Oncol. Hematol. 2016, 99, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Crowson, A.N.; Magro, C.M.; Mihm, M.C. Prognosticators of melanoma, the melanoma report, and the sentinel lymph node. Mod. Pathol. 2006, 19, S71–S87. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef]
- Herlyn, M.; Thurin, J.; Balaban, G.; Bennicelli, J.L.; Bondi, E.; Guerry, D.; Nowell, P.; Clark, W.H.; Koprowski, H. Characteristics of Cultured Human Melanocytes Isolated from Different Stages of Tumor Progression. Cancer Res. 1985, 45, 8. [Google Scholar]
- Liu, J.; Fukunaga-Kalabis, M.; Li, L.; Herlyn, M. Developmental pathways activated in melanocytes and melanoma. Arch. Biochem. Biophys. 2014, 563, 13–21. [Google Scholar] [CrossRef]
- Hsu, M.-Y.; Meier, F.; Herlyn, M. Melanoma development and progression: A conspiracy between tumor and host. Differentiation 2002, 70, 522–536. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Murphy, G. The Genetic Evolution of Melanoma. N. Engl. J. Med. 2016, 374, 993–996. [Google Scholar] [CrossRef]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Cao, K.; Tang, Y.; Liu, J.; Li, J.; Chen, J.; Wang, S.; Chen, Z.; Zhou, J. C16:0 ceramide effect on melanoma malignant behavior and glycolysis depends on its intracellular or exogenous location. Am. J. Transl. Res. 2020, 12, 1123–1135. [Google Scholar] [PubMed]
- Borodzicz, S.; Rudnicka, L.; Mirowska-Guzel, D.; Cudnoch-Jedrzejewska, A. The role of epidermal sphingolipids in dermatologic diseases. Lipids Health Dis. 2016, 15, 13. [Google Scholar] [CrossRef]
- Garandeau, D.; Mrad, M.; Levade, T.; Perrotta, C.; Andrieu-Abadie, N.; Diab-Assaf, M. Dysregulation of Sphingolipid Metabolism in Melanoma: Roles in Pigmentation, Cell Survival and Tumor Progression. In Bioactive Sphingolipids in Cancer Biology and Therapy; Hannun, Y.A., Luberto, C., Mao, C., Obeid, L.M., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 123–139. ISBN 978-3-319-20749-0. [Google Scholar]
- Tang, Y.; Cao, K.; Wang, Q.; Chen, J.; Liu, R.; Wang, S.; Zhou, J.; Xie, H. Silencing of CerS6 increases the invasion and glycolysis of melanoma WM35, WM451 and SK28 cell lines via increased GLUT1-induced downregulation of WNT5A. Oncol. Rep. 2016, 35, 2907–2915. [Google Scholar] [CrossRef]
- Realini, N.; Palese, F.; Pizzirani, D.; Pontis, S.; Basit, A.; Bach, A.; Ganesan, A.; Piomelli, D. Acid Ceramidase in Melanoma: EXPRESSION, LOCALIZATION, AND EFFECTS OF PHARMACOLOGICAL INHIBITION. J. Biol. Chem. 2016, 291, 2422–2434. [Google Scholar] [CrossRef]
- Leclerc, J.; Garandeau, D.; Pandiani, C.; Gaudel, C.; Bille, K.; Nottet, N.; Garcia, V.; Colosetti, P.; Pagnotta, S.; Bahadoran, P.; et al. Lysosomal acid ceramidase ASAH1 controls the transition between invasive and proliferative phenotype in melanoma cells. Oncogene 2019, 38, 1282–1295. [Google Scholar] [CrossRef]
- Madhunapantula, S.V.; Hengst, J.; Gowda, R.; Fox, T.E.; Yun, J.K.; Robertson, G.P. Targeting sphingosine kinase-1 to inhibit melanoma: Targeting SPHK1 in melanomas. Pigment Cell Melanoma Res. 2012, 25, 259–274. [Google Scholar] [CrossRef]
- Albinet, V.; Bats, M.-L.; Huwiler, A.; Rochaix, P.; Chevreau, C.; Ségui, B.; Levade, T.; Andrieu-Abadie, N. Dual role of sphingosine kinase-1 in promoting the differentiation of dermal fibroblasts and the dissemination of melanoma cells. Oncogene 2014, 33, 3364–3373. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.d.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Colie, S.; Van Veldhoven, P.P.; Kedjouar, B.; Bedia, C.; Albinet, V.; Sorli, S.-C.; Garcia, V.; Djavaheri-Mergny, M.; Bauvy, C.; Codogno, P.; et al. Disruption of Sphingosine 1-Phosphate Lyase Confers Resistance to Chemotherapy and Promotes Oncogenesis through Bcl-2/Bcl-xL Upregulation. Cancer Res. 2009, 69, 9346–9353. [Google Scholar] [CrossRef] [PubMed]
- Portoukalian, J.; Zwingelstein, G.; Doré, J.F. Lipid composition of human malignant melanoma tumors at various levels of malignant growth. Eur. J. Biochem. 1979, 94, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Dosik, J.S.; Zhao, Y.; Vidal, M.J.; Nanus, D.M.; Sudol, M.; Albino, A.P. Elevated expression of protein tyrosine kinase c-Yes, but not c-Src, in human malignant melanoma. Oncogene 1993, 8, 2637–2644. [Google Scholar] [PubMed]
- Hamamura, K.; Tsuji, M.; Hotta, H.; Ohkawa, Y.; Takahashi, M.; Shibuya, H.; Nakashima, H.; Yamauchi, Y.; Hashimoto, N.; Hattori, H.; et al. Functional Activation of Src Family Kinase Yes Protein Is Essential for the Enhanced Malignant Properties of Human Melanoma Cells Expressing Ganglioside GD3. J. Biol. Chem. 2011, 286, 18526–18537. [Google Scholar] [CrossRef] [PubMed]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.H.M.; Holthuis, J.C.M. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004, 23, 33–44. [Google Scholar] [CrossRef]
- Yamaoka, S.; Miyaji, M.; Kitano, T.; Umehara, H.; Okazaki, T. Expression Cloning of a Human cDNA Restoring Sphingomyelin Synthesis and Cell Growth in Sphingomyelin Synthase-defective Lymphoid Cells. J. Biol. Chem. 2004, 279, 18688–18693. [Google Scholar] [CrossRef]
- Bilal, F.; Montfort, A.; Gilhodes, J.; Garcia, V.; Riond, J.; Carpentier, S.; Filleron, T.; Colacios, C.; Levade, T.; Daher, A.; et al. Sphingomyelin Synthase 1 (SMS1) Downregulation Is Associated With Sphingolipid Reprogramming and a Worse Prognosis in Melanoma. Front. Pharmacol. 2019, 10, 443. [Google Scholar] [CrossRef]
- Higuchi, K.; Kawashima, M.; Ichikawa, Y.; Imokawa, G. Sphingosylphosphorylcholine is a Melanogenic Stimulator for Human Melanocytes. Pigment Cell Res. 2003, 16, 670–678. [Google Scholar] [CrossRef]
- Kim, D.-S.; Park, S.-H.; Kwon, S.-B.; Park, E.-S.; Huh, C.-H.; Youn, S.-W.; Park, K.-C. Sphingosylphosphorylcholine-induced ERK activation inhibits melanin synthesis in human melanocytes. Pigment Cell Res. 2006, 19, 146–153. [Google Scholar] [CrossRef]
- Jeong, H.-S.; Lee, S.H.; Yun, H.-Y.; Baek, K.J.; Kwon, N.S.; Park, K.-C.; Kim, D.-S. Involvement of mTOR signaling in sphingosylphosphorylcholine-induced hypopigmentation effects. J. Biomed. Sci. 2011, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.-S.; Park, K.-C.; Kim, D.-S. PP2A and DUSP6 are involved in sphingosylphosphorylcholine-induced hypopigmentation. Mol. Cell. Biochem. 2012, 367, 43–49. [Google Scholar] [CrossRef]
- Bizzozero, L.; Cazzato, D.; Cervia, D.; Assi, E.; Simbari, F.; Pagni, F.; De Palma, C.; Monno, A.; Verdelli, C.; Querini, P.R.; et al. Acid sphingomyelinase determines melanoma progression and metastatic behaviour via the microphtalmia-associated transcription factor signalling pathway. Cell Death Differ. 2014, 21, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Kim, S.-Y.; Chung, J.-H.; Kim, K.-H.; Eun, H.-C.; Park, K.-C. Delayed ERK activation by ceramide reduces melanin synthesis in human melanocytes. Cell. Signal. 2002, 14, 779–785. [Google Scholar] [CrossRef]
- Amos, C.I.; Wang, L.-E.; Lee, J.E.; Gershenwald, J.E.; Chen, W.V.; Fang, S.; Kosoy, R.; Zhang, M.; Qureshi, A.A.; Vattathil, S.; et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma†. Hum. Mol. Genet. 2011, 20, 5012–5023. [Google Scholar] [CrossRef]
- Astudillo, L.; Therville, N.; Colacios, C.; Ségui, B.; Andrieu-Abadie, N.; Levade, T. Glucosylceramidases and malignancies in mammals. Biochimie 2016, 125, 267–280. [Google Scholar] [CrossRef]
- Dubot, P.; Astudillo, L.; Therville, N.; Sabourdy, F.; Stirnemann, J.; Levade, T.; Andrieu-Abadie, N. Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses. Cancers 2020, 12, 475. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Flanagan, J.; Ranes, B.; Brignol, N.; Hamler, R.; Clark, S. The origins of glucosylsphingosine in Gaucher disease. Mol. Genet. Metab. 2013, 108, S40–S41. [Google Scholar] [CrossRef]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Pistello, M. Sphingolipid/Ceramide Pathways and Autophagy in the Onset and Progression of Melanoma: Novel Therapeutic Targets and Opportunities. Int. J. Mol. Sci. 2019, 20, 3436. [Google Scholar] [CrossRef]
- Jiang, W.; Ogretmen, B. Autophagy paradox and ceramide. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2014, 1841, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.-H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Miller, D.; Park, T.S.; Patel, B.; Azevedo, J.L.; Awad, O.; Masood, M.A.; Veenstra, T.D.; Goldin, E.; Stubblefield, B.K.; et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl. Acad. Sci. USA 2012, 109, 18054–18059. [Google Scholar] [CrossRef]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Model. Mech. 2019, 12, dmm038596. [Google Scholar] [CrossRef]
- Pópulo, H.; Soares, P.; Faustino, A.; Rocha, A.S.; Silva, P.; Azevedo, F.; Lopes, J.M. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics: Letter to the Editor. Pigment Cell Melanoma Res. 2011, 24, 254–257. [Google Scholar] [CrossRef]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.-B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Smalley, K.S.M. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int. J. Cancer 2003, 104, 527–532. [Google Scholar] [CrossRef]
- Gorden, A.; Osman, I.; Gai, W.; He, D.; Huang, W.; Davidson, A.; Houghton, A.N.; Busam, K.; Polsky, D. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. 2003, 63, 3955–3957. [Google Scholar]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Patel, H.N.; Cho, K.-H.; LeBoit, P.E. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 13. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Daud, A.; Pavlick, A.C.; Gonzalez, R.; Lewis, K.D.; Hamid, O.; Gajewski, T.F.; Puzanov, I.; Wongchenko, M.; Rooney, I.; et al. Extended 5-Year Follow-up Results of a Phase Ib Study (BRIM7) of Vemurafenib and Cobimetinib in BRAF -Mutant Melanoma. Clin. Cancer Res. 2020, 26, 46–53. [Google Scholar] [CrossRef]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef]
- Pitson, S.M.; Xia, P.; Leclercq, T.M.; Moretti, P.A.B.; Zebol, J.R.; Lynn, H.E.; Wattenberg, B.W.; Vadas, M.A. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J. Exp. Med. 2005, 201, 49–54. [Google Scholar] [CrossRef]
- Leclercq, T.; Pitson, S. Cellular signalling by sphingosine kinase and sphingosine 1-phosphate. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2006, 58, 467–472. [Google Scholar] [CrossRef]
- Francy, J.M.; Nag, A.; Conroy, E.J.; Hengst, J.A.; Yun, J.K. Sphingosine kinase 1 expression is regulated by signaling through PI3K, AKT2, and mTOR in human coronary artery smooth muscle cells. Biochim. Biophys. Acta BBA Gene Struct. Expr. 2007, 1769, 253–265. [Google Scholar] [CrossRef]
- Mrad, M.; Imbert, C.; Garcia, V.; Rambow, F.; Therville, N.; Carpentier, S.; Ségui, B.; Levade, T.; Azar, R.; Marine, J.-C.; et al. Downregulation of sphingosine kinase-1 induces protective tumor immunity by promoting M1 macrophage response in melanoma. Oncotarget 2016, 7, 71873–71886. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Garandeau, D.; Noujarède, J.; Leclerc, J.; Imbert, C.; Garcia, V.; Bats, M.-L.; Rambow, F.; Gilhodes, J.; Filleron, T.; Meyer, N.; et al. Targeting the Sphingosine 1-Phosphate Axis Exerts Potent Antitumor Activity in BRAFi-Resistant Melanomas. Mol. Cancer Ther. 2019, 18, 289–300. [Google Scholar] [CrossRef]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 7411. [Google Scholar] [CrossRef]
- Bedia, C.; Casas, J.; Andrieu-Abadie, N.; Fabriàs, G.; Levade, T. Acid Ceramidase Expression Modulates the Sensitivity of A375 Melanoma Cells to Dacarbazine. J. Biol. Chem. 2011, 286, 28200–28209. [Google Scholar] [CrossRef] [PubMed]
- Han, W.S.; Yoo, J.Y.; Youn, S.W.; Kim, D.S.; Park, C.; Kim, S.Y.; Kim, K.H. Effects of C2-ceramide on the Malme-3M melanoma cell line. J. Dermatol. Sci. 2002, 10. [Google Scholar] [CrossRef]
- Deng, W.; Li, R.; Guerrera, M.; Liu, Y.; Ladisch, S. Transfection of glucosylceramide synthase antisense inhibits mouse melanoma formation. Glycobiology 2002, 12, 145–152. [Google Scholar] [CrossRef][Green Version]
- Weiss, M.; Hettmer, S.; Smith, P.; Ladisch, S. Inhibition of melanoma tumor growth by a novel inhibitor of glucosylceramide synthase. Cancer Res. 2003, 63, 3654–3658. [Google Scholar] [PubMed]
- Sorli, S.; Colié, S.; Albinet, V.; Dubrac, A.; Touriol, C.; Guilbaud, N.; Bedia, C.; Fabriàs, G.; Casas, J.; Ségui, B.; et al. The nonlysosomal β-glucosidase GBA2 promotes endoplasmic reticulum stress and impairs tumorigenicity of human melanoma cells. FASEB J. 2013, 27, 489–498. [Google Scholar] [CrossRef]
- Nakano, J.; Raj, B.K.; Asagami, C.; Lloyd, K.O. Human melanoma cell lines deficient in GD3 ganglioside expression exhibit altered growth and tumorigenic characteristics. J. Invest. Dermatol. 1996, 107, 543–548. [Google Scholar] [CrossRef][Green Version]
- Hamamura, K.; Furukawa, K.; Hayashi, T.; Hattori, T.; Nakano, J.; Nakashima, H.; Okuda, T.; Mizutani, H.; Hattori, H.; Ueda, M.; et al. Ganglioside GD3 promotes cell growth and invasion through p130Cas and paxillin in malignant melanoma cells. Proc. Natl. Acad. Sci. USA 2005, 102, 11041–11046. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Kambe, M.; Miyata, M.; Ohkawa, Y.; Tajima, O.; Furukawa, K. Ganglioside GD3 induces convergence and synergism of adhesion and hepatocyte growth factor/Met signals in melanomas. Cancer Sci. 2014, 105, 52–63. [Google Scholar] [CrossRef]
- Li, G.; Satyamoorthy, K.; Herlyn, M. D ynamics of C ell I nteractions and C ommunications during M elanoma D evelopment. Crit. Rev. Oral Biol. Med. 2002, 13, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Haass, N.K.; Smalley, K.S.M.; Herlyn, M. The Role of Altered Cell–Cell Communication in Melanoma Progression. J. Mol. Histol. 2003, 35, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Haass, N.K.; Smalley, K.S.M.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef]
- Hsu, M.-Y.; Meier, F.E.; Nesbit, M.; Hsu, J.-Y.; Van Belle, P.; Elder, D.E.; Herlyn, M. E-Cadherin Expression in Melanoma Cells Restores Keratinocyte-Mediated Growth Control and Down-Regulates Expression of Invasion-Related Adhesion Receptors. Am. J. Pathol. 2000, 156, 1515–1525. [Google Scholar] [CrossRef]
- Tamashiro, P.M.; Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingosine kinase 1 mediates head & neck squamous cell carcinoma invasion through sphingosine 1-phosphate receptor 1. Cancer Cell Int. 2014, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, Y.; He, H.-W.; Zhao, W.-L.; Shao, R.-G. SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy 2017, 13, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Navarro, R.; Juan, G.; Peiró, T.; Serrano, A.; Ramón, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef]
- Kono, Y.; Nishiuma, T.; Nishimura, Y.; Kotani, Y.; Okada, T.; Nakamura, S.; Yokoyama, M. Sphingosine Kinase 1 Regulates Differentiation of Human and Mouse Lung Fibroblasts Mediated by TGF-β1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 395–404. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kitayama, J.; Takuwa, N.; Arikawa, K.; Inoki, I.; Takehara, K.; Nagawa, H.; Takuwa, Y. Sphingosine-1-phosphate receptor subtype-specific positive and negative regulation of Rac and haematogenous metastasis of melanoma cells. Biochem. J. 2003, 374, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Braga, V.M.M.; Machesky, L.M.; Hall, A.; Hotchin, N.A. The Small GTPases Rho and Rac Are Required for the Establishment of Cadherin-dependent Cell–Cell Contacts. J. Cell Biol. 1997, 137, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, Y.; Miyazaki, S.; Hamamura, K.; Kambe, M.; Miyata, M.; Tajima, O.; Ohmi, Y.; Yamauchi, Y.; Furukawa, K.; Furukawa, K. Ganglioside GD3 Enhances Adhesion Signals and Augments Malignant Properties of Melanoma Cells by Recruiting Integrins to Glycolipid-enriched Microdomains. J. Biol. Chem. 2010, 285, 27213–27223. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Kambe, M.; Ohkawa, Y.; Hamamura, K.; Tajima, O.; Takeuchi, R.; Furukawa, K.; Furukawa, K. Differential roles of gangliosides in malignant properties of melanomas. PLoS ONE 2018, 13, e0206881. [Google Scholar] [CrossRef] [PubMed]
- Hodorogea, A.; Calinescu, A.; Antohe, M.; Balaban, M.; Nedelcu, R.I.; Turcu, G.; Ion, D.A.; Badarau, I.A.; Popescu, C.M.; Popescu, R.; et al. Epithelial-Mesenchymal Transition in Skin Cancers: A Review. Anal. Cell. Pathol. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF—The first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef]
- Levy, C.; Lee, Y.-N.; Nechushtan, H.; Schueler-Furman, O.; Sonnenblick, A.; Hacohen, S.; Razin, E. Identifying a common molecular mechanism for inhibition of MITF and STAT3 by PIAS3. Blood 2006, 107, 2839–2845. [Google Scholar] [CrossRef]
- Yasumoto, K.; Takeda, K.; Saito, H.; Watanabe, K.; Takahashi, K.; Shibahara, S. Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J. 2002, 21, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, E.M.; Roberts, P.C.; Kustin, E.M.; Lemonnier, L.A.; Sullards, M.C.; Dillehay, D.L.; Merrill, A.H. Modulation of intracellular beta-catenin localization and intestinal tumorigenesis in vivo and in vitro by sphingolipids. Cancer Res. 2001, 61, 6723–6729. [Google Scholar] [PubMed]
- Liu, H.; Zhang, C.-X.; Ma, Y.; He, H.-W.; Wang, J.-P.; Shao, R.-G. SphK1 inhibitor SKI II inhibits the proliferation of human hepatoma HepG2 cells via the Wnt5A/β-catenin signaling pathway. Life Sci. 2016, 151, 23–29. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, S.Y.; Jeong, Y.-M.; Yun, H.-Y.; Baek, K.J.; Kwon, N.S.; Park, K.-C.; Kim, D.-S. The regulatory mechanism of melanogenesis by FTY720, a sphingolipid analogue: The regulation of melanogenesis by FTY720. Exp. Dermatol. 2011, 20, 237–241. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Lu, P.; White-Gilbertson, S.; Nganga, R.; Kester, M.; Voelkel-Johnson, C. Expression of the SNAI2 transcriptional repressor is regulated by C 16 -ceramide. Cancer Biol. Ther. 2019, 20, 922–930. [Google Scholar] [CrossRef]
- Edmond, V.; Dufour, F.; Poiroux, G.; Shoji, K.; Malleter, M.; Fouqué, A.; Tauzin, S.; Rimokh, R.; Sergent, O.; Penna, A.; et al. Downregulation of ceramide synthase-6 during epithelial-to-mesenchymal transition reduces plasma membrane fluidity and cancer cell motility. Oncogene 2015, 34, 996–1005. [Google Scholar] [CrossRef]
- Zheng, K.; Chen, Z.; Feng, H.; Chen, Y.; Zhang, C.; Yu, J.; Luo, Y.; Zhao, L.; Jiang, X.; Shi, F. Sphingomyelin synthase 2 promotes an aggressive breast cancer phenotype by disrupting the homoeostasis of ceramide and sphingomyelin. Cell Death Dis. 2019, 10, 157. [Google Scholar] [CrossRef]
- Levade, T.; Andrieu-Abadie, N.; Micheau, O.; Legembre, P.; Ségui, B. Sphingolipids modulate the epithelial-mesenchymal transition in cancer. Cell Death Discov. 2015, 1, 15001. [Google Scholar] [CrossRef]
- Mathow, D.; Chessa, F.; Rabionet, M.; Kaden, S.; Jennemann, R.; Sandhoff, R.; Gröne, H.-J.; Feuerborn, A. Zeb1 affects epithelial cell adhesion by diverting glycosphingolipid metabolism. EMBO Rep. 2015, 16, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.-Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.-Y.; Khoo, K.-H.; Li, S.-C.; Li, Y.-T.; et al. Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar] [CrossRef] [PubMed]
- Hosain, S.B.; Khiste, S.K.; Uddin, M.B.; Vorubindi, V.; Ingram, C.; Zhang, S.; Hill, R.A.; Gu, X.; Liu, Y.-Y. Inhibition of glucosylceramide synthase eliminates the oncogenic function of p53 R273H mutant in the epithelial-mesenchymal transition and induced pluripotency of colon cancer cells. Oncotarget 2016, 7, 60575–60592. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Ren, S.; Kleuser, B.; Shabahang, S.; Eberhardt, W.; Radeke, H.; Schäfer-Korting, M.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-Phosphate Cross-activates the Smad Signaling Cascade and Mimics Transforming Growth Factor-β-induced Cell Responses. J. Biol. Chem. 2004, 279, 35255–35262. [Google Scholar] [CrossRef] [PubMed]
- Sauer, B.; Vogler, R.; von Wenckstern, H.; Fujii, M.; Anzano, M.B.; Glick, A.B.; Schäfer-Korting, M.; Roberts, A.B.; Kleuser, B. Involvement of Smad Signaling in Sphingosine 1-Phosphate-mediated Biological Responses of Keratinocytes. J. Biol. Chem. 2004, 279, 38471–38479. [Google Scholar] [CrossRef]
- Radeke, H.H.; von Wenckstern, H.; Stoidtner, K.; Sauer, B.; Hammer, S.; Kleuser, B. Overlapping Signaling Pathways of Sphingosine 1-Phosphate and TGF-β in the Murine Langerhans Cell Line XS52. J. Immunol. 2005, 174, 2778–2786. [Google Scholar] [CrossRef]
- Zeng, Y.; Yao, X.; Chen, L.; Yan, Z.; Liu, J.; Zhang, Y.; Feng, T.; Wu, J.; Liu, X. Sphingosine-1-phosphate induced epithelial-mesenchymal transition of hepatocellular carcinoma via an MMP-7/syndecan-1/TGF-β autocrine loop. Oncotarget 2016, 7, 63324–63337. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Zhang, H.; Zhang, L.; Cai, T.-T.; Huang, D.-J.; He, J.; Ni, H.-H.; Zhou, F.-J.; Zhang, X.-S.; Li, J. Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: Leading to poor survival in bladder cancer. Cell Death Dis. 2019, 10, 50. [Google Scholar] [CrossRef]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine Kinases and Sphingosine-1-Phosphate Are Critical for Transforming Growth Factor β-Induced Extracellular Signal-Regulated Kinase 1 and 2 Activation and Promotion of Migration and Invasion of Esophageal Cancer Cells. Mol. Cell. Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine Kinase 1 (SPHK1) Is Induced by Transforming Growth Factor-β and Mediates TIMP-1 Up-regulation. J. Biol. Chem. 2004, 279, 53994–54001. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed]
- Nallet-Staub, F.; Marsaud, V.; Li, L.; Gilbert, C.; Dodier, S.; Bataille, V.; Sudol, M.; Herlyn, M.; Mauviel, A. Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J. Invest. Dermatol. 2014, 134, 123–132. [Google Scholar] [CrossRef]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.C.; Liu, J.; Peters, E.C.; Wu, X. Identification of Serum-Derived Sphingosine-1-Phosphate as a Small Molecule Regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Pors, S.E.; Harðardóttir, L.; Olesen, H.Ø.; Riis, M.L.; Jensen, L.B.; Andersen, A.S.; Cadenas, J.; Grønning, A.P.; Colmorn, L.B.; Dueholm, M.; et al. Effect of sphingosine-1-phosphate on activation of dormant follicles in murine and human ovarian tissue. Mol. Hum. Reprod. 2020, gaaa022. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.-K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef]
- Kemppainen, K.; Wentus, N.; Lassila, T.; Laiho, A.; Törnquist, K. Sphingosylphosphorylcholine regulates the Hippo signaling pathway in a dual manner. Cell. Signal. 2016, 28, 1894–1903. [Google Scholar] [CrossRef]
- Arikawa, K.; Takuwa, N.; Yamaguchi, H.; Sugimoto, N.; Kitayama, J.; Nagawa, H.; Takehara, K.; Takuwa, Y. Ligand-dependent Inhibition of B16 Melanoma Cell Migration and Invasion via Endogenous S1P 2 G Protein-coupled Receptor: REQUIREMENT OF INHIBITION OF CELLULAR RAC ACTIVITY. J. Biol. Chem. 2003, 278, 32841–32851. [Google Scholar] [CrossRef]
- Carpinteiro, A.; Becker, K.A.; Japtok, L.; Hessler, G.; Keitsch, S.; Požgajovà, M.; Schmid, K.W.; Adams, C.; Müller, S.; Kleuser, B.; et al. Regulation of hematogenous tumor metastasis by acid sphingomyelinase. EMBO Mol. Med. 2015, 7, 714–734. [Google Scholar] [CrossRef]
- Tsuchida, T.; Saxton, R.E.; Morton, D.L.; Irie, R.F. Gangliosides of human melanoma. J. Natl. Cancer Inst. 1987, 78, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Moriya, S.; Shineha, R.; Satomi, S.; Miyagi, T. Comparative study of sialidase activity and G(M3) content in B16 melanoma variants with different metastatic potential. Acta Biochim. Pol. 1998, 45, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Mohanty, K.C. Enhancement of metastatic potential of mouse B16-melanoma cells to lung after treatment with gangliosides of B-16-melanoma cells of higher metastatic potential to lung. Indian J. Exp. Biol. 2003, 41, 1253–1258. [Google Scholar] [PubMed]
- Liu, J.-W.; Sun, P.; Yan, Q.; Paller, A.S.; Gerami, P.; Ho, N.; Vashi, N.; Le Poole, I.C.; Wang, X.-Q. De-N-acetyl GM3 Promotes Melanoma Cell Migration and Invasion through Urokinase Plasminogen Activator Receptor Signaling-Dependent MMP-2 Activation. Cancer Res. 2009, 69, 8662–8669. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma cell–derived exosomes promote epithelial–mesenchymal transition in primary melanocytes through paracrine/autocrine signaling in the tumor microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- López-Montero, I.; Vélez, M.; Devaux, P.F. Surface tension induced by sphingomyelin to ceramide conversion in lipid membranes. Biochim. Biophys. Acta BBA Biomembr. 2007, 1768, 553–561. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory Mechanisms and Intercellular Transfer of MicroRNAs in Living Cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral Sphingomyelinase 2 (nSMase2)-dependent Exosomal Transfer of Angiogenic MicroRNAs Regulate Cancer Cell Metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Miya, S.; Zhang, L.; Nakamura, S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat. Commun. 2013, 4, 2712. [Google Scholar] [CrossRef]
- Mohamed, N.N.I.; Okada, T.; Kajimoto, T.; Nakamura, S.-I. Essential Role of Sphingosine Kinase 2 in the Regulation of Cargo Contents in the Exosomes from K562 Cells. Kobe J. Med. Sci. 2018, 63, E123–E129. [Google Scholar]
- Kajimoto, T.; Mohamed, N.N.I.; Badawy, S.M.M.; Matovelo, S.A.; Hirase, M.; Nakamura, S.; Yoshida, D.; Okada, T.; Ijuin, T.; Nakamura, S. Involvement of Gβγ subunits of G i protein coupled with S1P receptor on multivesicular endosomes in F-actin formation and cargo sorting into exosomes. J. Biol. Chem. 2018, 293, 245–253. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.; Hadju, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.-J.; Card, D. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Resop, R.S.; Douaisi, M.; Craft, J.; Jachimowski, L.C.M.; Blom, B.; Uittenbogaart, C.H. Sphingosine-1-phosphate/sphingosine-1-phosphate receptor 1 signaling is required for migration of naive human T cells from the thymus to the periphery. J. Allergy Clin. Immunol. 2016, 138, 551–557.e8. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.G.; Xu, Y.; Proia, R.L.; Cyster, J.G. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J. Exp. Med. 2005, 201, 291–301. [Google Scholar] [CrossRef]
- Bankovich, A.J.; Shiow, L.R.; Cyster, J.G. CD69 Suppresses Sphingosine 1-Phosophate Receptor-1 (S1P 1 ) Function through Interaction with Membrane Helix 4. J. Biol. Chem. 2010, 285, 22328–22337. [Google Scholar] [CrossRef]
- Shiow, L.R.; Rosen, D.B.; Brdičková, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting Edge: CD69 Interference with Sphingosine-1-Phosphate Receptor Function Regulates Peripheral T Cell Retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef]
- Skon, C.N.; Lee, J.-Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef] [PubMed]
- Hochheiser, K.; Aw Yeang, H.X.; Wagner, T.; Tutuka, C.; Behren, A.; Waithman, J.; Angel, C.; Neeson, P.J.; Gebhardt, T.; Gyorki, D.E. Accumulation of CD103 + CD8 + T cells in a cutaneous melanoma micrometastasis. Clin. Transl. Immunol. 2019, 8. [Google Scholar] [CrossRef]
- Drouillard, A.; Neyra, A.; Mathieu, A.-L.; Marçais, A.; Wencker, M.; Marvel, J.; Belot, A.; Walzer, T. Human Naive and Memory T Cells Display Opposite Migratory Responses to Sphingosine-1 Phosphate. J. Immunol. 2018, 200, 551–557. [Google Scholar] [CrossRef]
- Sic, H.; Kraus, H.; Madl, J.; Flittner, K.-A.; von Münchow, A.L.; Pieper, K.; Rizzi, M.; Kienzler, A.-K.; Ayata, K.; Rauer, S.; et al. Sphingosine-1-phosphate receptors control B-cell migration through signaling components associated with primary immunodeficiencies, chronic lymphocytic leukemia, and multiple sclerosis. J. Allergy Clin. Immunol. 2014, 134, 420–428.e15. [Google Scholar] [CrossRef]
- Walzer, T.; Chiossone, L.; Chaix, J.; Calver, A.; Carozzo, C.; Garrigue-Antar, L.; Jacques, Y.; Baratin, M.; Tomasello, E.; Vivier, E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol. 2007, 8, 1337–1344. [Google Scholar] [CrossRef]
- Drouillard, A.; Mathieu, A.-L.; Marçais, A.; Belot, A.; Viel, S.; Mingueneau, M.; Guckian, K.; Walzer, T. S1PR5 is essential for human natural killer cell migration toward sphingosine-1 phosphate. J. Allergy Clin. Immunol. 2018, 141, 2265–2268.e1. [Google Scholar] [CrossRef]
- Jenne, C.N.; Enders, A.; Rivera, R.; Watson, S.R.; Bankovich, A.J.; Pereira, J.P.; Xu, Y.; Roots, C.M.; Beilke, J.N.; Banerjee, A.; et al. T-bet–dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J. Exp. Med. 2009, 206, 2469–2481. [Google Scholar] [CrossRef]
- Mayol, K.; Biajoux, V.; Marvel, J.; Balabanian, K.; Walzer, T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood 2011, 118, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, K.; Burns, S.; Shrestha, S.; Chi, H. The S1P1-mTOR axis directs the reciprocal differentiation of TH1 and Treg cells. Nat. Immunol. 2010, 11, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-Survival Lipid Sphingosine-1-Phosphate Metabolically Programs T Cells to Limit Anti-tumor Activity. Cell Rep. 2019, 28, 1879–1893.e7. [Google Scholar] [CrossRef] [PubMed]
- Sanger Mouse Genetics Project; van der Weyden, L.; Arends, M.J.; Campbell, A.D.; Bald, T.; Wardle-Jones, H.; Griggs, N.; Velasco-Herrera, M.D.C.; Tüting, T.; Sansom, O.J.; et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 2017, 541, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Assi, E.; Cervia, D.; Bizzozero, L.; Capobianco, A.; Pambianco, S.; Morisi, F.; De Palma, C.; Moscheni, C.; Pellegrino, P.; Clementi, E.; et al. Modulation of Acid Sphingomyelinase in Melanoma Reprogrammes the Tumour Immune Microenvironment. Mediators Inflamm. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Coerdt, I.; Full-Scharrer, G.; Hösl, E. Long-term results in surgical treatment of cleft lips and palates. Prog. Pediatr. Surg. 1977, 10, 1–3. [Google Scholar]
- Nakagawa, R.; Serizawa, I.; Motoki, K.; Sato, M.; Ueno, H.; Iijima, R.; Nakamura, H.; Shimosaka, A.; Koezuka, Y. Antitumor activity of alpha-galactosylceramide, KRN7000, in mice with the melanoma B16 hepatic metastasis and immunohistological study of tumor infiltrating cells. Oncol. Res. 2000, 12, 51–58. [Google Scholar] [CrossRef]
- Ghosh, S.; Juin, S.K.; Nandi, P.; Majumdar, S.B.; Bose, A.; Baral, R.; Sil, P.C.; Majumdar, S. PKCζ mediated anti-proliferative effect of C2 ceramide on neutralization of the tumor microenvironment and melanoma regression. Cancer Immunol. Immunother. CII 2020, 69, 611–627. [Google Scholar] [CrossRef]
- Tiwary, S.; Berzofsky, J.A.; Terabe, M. Altered Lipid Tumor Environment and Its Potential Effects on NKT Cell Function in Tumor Immunity. Front. Immunol. 2019, 10, 2187. [Google Scholar] [CrossRef]
- Bay, S.; Fort, S.; Birikaki, L.; Ganneau, C.; Samain, E.; Coïc, Y.-M.; Bonhomme, F.; Dériaud, E.; Leclerc, C.; Lo-Man, R. Induction of a melanoma-specific antibody response by a monovalent, but not a divalent, synthetic GM2 neoglycopeptide. ChemMedChem 2009, 4, 582–587. [Google Scholar] [CrossRef]
- Fernandez, L.E.; Gabri, M.R.; Guthmann, M.D.; Gomez, R.E.; Gold, S.; Fainboim, L.; Gomez, D.E.; Alonso, D.F. NGcGM3 ganglioside: A privileged target for cancer vaccines. Clin. Dev. Immunol. 2010, 2010, 814397. [Google Scholar] [CrossRef]
- Pérez, K.; Osorio, M.; Hernández, J.; Carr, A.; Fernández, L.E. NGcGM3/VSSP vaccine as treatment for melanoma patients. Hum. Vaccines Immunother. 2013, 9, 1237–1240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernhard, H.; Meyer zum Büschenfelde, K.H.; Dippold, W.G. Ganglioside GD3 shedding by human malignant melanoma cells. Int. J. Cancer 1989, 44, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Portoukalian, J.; Zwingelstein, G.; Abdul-Malak, N.; Doré, J.F. Alteration of gangliosides in plasma and red cells of humans bearing melanoma tumors. Biochem. Biophys. Res. Commun. 1978, 85, 916–920. [Google Scholar] [CrossRef]
- Péguet-Navarro, J.; Sportouch, M.; Popa, I.; Berthier, O.; Schmitt, D.; Portoukalian, J. Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J. Immunol. Baltim. Md 1950 2003, 170, 3488–3494. [Google Scholar] [CrossRef]
- Bennaceur, K.; Popa, I.; Chapman, J.A.; Migdal, C.; Péguet-Navarro, J.; Touraine, J.-L.; Portoukalian, J. Different mechanisms are involved in apoptosis induced by melanoma gangliosides on human monocyte-derived dendritic cells. Glycobiology 2009, 19, 576–582. [Google Scholar] [CrossRef]
- Tsao, C.-Y.; Sabbatino, F.; Cheung, N.-K.V.; Hsu, J.C.-F.; Villani, V.; Wang, X.; Ferrone, S. Anti-proliferative and pro-apoptotic activity of GD2 ganglioside-specific monoclonal antibody 3F8 in human melanoma cells. Oncoimmunology 2015, 4, e1023975. [Google Scholar] [CrossRef]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.-K. Humanized 3F8 Anti-GD2 Monoclonal Antibody Dosing With Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol. 2018, 4, 1729–1735. [Google Scholar] [CrossRef]
- Kramer, K.; Pandit-Taskar, N.; Humm, J.L.; Zanzonico, P.B.; Haque, S.; Dunkel, I.J.; Wolden, S.L.; Donzelli, M.; Goldman, D.A.; Lewis, J.S.; et al. A phase II study of radioimmunotherapy with intraventricular 131 I-3F8 for medulloblastoma. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef]
- Yu, J.; Wu, X.; Yan, J.; Yu, H.; Xu, L.; Chi, Z.; Sheng, X.; Si, L.; Cui, C.; Dai, J.; et al. Anti-GD2/4-1BB chimeric antigen receptor T cell therapy for the treatment of Chinese melanoma patients. J. Hematol. Oncol. J. Hematol. Oncol. 2018, 11, 1. [Google Scholar] [CrossRef]
- Albertini, M.R.; Yang, R.K.; Ranheim, E.A.; Hank, J.A.; Zuleger, C.L.; Weber, S.; Neuman, H.; Hartig, G.; Weigel, T.; Mahvi, D.; et al. Pilot trial of the hu14.18-IL2 immunocytokine in patients with completely resectable recurrent stage III or stage IV melanoma. Cancer Immunol. Immunother. CII 2018, 67, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, W.; McGinley, E.C.; Klinke, D.J. Melanoma exosomes deliver a complex biological payload that upregulates PTPN11 to suppress T lymphocyte function. Pigment Cell Melanoma Res. 2017, 30, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4+ T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Düchler, M.; Czernek, L.; Peczek, L.; Cypryk, W.; Sztiller-Sikorska, M.; Czyz, M. Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance. Cells 2019, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Vignard, V.; Labbé, M.; Marec, N.; André-Grégoire, G.; Jouand, N.; Fonteneau, J.-F.; Labarrière, N.; Fradin, D. MicroRNAs in Tumor Exosomes Drive Immune Escape in Melanoma. Cancer Immunol. Res. 2020, 8, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef]
- Falkson, C.I.; Ibrahim, J.; Kirkwood, J.M.; Coates, A.S.; Atkins, M.B.; Blum, R.H. Phase III trial of dacarbazine versus dacarbazine with interferon alpha-2b versus dacarbazine with tamoxifen versus dacarbazine with interferon alpha-2b and tamoxifen in patients with metastatic malignant melanoma: An Eastern Cooperative Oncology Group study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 1743–1751. [Google Scholar] [CrossRef]
- Middleton, M.R.; Grob, J.J.; Aaronson, N.; Fierlbeck, G.; Tilgen, W.; Seiter, S.; Gore, M.; Aamdal, S.; Cebon, J.; Coates, A.; et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 158–166. [Google Scholar] [CrossRef]
- Avril, M.F.; Aamdal, S.; Grob, J.J.; Hauschild, A.; Mohr, P.; Bonerandi, J.J.; Weichenthal, M.; Neuber, K.; Bieber, T.; Gilde, K.; et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: A phase III study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e920957. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Postow, M.A. Managing immune checkpoint-blocking antibody side effects. Am. Soc. Clin. Oncol. Educ. Book 2015, 76–83. [Google Scholar] [CrossRef]
- Suo, A.; Chan, Y.; Beaulieu, C.; Kong, S.; Cheung, W.Y.; Monzon, J.G.; Smylie, M.; Walker, J.; Morris, D.; Cheng, T. Anti-PD1-Induced Immune-Related Adverse Events and Survival Outcomes in Advanced Melanoma. Oncologist 2020, 25, 438–446. [Google Scholar] [CrossRef]
- Cervia, D.; Assi, E.; De Palma, C.; Giovarelli, M.; Bizzozero, L.; Pambianco, S.; Di Renzo, I.; Zecchini, S.; Moscheni, C.; Vantaggiato, C.; et al. Essential role for acid sphingomyelinase-inhibited autophagy in melanoma response to cisplatin. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Ishitsuka, A.; Fujine, E.; Mizutani, Y.; Tawada, C.; Kanoh, H.; Banno, Y.; Seishima, M. FTY720 and cisplatin synergistically induce the death of cisplatin-resistant melanoma cells through the downregulation of the PI3K pathway and the decrease in epidermal growth factor receptor expression. Int. J. Mol. Med. 2014, 34, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Jolly, P.S.; Müller, C.; Eberle, J.; Spiegel, S.; Geilen, C.C. Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: Role of Bcl-2 expression. Oncogene 2005, 24, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Abe, N.; Kanoh, H.; Banno, Y.; Seishima, M. Synergistic effects of vemurafenib and fingolimod (FTY720) in vemurafenib-resistant melanoma cell lines. Mol. Med. Rep. 2018, 18, 5151–5158. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Yang, Y.-L.; He, L.; Gu, B.; Xia, J.-P.; Sun, W.-L.; Su, Z.-L.; Chen, B.; Bi, Z.-G. Increasing ceramides sensitizes genistein-induced melanoma cell apoptosis and growth inhibition. Biochem. Biophys. Res. Commun. 2012, 421, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.A.; Smith, C.D.; Kester, M.; Robertson, G.P. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 3571–3581. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, P.; Fu, C.; Hu, Y.; Dong, C.; Song, Y.; Song, E. C6-ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCζ tumor-suppressive activities and regulating integrin affinity modulation. Sci. Rep. 2015, 5, 9275. [Google Scholar] [CrossRef]
- Kobayashi, E.; Motoki, K.; Uchida, T.; Fukushima, H.; Koezuka, Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol. Res. 1995, 7, 529–534. [Google Scholar]
- Guthmann, M.D.; Bitton, R.J.; Carnero, A.J.L.; Gabri, M.R.; Cinat, G.; Koliren, L.; Lewi, D.; Fernandez, L.E.; Alonso, D.F.; Gómez, D.E.; et al. Active specific immunotherapy of melanoma with a GM3 ganglioside-based vaccine: A report on safety and immunogenicity. J. Immunother. 2004, 27, 442–451. [Google Scholar] [CrossRef]
- Irie, R.F.; Ollila, D.W.; O’Day, S.; Morton, D.L. Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol. Immunother. CII 2004, 53, 110–117. [Google Scholar] [CrossRef]
- Musumarra, G.; Barresi, V.; Condorelli, D.F.; Scirè, S. A bioinformatic approach to the identification of candidate genes for the development of new cancer diagnostics. Biol. Chem. 2003, 384, 321–327. [Google Scholar] [CrossRef]
- Ene (Nicolae), C.-D.; Nicolae, I. Gangliosides and Antigangliosides in Malignant Melanoma. In Melanoma—Current Clinical Management and Future Therapeutics; Murph, M., Ed.; Intech: London, UK, 2015; ISBN 978-953-51-2036-0. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SL or SL-Metabolising Enzymes | Dysregulation | Cell Lines or Patients | Effects | Refs |
|---|---|---|---|---|
| CerS6 | Decreased | WM35, WM451 and SKMEL28 human melanoma cells | Malignant behaviour | [17] |
| AC | Decreased | Proliferative and invasive human melanoma cells | Pro-invasive | [18,19] |
| SphK1 | Increased | Murine and human melanoma cells and biopsies | Pro-tumoral Immunosuppressive signature | [20,21,22] |
| SPL | Decreased | Human melanoma cells | Resistance to chemotherapy Increased proliferation | [23] |
| GD3 | Increased | GD3+ human melanoma cells with c-Yes inhibition | Reduced malignancy | [24,25,26] |
| SMS1 | Decreased | Human biopsies | Worse prognosis | [29] |
| SPC | Increased | Mel-Ab and human melanocytes | Stimulate melanomagenesis Hypopigmentation in melanocytes | [30,31,32,33] |
| A-SMase | Decreased | Primary melanomas and lymph node metastases Pigmented murine and human melanomas | Inverse correlation with melanin content | [34,35] |
| Targeted SL-Metabolising Enzyme | Melanoma Cells | Experimental Strategy | Treatment | Effects on Drug Sensitivity | Refs |
|---|---|---|---|---|---|
| A-SMase | B16-W6_pSIL10 | shRNA | Cisplatin (chemotherapy) | Low A-SMase is associated with reduced mTOR-related autophagy and resistance to cisplatin | [201] |
| AC | A375 | AC overexpression | Dacarbazine (chemotherapy) | AC overexpression confers resistance to dacarbazine | [66] |
| AC | G361 A375 | ARN14988 ARN398 (AC inhibitor) | 5-FU (chemotherapy) | AC inhibition sensitises G361 cells (proliferative phenotype) but not A375 cells (invasive phenotype) to chemotherapeutic drugs | [18] |
| SphK1 | SK-Mel-28 A375 | FTY720 | Cisplatin (chemotherapy) | SK1 inhibition increases cisplatin-induced apoptosis through a downregulation of the PI3K/AKT/mTOR pathway and decreases EGFR expression | [202] |
| SphK1 | UACC 903 | siRNA | Staurosporine (Apoptosis inducing agent) | Downregulation of Sphk1 sensitises cells to staurosporine-induced apoptosis through AKT inhibition, and G0/G1 phase cell cycle arrest | [20] |
| SphK1 | A375 (overexpression) Mel-2a (downregulation) | SphK1 overexpression or downregulation | Doxorubicin (chemotherapy) | Sphk1 overexpression induces resistance to doxorubicin-induced apoptosis whereas its downregulation by siRNA increases melanoma cell sensitivity to the treatment | [203] |
| SphK1 | WM115 SK-Mel-28 | FTY720 | Vemurafenib (BRAF inhibitor) | SK1 inhibition increases vemurafenib-induced apoptosis | [204] |
| SphK1 | WM9 | SKI-I | Vemurafenib (BRAF inhibitor) | Sphk1 inhibition blocks BRAFi-resistant melanoma cell growth by reducing MITF and Bcl-2 expression | [64] |
| SphK1 | B16-F10 | PF-543 | ICB Adoptive transfer of melanoma antigen-specific T cells | Sphk1 inhibition in T cells maintains Tcm phenotype, reduces Treg induction and synergises with anti-PD1 treatment | [164] |
| SphK1 | Yumm 1.7 | shRNA | ICB | SphK1 downregulation enhances ICB therapy efficacy by reducing Treg infiltration | [22] |
| GCS | B16 | PDMP | Genistein (Apoptosis inducing agent) | Ceramide accumulation enhances genistein-induced apoptosis and growth inhibition through JNK activation and AKT inhibition | [205] |
| SL-Related Treatment | Models | Associated Drug | Effects | Refs |
|---|---|---|---|---|
| Nanoliposomal ceramide | UACC 903 cells 1205 Lu cells Xenografts in nude mice | Sorafenib | Inhibition of melanoma cell growth by targeting both PI3K and MAPK signalling | [206] |
| Nanoliposomal ceramide | 1205 Lu cells In vitro experiments | None | Reduction of integrin affinity and inhibition of melanoma cell migration through PI3K and PKCζ tumour-suppressive activities | [207] |
| KRN7000 | B16 melanoma cell graft intravenously injected in mice | None | Increase of lifespan of mice | [208] |
| OGT2378 (GCS inhibitor) | B16 derived MEB4 melanoma cell graft in female C57BL/6 mice | None | Inhibition of tumour growth and reduction of established tumours | [69] |
| Intra-muscular GM3/VSSP vaccine | Phase I clinical trial: 26 patients with advanced (stage III and IV) melanoma | Adjuvant Montanide Isa 51 | GM3/VSSP vaccine induces anti-GM3 IgM response in 44% of patients. Serum reactivity against melanoma cells and tumour biopsies is reported | [209] |
| L612-HuMAb (Human monoclonal antibody that binds to GM3) | Phase I clinical trial: 9 patients with advanced (stage IV) melanoma | None | L612 HuMAb induces significant antitumour activity in melanoma patients | [210] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrié, L.; Virazels, M.; Dufau, C.; Montfort, A.; Levade, T.; Ségui, B.; Andrieu-Abadie, N. New Insights into the Role of Sphingolipid Metabolism in Melanoma. Cells 2020, 9, 1967. https://doi.org/10.3390/cells9091967
Carrié L, Virazels M, Dufau C, Montfort A, Levade T, Ségui B, Andrieu-Abadie N. New Insights into the Role of Sphingolipid Metabolism in Melanoma. Cells. 2020; 9(9):1967. https://doi.org/10.3390/cells9091967
Chicago/Turabian StyleCarrié, Lorry, Mathieu Virazels, Carine Dufau, Anne Montfort, Thierry Levade, Bruno Ségui, and Nathalie Andrieu-Abadie. 2020. "New Insights into the Role of Sphingolipid Metabolism in Melanoma" Cells 9, no. 9: 1967. https://doi.org/10.3390/cells9091967
APA StyleCarrié, L., Virazels, M., Dufau, C., Montfort, A., Levade, T., Ségui, B., & Andrieu-Abadie, N. (2020). New Insights into the Role of Sphingolipid Metabolism in Melanoma. Cells, 9(9), 1967. https://doi.org/10.3390/cells9091967