Signalling, Metabolic Pathways and Iron Homeostasis in Endothelial Cells in Health, Atherosclerosis and Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Overview of the Endothelial Function
1.2. Endothelial Response to Flow
1.3. Flow Mechanosensors in Endothelial Cells
2. Angiogenesis in Development and Diseases
2.1. Drivers of Angiogenesis
2.2. Sprouting in Angiogenesis
2.3. VEGF as a Therapeutic Target in Pathological Angiogenesis
3. Neuropilin-1 Signalling in Endothelial Cells
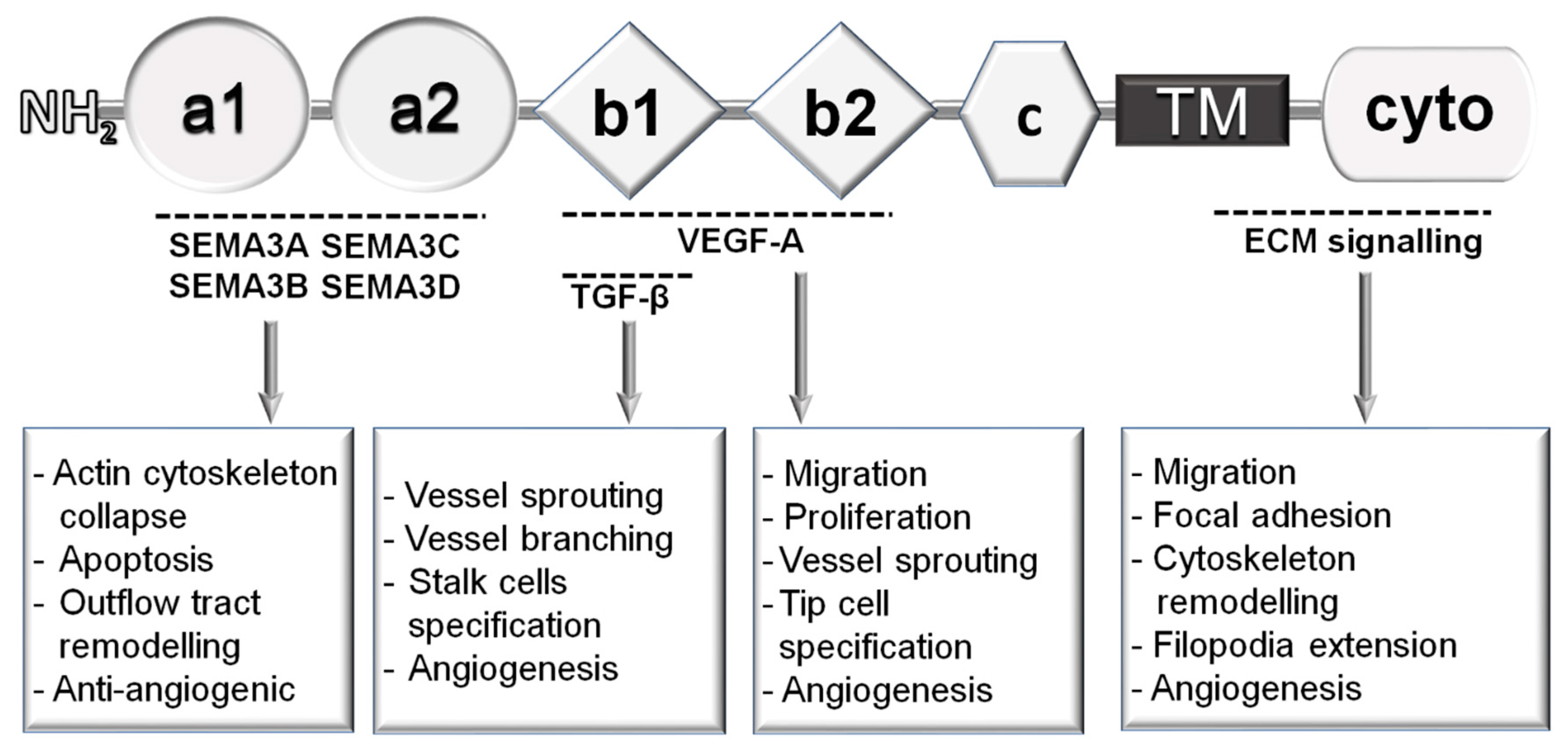
3.1. Neuropilin-1-Dependent Semaphorin Signallings
3.2. Role of Neuropilin-1 in VEGF Signalling
3.3. VEGF-Independent Role of Neuropilin-1 in Angiogenesis
3.4. Role of Neuropilin-1 in Integrin and TGFβ-Mediated Signals
4. Metabolism and Endothelial Function
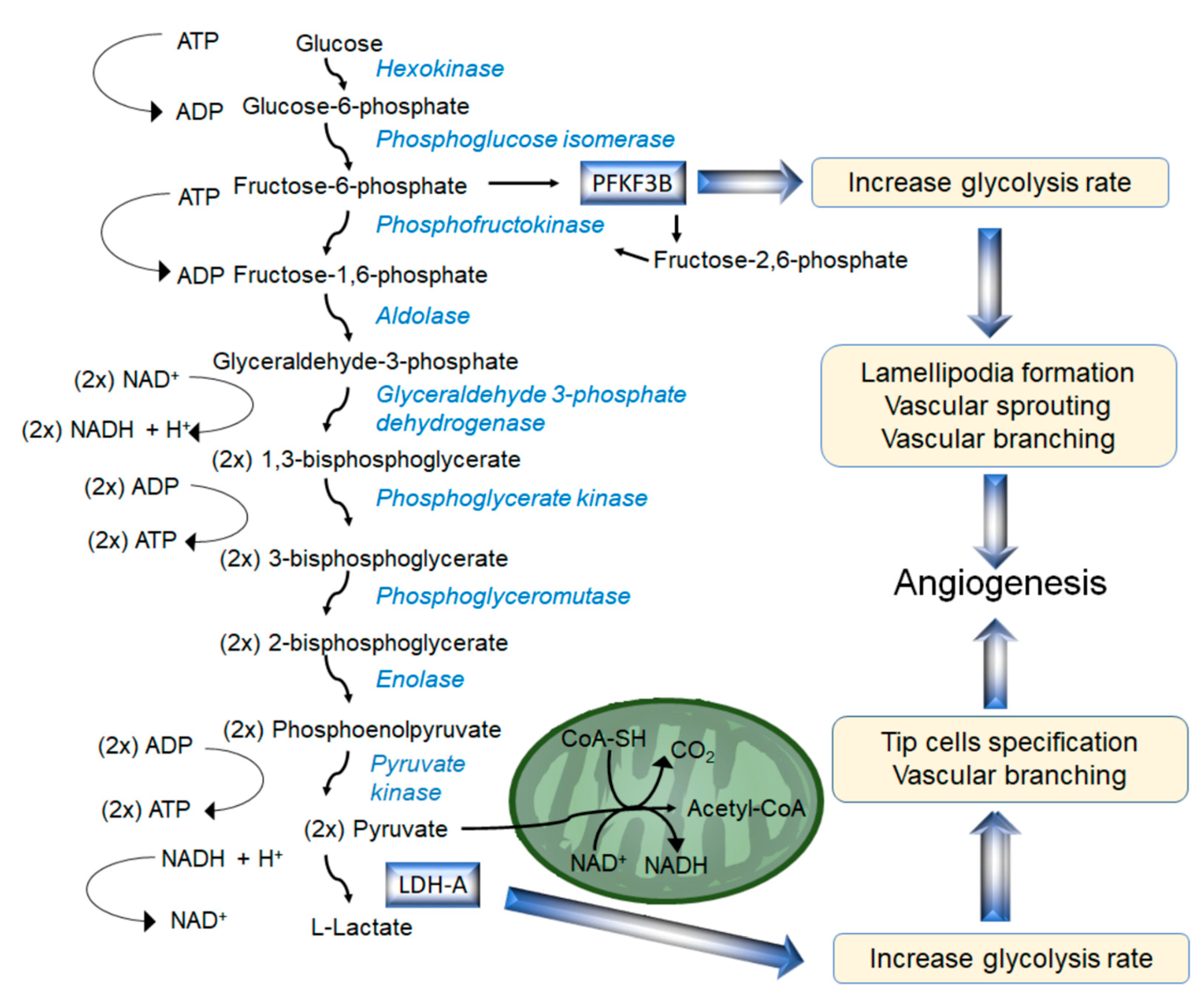
4.1. Glycolytic Flux and Angiogenesis
4.2. 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase (PFKFB3) in Physiological and Pathological Angiogenesis
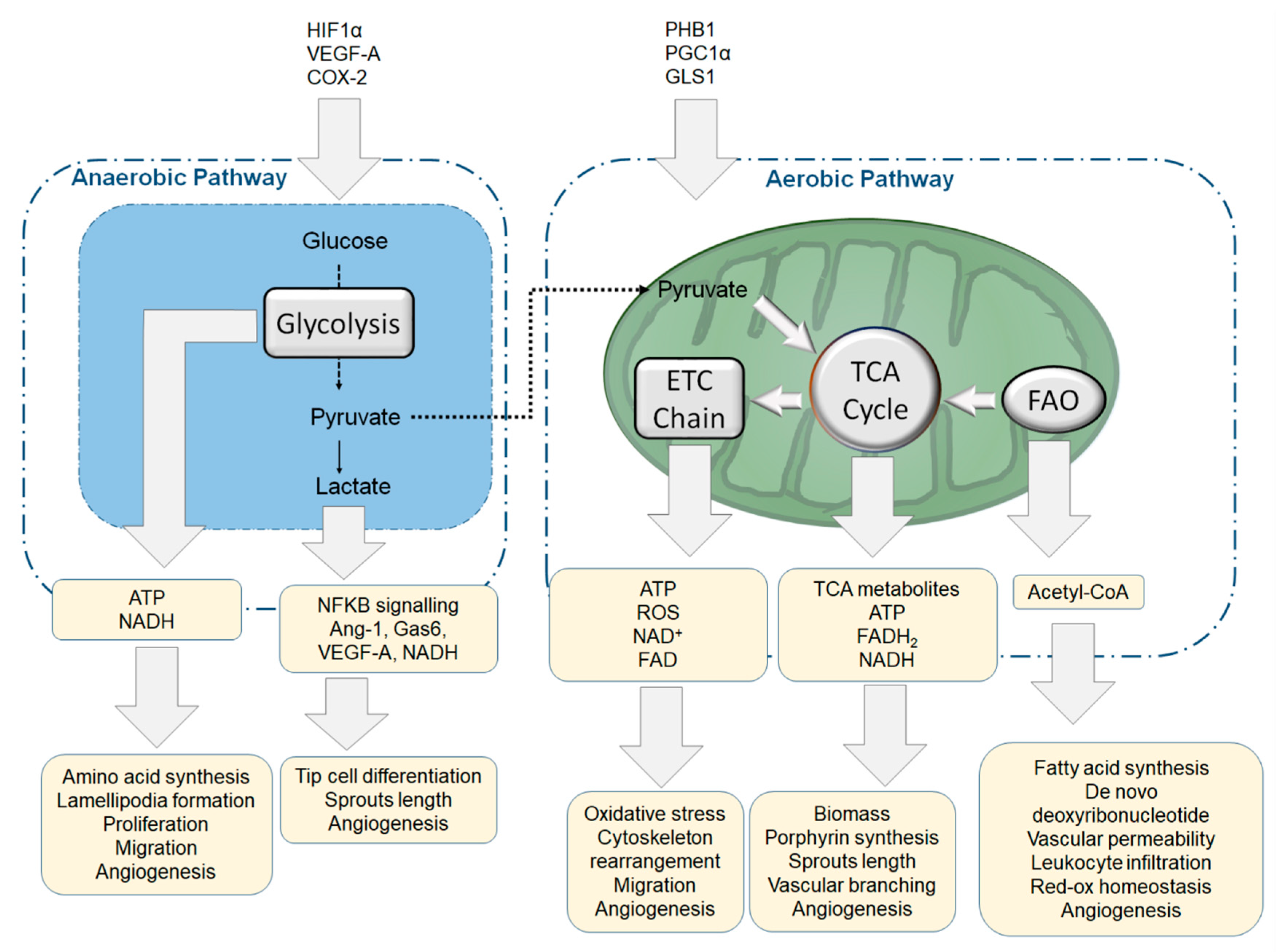
4.3. Aerobic, Anaerobic Respiration and Mitochondria Homeostasis in ECs
4.4. Biosynthetic Pathways and Anaplerosis Modulate Endothelial Function
5. Reactive Oxygen Species (ROS)
5.1. Mitochondrial ROS Production and Detoxification
5.2. ROS: Double-Edged Modulators of Endothelial Function
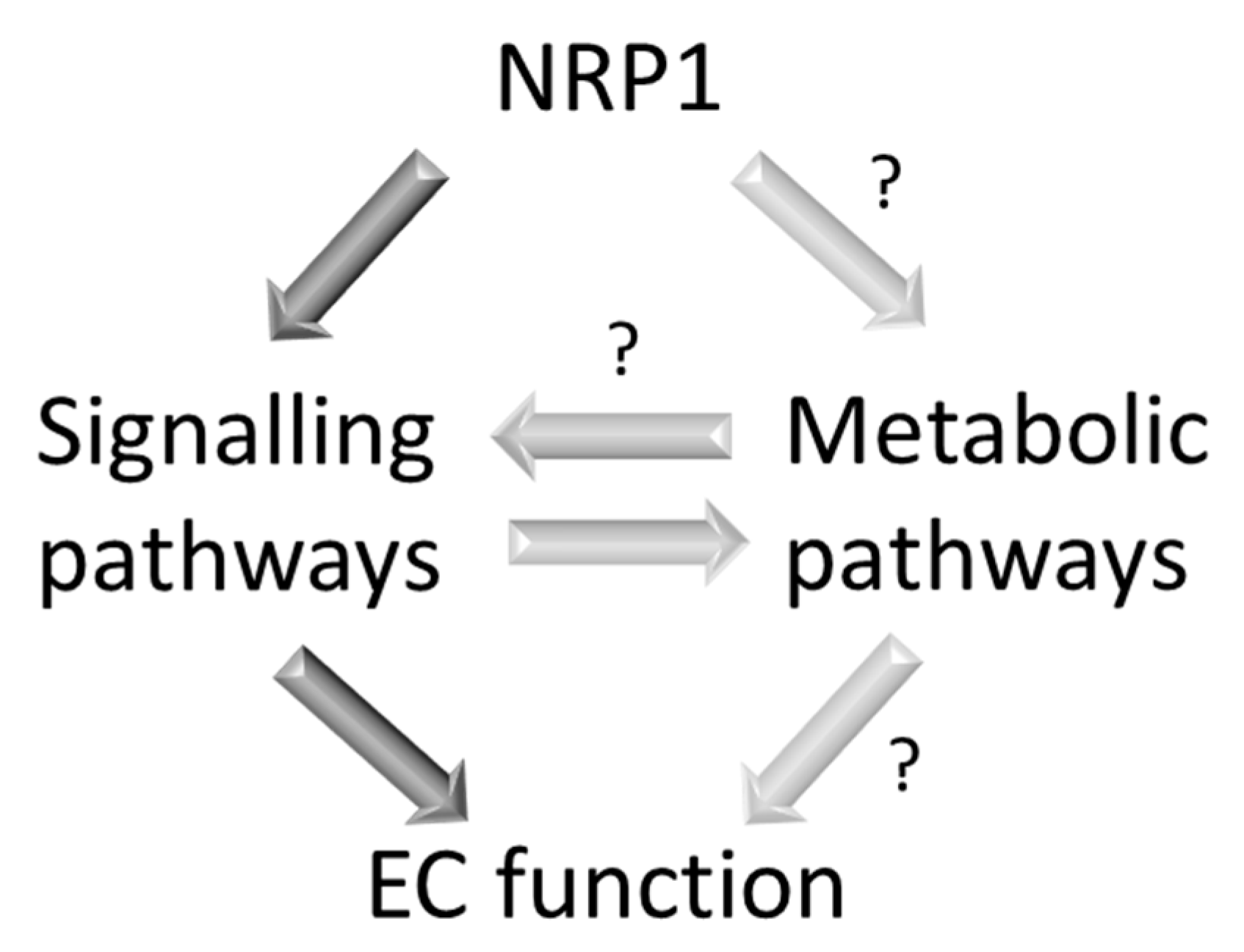
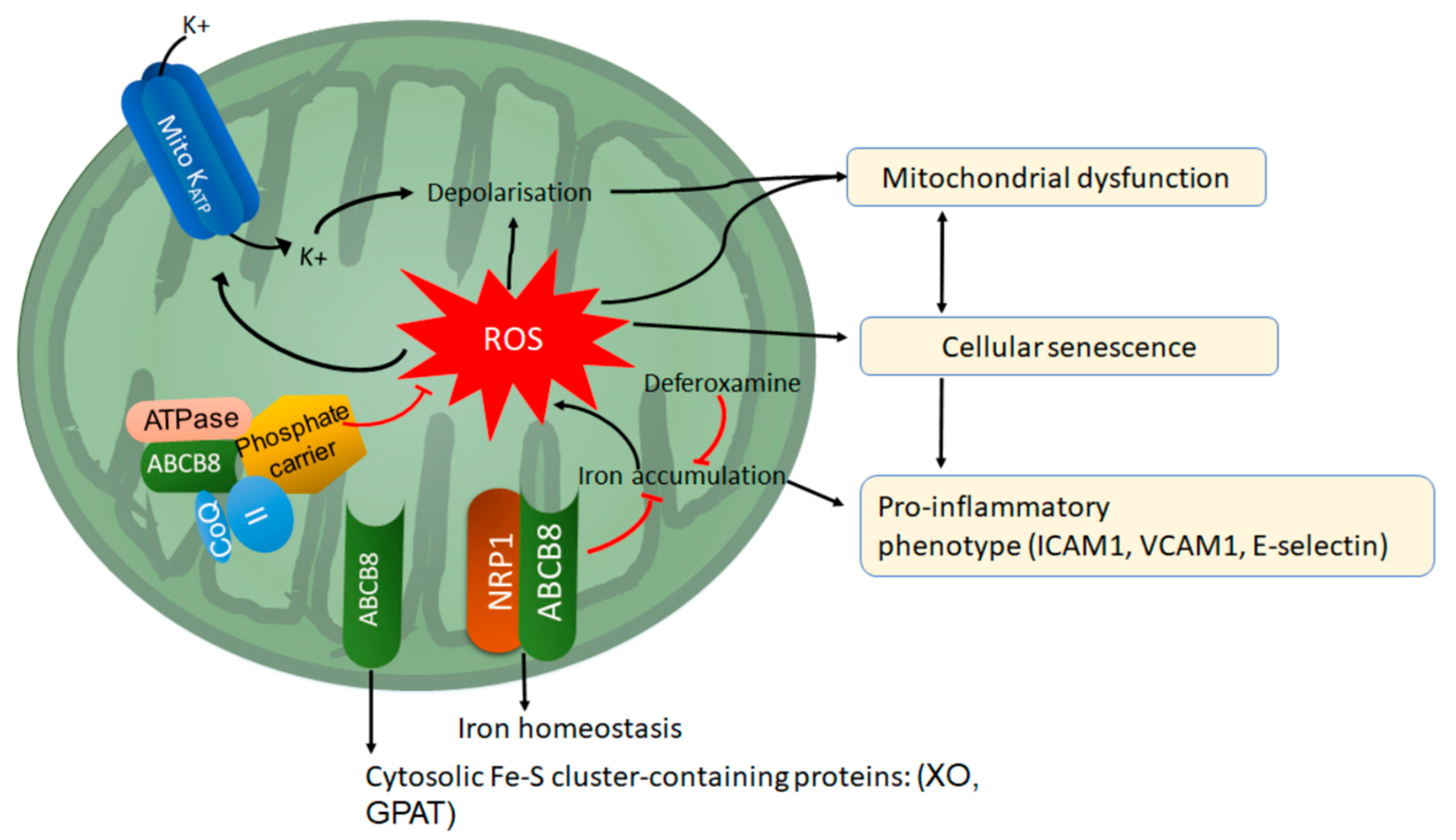
6. Neuropilin-1 and ATP Binding Cassette Subfamily B Member 8 (ABCB8): Two Modulators of Mitochondrial Function in ECs
6.1. ABC Transporters
6.2. ABCB8
7. Iron Metabolism and Homeostasis
7.1. Role of Endothelial Cells in Iron Metabolism
7.2. Effects of Iron Levels on Endothelial Function
8. Role of EC Dysfunction in Pathology
8.1. Atherosclerosis
8.1.1. Role of Iron in Atherosclerosis
8.1.2. Atherosclerosis and Alzheimer’s Disease (AD)
8.2. Alzheimer’s Disease (AD)
8.2.1. Blood–Brain Barrier Dysfunction and Alzheimer’s Disease
8.2.2. The Role of Iron in Alzheimer’s Disease
8.2.3. Mitochondrial Dysfunction and Alzheimer’s Disease
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Augustin, H. Endothelial Chemotaxis and Chemokinesis Assays. In Methods in Endothelial Cell Biology; Augustin, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 145–156. [Google Scholar]
- Van Belle, E.; Bauters, C.; Asahara, T.; Isner, J.M. Endothelial Regrowth after Arterial Injury: From Vascular Repair to Therapeutics. Cardiovasc. Res. 1998, 38, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Asahara, T.; Bauters, C.; Pastore, C.; Kearney, M.; Rossow, S.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Local Delivery of Vascular Endothelial Growth Factor Accelerates Reendothelialization and Attenuates Intimal Hyperplasia in Balloon-Injured Rat Carotid Artery. Circulation 1995, 91, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.R. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr. Med. Chem. 2010, 17, 2059–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. von Willebrand factor biosynthesis, secretion, and clearance: Connecting the far ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randi, A.M.; Laffan, M.A. Von Willebrand factor and angiogenesis: Basic and applied issues. J. Thromb. Haemost. 2017, 15, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kapil, V.; Khambata, R.S.; Robertson, A.; Caulfield, M.J.; Ahluwalia, A. Dietary nitrate provides sustained blood pressure lowering in hypertensive patients: A randomized, phase 2, double-blind, placebo-controlled study. Hypertension 2015, 65, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Warboys, C.M.; Ghim, M.; Weinberg, P.D. Understanding mechanobiology in cultured endothelium: A review of the orbital shaker method. Atherosclerosis 2019, 285, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Bryan, M.T.; Duckles, H.; Feng, S.; Hsiao, S.T.; Kim, H.R.; Serbanovic-Canic, J.; Evans, P.C. Mechanoresponsive Networks Controlling Vascular Inflammation. Arterioscl. Throm. Vas. 2014, 34, 2199–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahnsen, E.D.; Trindade, A.; Zaun, H.C.; Lehoux, S.; Duarte, A.; Jones, E.A. Notch1 is pan-endothelial at the onset of flow and regulated by flow. PLoS ONE 2015, 10, e0122622. [Google Scholar] [CrossRef] [Green Version]
- Urschel, K.; Cicha, I. TNF-α in the cardiovascular system: From physiology to therapy. Int. J. Interf. Cytokine Mediat. Res. 2015, 7, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Huang, R.T.; Hamanaka, R.B.; Krause, M.; Oh, M.J.; Kuo, C.H.; Nigdelioglu, R.; Meliton, A.Y.; Witt, L.; Dai, G.; et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife 2017, 6. [Google Scholar] [CrossRef]
- Yurdagul, A., Jr.; Orr, A.W. Blood Brothers: Hemodynamics and Cell-Matrix Interactions in Endothelial Function. Antioxid. Redox. Signal. 2016, 25, 415–434. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Tzima, E.; Del Pozo, M.A.; Kiosses, W.B.; Mohamed, S.A.; Li, S.; Chien, S.; Schwartz, M.A. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002, 21, 6791–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzima, E.; del Pozo, M.A.; Shattil, S.J.; Chien, S.; Schwartz, M.A. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001, 20, 4639–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Kiosses, W.B.; del Pozo, M.A.; Schwartz, M.A. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J. Biol. Chem. 2003, 278, 31020–31023. [Google Scholar] [CrossRef] [Green Version]
- Lehoux, S.; Castier, Y.; Tedgui, A. Molecular mechanisms of the vascular responses to haemodynamic forces. J. Intern. Med. 2006, 259, 381–392. [Google Scholar] [CrossRef]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox. Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Baker, B.M.; Chen, C.S.; Schwartz, M.A. Endothelial cell sensing of flow direction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2130–2136. [Google Scholar] [CrossRef] [Green Version]
- Lucitti, J.L.; Jones, E.A.; Huang, C.; Chen, J.; Fraser, S.E.; Dickinson, M.E. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 2007, 134, 3317–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culver, J.C.; Dickinson, M.E. The effects of hemodynamic force on embryonic development. Microcirculation 2010, 17, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Pipp, F.; Boehm, S.; Cai, W.J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W.; et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, F.; Paye, J.; Mac Gabhann, F.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell-dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.W.; Ginsberg, M.H.; Shattil, S.J.; Deckmyn, H.; Schwartz, M.A. Matrix-specific suppression of integrin activation in shear stress signaling. Mol. Biol. Cell 2006, 17, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Honda, K.; Ohata, H. Modulation of Ca2+ transients in cultured endothelial cells in response to fluid flow through alphav integrin. Life Sci. 2007, 81, 1421–1430. [Google Scholar] [CrossRef]
- Yamamoto, K.; Korenaga, R.; Kamiya, A.; Ando, J. Fluid shear stress activates Ca(2+) influx into human endothelial cells via P2X4 purinoceptors. Circ. Res. 2000, 87, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Ohata, H.; Yamamoto, M.; Momose, K. Spontaneous and flow-induced Ca2+ transients in retracted regions in endothelial cells. Biochem. Biophys. Res. Commun. 2001, 281, 172–179. [Google Scholar] [CrossRef]
- Sathanoori, R.; Sward, K.; Olde, B.; Erlinge, D. The ATP Receptors P2X7 and P2X4 Modulate High Glucose and Palmitate-Induced Inflammatory Responses in Endothelial Cells. PLoS ONE 2015, 10, e0125111. [Google Scholar] [CrossRef] [Green Version]
- Coon, B.G.; Baeyens, N.; Han, J.; Budatha, M.; Ross, T.D.; Fang, J.S.; Yun, S.; Thomas, J.L.; Schwartz, M.A. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 2015, 208, 975–986. [Google Scholar] [CrossRef]
- Liu, Y.; Sweet, D.T.; Irani-Tehrani, M.; Maeda, N.; Tzima, E. Shc coordinates signals from intercellular junctions and integrins to regulate flow-induced inflammation. J. Cell Biol. 2008, 182, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.T.; Chen, Z.; Givens, C.S.; Owens, A.P.; Rojas, M.; Tzima, E. Endothelial Shc regulates arteriogenesis through dual control of arterial specification and inflammation via the notch and nuclear factor-kappa-light-chain-enhancer of activated B-cell pathways. Circ. Res. 2013, 113, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeyens, N.; Nicoli, S.; Coon, B.G.; Ross, T.D.; Van den Dries, K.; Han, J.; Lauridsen, H.M.; Mejean, C.O.; Eichmann, A.; Thomas, J.L.; et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Jeong, C.H.; Koo, S.Y.; Son, M.J.; Song, H.S.; Bae, S.K.; Raleigh, J.A.; Chung, H.Y.; Yoo, M.A.; Kim, K.W. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: A possible signal for vessel development. Dev. Dyn. 2001, 220, 175–186. [Google Scholar] [CrossRef]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef]
- Drake, C.J.; Fleming, P.A. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood 2000, 95, 1671–1679. [Google Scholar] [CrossRef]
- Swift, M.R.; Weinstein, B.M. Arterial-venous specification during development. Circ. Res. 2009, 104, 576–588. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Schuermann, A.; Helker, C.S.; Herzog, W. Angiogenesis in zebrafish. Semin. Cell Dev. Biol. 2014, 31, 106–114. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Groep, P.; Kemming, D.; Shvarts, A.; Semenza, G.L.; Meijer, G.A.; van de Wiel, M.A.; Belien, J.A.; van Diest, P.J.; van der Wall, E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J. Pathol. 2005, 206, 291–304. [Google Scholar] [CrossRef]
- Simon, M.P.; Tournaire, R.; Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell Physiol. 2008, 217, 809–818. [Google Scholar] [CrossRef]
- Oladipupo, S.; Hu, S.; Kovalski, J.; Yao, J.; Santeford, A.; Sohn, R.E.; Shohet, R.; Maslov, K.; Wang, L.V.; Arbeit, J.M. VEGF is essential for hypoxia-inducible factor-mediated neovascularization but dispensable for endothelial sprouting. Proc. Natl. Acad. Sci. USA 2011, 108, 13264–13269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.M.; Waltenberger, J.; Yachechko, R.; Mirzapoiazova, T.; Sham, J.S.; Lee, C.G.; Elias, J.A.; Verin, A.D. Neuropilin-1 regulates vascular endothelial growth factor-mediated endothelial permeability. Circ. Res. 2005, 96, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, S.; Leppanen, V.M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Evans, I.M.; Yamaji, M.; Britton, G.; Pellet-Many, C.; Lockie, C.; Zachary, I.C.; Frankel, P. Neuropilin-1 signaling through p130Cas tyrosine phosphorylation is essential for growth factor-dependent migration of glioma and endothelial cells. Mol. Cell Biol. 2011, 31, 1174–1185. [Google Scholar] [CrossRef] [Green Version]
- Abedi, H.; Zachary, I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J. Biol. Chem. 1997, 272, 15442–15451. [Google Scholar] [CrossRef] [Green Version]
- Lamalice, L.; Houle, F.; Jourdan, G.; Huot, J. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 2004, 23, 434–445. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, R.; Kwon, J.; Li, X.; Wang, E.; Patra, S.; Bida, J.P.; Bajzer, Z.; Claesson-Welsh, L.; Mukhopadhyay, D. Distinct role of PLCbeta3 in VEGF-mediated directional migration and vascular sprouting. J. Cell Sci. 2009, 122, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanahan, A.A.; Hermans, K.; Claes, F.; Kerley-Hamilton, J.S.; Zhuang, Z.W.; Giordano, F.J.; Carmeliet, P.; Simons, M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev. Cell 2010, 18, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Cantu Gutierrez, M.; Dang, L.T.; Khyzha, N.; Chen, Z.; Veitch, S.; Cheng, H.S.; Khor, M.; Antounians, L.; Njock, M.S.; et al. Dynamic regulation of VEGF-inducible genes by an ERK/ERG/p300 transcriptional network. Development 2017, 144, 2428–2444. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Chanthery, Y.; Liang, W.C.; Stawicki, S.; Mak, J.; Rathore, N.; Tong, R.K.; Kowalski, J.; Yee, S.F.; Pacheco, G.; et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 2007, 11, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, M.R.; Guo, S.; Minami, T.; Spokes, K.C.; Ueki, K.; Skurk, C.; Walsh, K.; Aird, W.C. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Fruttiger, M. Development of the mouse retinal vasculature: Angiogenesis versus vasculogenesis. Investig. Ophthalmol. Vis. Sci. 2002, 43, 522–527. [Google Scholar]
- Stenzel, D.; Lundkvist, A.; Sauvaget, D.; Busse, M.; Graupera, M.; van der Flier, A.; Wijelath, E.S.; Murray, J.; Sobel, M.; Costell, M.; et al. Integrin-dependent and -independent functions of astrocytic fibronectin in retinal angiogenesis. Development 2011, 138, 4451–4463. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, C.; Fantin, A.; Lampropoulou, A.; Denti, L.; Chikh, A.; Ruhrberg, C. Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J. Exp. Med. 2014, 211, 1167–1183. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef]
- Barlow, K.D.; Sanders, A.M.; Soker, S.; Ergun, S.; Metheny-Barlow, L.J. Pericytes on the tumor vasculature: Jekyll or hyde? Cancer Microenviron. 2013, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ruhrberg, C.; Gerhardt, H.; Golding, M.; Watson, R.; Ioannidou, S.; Fujisawa, H.; Betsholtz, C.; Shima, D.T. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002, 16, 2684–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Brash, J.T.; Fantin, A.; Ruhrberg, C. NRP1 function and targeting in neurovascular development and eye disease. Prog. Retin. Eye Res. 2016, 52, 64–83. [Google Scholar] [CrossRef] [Green Version]
- Claxton, S.; Fruttiger, M. Periodic Delta-like 4 expression in developing retinal arteries. Gene Expr. Patterns. 2004, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Mansoor, S.; Sharma, A.; Sapkal, A.; Sheth, J.; Falatoonzadeh, P.; Kuppermann, B.; Kenney, M. Diabetic retinopathy and VEGF. Open Ophthalmol. J. 2013, 7, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef]
- Smith, R.O.; Ninchoji, T.; Gordon, E.; Andre, H.; Dejana, E.; Vestweber, D.; Kvanta, A.; Claesson-Welsh, L. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. Elife 2020, 9. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef]
- Ferrara, N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am. J. Physiol. Cell Physiol. 2001, 280, C1358–C1366. [Google Scholar] [CrossRef] [PubMed]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Shapiro, H.; Tuomi, L.; Webster, M.; Elledge, J.; Blodi, B. Characteristics of patients losing vision after 2 years of monthly dosing in the phase III ranibizumab clinical trials. Ophthalmology 2011, 118, 523–530. [Google Scholar] [CrossRef]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K.; Group, S.-U.S. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef]
- Usui-Ouchi, A.; Friedlander, M. Anti-VEGF therapy: Higher potency and long-lasting antagonism are not necessarily better. J. Clin. Investig. 2019, 129, 3032–3034. [Google Scholar] [CrossRef]
- Ebos, J.M.; Kerbel, R.S. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ohga, N.; Morishita, Y.; Hida, K.; Miyazono, K.; Watabe, T. BMP-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J. Cell Sci. 2010, 123, 1684–1692. [Google Scholar] [CrossRef] [Green Version]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Somanath, P.R.; Kozak, A.; Goc, A.; El-Remessy, A.B.; Ergul, A.; Johnson, M.H.; Alhusban, A.; Soliman, S.; Fagan, S.C. Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS ONE 2011, 6, e24551. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.G.; Zhang, L.; Tsang, W.; Soltanian-Zadeh, H.; Morris, D.; Zhang, R.; Goussev, A.; Powers, C.; Yeich, T.; Chopp, M. Correlation of VEGF and angiopoietin expression with disruption of blood-brain barrier and angiogenesis after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Margaritescu, O.; Pirici, D.; Margaritescu, C. VEGF expression in human brain tissue after acute ischemic stroke. Rom. J. Morphol. Embryol. 2011, 52, 1283–1292. [Google Scholar]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience 2002, 110, 589–604. [Google Scholar] [CrossRef]
- Dzietko, M.; Derugin, N.; Wendland, M.F.; Vexler, Z.S.; Ferriero, D.M. Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl. Stroke Res. 2013, 4, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Invest. 2003, 111, 1843–1851. [Google Scholar] [CrossRef]
- Kaya, D.; Gursoy-Ozdemir, Y.; Yemisci, M.; Tuncer, N.; Aktan, S.; Dalkara, T. VEGF protects brain against focal ischemia without increasing blood—Brain permeability when administered intracerebroventricularly. J. Cereb. Blood Flow Metab. 2005, 25, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Kitsukawa, T.; Shimizu, M.; Sanbo, M.; Hirata, T.; Taniguchi, M.; Bekku, Y.; Yagi, T.; Fujisawa, H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 1997, 19, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kitsukawa, T.; Bekku, Y.; Matsuda, Y.; Sanbo, M.; Yagi, T.; Fujisawa, H. A requirement for neuropilin-1 in embryonic vessel formation. Development 1999, 126, 4895–4902. [Google Scholar]
- Schwarz, Q.; Ruhrberg, C. Neuropilin, you gotta let me know: Should I stay or should I go? Cell Adh. Migr. 2010, 4, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C. Neuropilin-1 enforces extracellular matrix signalling via ABL1 to promote angiogenesis. Biochem. Soc. Trans. 2014, 42, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Bouvree, K.; Brunet, I.; Del Toro, R.; Gordon, E.; Prahst, C.; Cristofaro, B.; Mathivet, T.; Xu, Y.; Soueid, J.; Fortuna, V.; et al. Semaphorin3A, Neuropilin-1, and PlexinA1 are required for lymphatic valve formation. Circ. Res. 2012, 111, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, G.; Maby-El Hajjami, H.; Karaman, S.; Ochsenbein, A.M.; Alitalo, A.; Siddiqui, S.S.; Ochoa Pereira, C.; Petrova, T.V.; Detmar, M. An unexpected role of semaphorin3a-neuropilin-1 signaling in lymphatic vessel maturation and valve formation. Circ. Res. 2012, 111, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Ochsenbein, A.M.; Karaman, S.; Jurisic, G.; Detmar, M. The role of neuropilin-1/semaphorin 3A signaling in lymphatic vessel development and maturation. Adv. Anat. Embryol. Cell Biol. 2014, 214, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Valdembri, D.; Regano, D.; Maione, F.; Giraudo, E.; Serini, G. Class 3 semaphorins in cardiovascular development. Cell Adh. Migr. 2016, 10, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Rodriguez, E.R.; Reimert, D.V.; Shu, T.; Fritzsch, B.; Richards, L.J.; Kolodkin, A.L.; Ginty, D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 2003, 5, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Joza, S.; Wang, J.; Tseu, I.; Ackerley, C.; Post, M. Fetal, but not postnatal, deletion of semaphorin-neuropilin-1 signaling affects murine alveolar development. Am. J. Respir. Cell Mol. Biol. 2013, 49, 627–636. [Google Scholar] [CrossRef]
- Joza, S.; Wang, J.; Fox, E.; Hillman, V.; Ackerley, C.; Post, M. Loss of semaphorin-neuropilin-1 signaling causes dysmorphic vascularization reminiscent of alveolar capillary dysplasia. Am. J. Pathol. 2012, 181, 2003–2017. [Google Scholar] [CrossRef]
- Varshavsky, A.; Kessler, O.; Abramovitch, S.; Kigel, B.; Zaffryar, S.; Akiri, G.; Neufeld, G. Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res. 2008, 68, 6922–6931. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.J.; Hu, J.; Uemura, A.; Tetzlaff, F.; Augustin, H.G.; Fischer, A. Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med. 2015, 7, 1267–1284. [Google Scholar] [CrossRef]
- Plein, A.; Calmont, A.; Fantin, A.; Denti, L.; Anderson, N.A.; Scambler, P.J.; Ruhrberg, C. Neural crest-derived SEMA3C activates endothelial NRP1 for cardiac outflow tract septation. J. Clin. Investig. 2015, 125, 2661–2676. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, H.; Choi, C.; Ho, V.C.; Gupta, M.; Singh, M.K.; Epstein, J.A. Semaphorin 3d and semaphorin 3e direct endothelial motility through distinct molecular signaling pathways. J. Biol. Chem. 2014, 289, 17971–17979. [Google Scholar] [CrossRef] [Green Version]
- Mamluk, R.; Gechtman, Z.; Kutcher, M.E.; Gasiunas, N.; Gallagher, J.; Klagsbrun, M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002, 277, 24818–24825. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Limberg, B.J.; Whitaker, G.B.; Perman, B.; Leahy, D.J.; Rosenbaum, J.S.; Ginty, D.D.; Kolodkin, A.L. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 18069–18076. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Bae, J.; Shin, T.H.; Kang, S.H.; Jeong, M.; Han, Y.; Park, J.H.; Kim, S.K.; Kim, Y.S. Immunoglobulin Fc-fused, neuropilin-1-specific peptide shows efficient tumor tissue penetration and inhibits tumor growth via anti-angiogenesis. J. Control. Release 2015, 216, 56–68. [Google Scholar] [CrossRef]
- Ko, J.H.; Kwon, H.S.; Kim, B.; Min, G.; Shin, C.; Yang, S.W.; Lee, S.W.; Lee, Y.; Hong, D.; Kim, Y.S. Preclinical Efficacy and Safety of an Anti-Human VEGFA and Anti-Human NRP1 Dual-Targeting Bispecific Antibody (IDB0076). Biomolecules 2020, 10, 919. [Google Scholar] [CrossRef]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol. Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef]
- Fantin, A.; Herzog, B.; Mahmoud, M.; Yamaji, M.; Plein, A.; Denti, L.; Ruhrberg, C.; Zachary, I. Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 2014, 141, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Gelfand, M.V.; Hagan, N.; Tata, A.; Oh, W.J.; Lacoste, B.; Kang, K.T.; Kopycinska, J.; Bischoff, J.; Wang, J.H.; Gu, C. Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. Elife 2014, 3, e03720. [Google Scholar] [CrossRef]
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Valdembri, D.; Caswell, P.T.; Anderson, K.I.; Schwarz, J.P.; Konig, I.; Astanina, E.; Caccavari, F.; Norman, J.C.; Humphries, M.J.; Bussolino, F.; et al. Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol. 2009, 7, e25. [Google Scholar] [CrossRef]
- Yaqoob, U.; Cao, S.; Shergill, U.; Jagavelu, K.; Geng, Z.; Yin, M.; de Assuncao, T.M.; Cao, Y.; Szabolcs, A.; Thorgeirsson, S.; et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 2012, 72, 4047–4059. [Google Scholar] [CrossRef] [Green Version]
- Murga, M.; Fernandez-Capetillo, O.; Tosato, G. Neuropilin-1 regulates attachment in human endothelial cells independently of vascular endothelial growth factor receptor-2. Blood 2005, 105, 1992–1999. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Milo, R.; Kam, Z.; Geiger, B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 2007, 120, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Qin, R.; Schmid, H.; Munzberg, C.; Maass, U.; Krndija, D.; Adler, G.; Seufferlein, T.; Liedert, A.; Ignatius, A.; Oswald, F.; et al. Phosphorylation and turnover of paxillin in focal contacts is controlled by force and defines the dynamic state of the adhesion site. Cytoskeleton (Hoboken) 2015, 72, 101–112. [Google Scholar] [CrossRef]
- Fantin, A.; Lampropoulou, A.; Gestri, G.; Raimondi, C.; Senatore, V.; Zachary, I.; Ruhrberg, C. NRP1 Regulates CDC42 Activation to Promote Filopodia Formation in Endothelial Tip Cells. Cell Rep. 2015, 11, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Glinka, Y.; Prud’homme, G.J. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 2008, 84, 302–310. [Google Scholar] [CrossRef]
- Glinka, Y.; Stoilova, S.; Mohammed, N.; Prud’homme, G.J. Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 2011, 32, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Hirota, S.; Clements, T.P.; Tang, L.K.; Morales, J.E.; Lee, H.S.; Oh, S.P.; Rivera, G.M.; Wagner, D.S.; McCarty, J.H. Neuropilin 1 balances beta8 integrin-activated TGFbeta signaling to control sprouting angiogenesis in the brain. Development 2015, 142, 4363–4373. [Google Scholar] [CrossRef] [Green Version]
- Aspalter, I.M.; Gordon, E.; Dubrac, A.; Ragab, A.; Narloch, J.; Vizan, P.; Geudens, I.; Collins, R.T.; Franco, C.A.; Abrahams, C.L.; et al. Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat. Commun. 2015, 6, 7264. [Google Scholar] [CrossRef] [Green Version]
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Cantelmo, A.R.; Brajic, A.; Carmeliet, P. Endothelial Metabolism Driving Angiogenesis: Emerging Concepts and Principles. Cancer J. 2015, 21, 244–249. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Dobrina, A.; Rossi, F. Metabolic properties of freshly isolated bovine endothelial cells. Biochim. Biophys. Acta 1983, 762, 295–301. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Krutzfeldt, A.; Spahr, R.; Mertens, S.; Siegmund, B.; Piper, H.M. Metabolism of exogenous substrates by coronary endothelial cells in culture. J. Mol. Cell Cardiol. 1990, 22, 1393–1404. [Google Scholar] [CrossRef]
- Peters, K.; Kamp, G.; Berz, A.; Unger, R.E.; Barth, S.; Salamon, A.; Rychly, J.; Kirkpatrick, C.J. Changes in human endothelial cell energy metabolic capacities during in vitro cultivation. The role of aerobic glycolysis and proliferation. Cell Physiol. Biochem. 2009, 24, 483–492. [Google Scholar] [CrossRef]
- Parra-Bonilla, G.; Alvarez, D.F.; Al-Mehdi, A.B.; Alexeyev, M.; Stevens, T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L513–L522. [Google Scholar] [CrossRef] [Green Version]
- Tretyakov, A.V.; Farber, H.W. Endothelial cell tolerance to hypoxia. Potential role of purine nucleotide phosphates. J. Clin. Investig. 1995, 95, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Hue, L.; Rider, M.H. Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochem. J. 1987, 245, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W. H. Freeman and Co.: New York, NY, USA, 2001. [Google Scholar]
- Minchenko, A.; Leshchinsky, I.; Opentanova, I.; Sang, N.; Srinivas, V.; Armstead, V.; Caro, J. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem. 2002, 277, 6183–6187. [Google Scholar] [CrossRef] [Green Version]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef] [Green Version]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Ruan, G.X.; Kazlauskas, A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J. Biol. Chem. 2013, 288, 21161–21172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, T.; Guo, H.; Cui, H.; Li, P.; Feng, D.; Hu, E.; Huang, Q.; Yang, A.; Zhou, J.; et al. Lactate potentiates angiogenesis and neurogenesis in experimental intracerebral hemorrhage. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, S.; Li, L.; Chen, Z.; Yang, Y. COX2 inhibition in the endothelium induces glucose metabolism normalization and impairs tumor progression. Mol. Med. Rep. 2018, 17, 2937–2944. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yetkin-Arik, B.; Vogels, I.M.C.; Neyazi, N.; van Duinen, V.; Houtkooper, R.H.; van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Endothelial tip cells in vitro are less glycolytic and have a more flexible response to metabolic stress than non-tip cells. Sci Rep. 2019, 9, 10414. [Google Scholar] [CrossRef] [Green Version]
- Merchan, J.R.; Kovacs, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Pina, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef] [Green Version]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [Green Version]
- Goveia, J.; Stapor, P.; Carmeliet, P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 2014, 6, 1105–1120. [Google Scholar] [CrossRef]
- Coutelle, O.; Hornig-Do, H.T.; Witt, A.; Andree, M.; Schiffmann, L.M.; Piekarek, M.; Brinkmann, K.; Seeger, J.M.; Liwschitz, M.; Miwa, S.; et al. Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol. Med. 2014, 6, 624–639. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Okamoto, O.K. Combined effects of pericytes in the tumor microenvironment. Stem Cells Int. 2015, 2015, 868475. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.F.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Dromparis, P.; Michelakis, E.D. Mitochondria in vascular health and disease. Annu Rev. Physiol 2013, 75, 95–126. [Google Scholar] [CrossRef]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Lian, Q.; Chen, M.P.; Jiang, D.; Ho, J.T.K.; Cheung, Y.F.; Chan, G.C. Deferiprone inhibits iron overload-induced tissue factor bearing endothelial microparticle generation by inhibition oxidative stress induced mitochondrial injury, and apoptosis. Toxicol. Appl. Pharmacol. 2018, 338, 148–158. [Google Scholar] [CrossRef]
- Wilson, C.; Lee, M.D.; McCarron, J.G. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J. Physiol. 2016, 594, 7267–7307. [Google Scholar] [CrossRef]
- Wilson, C.; Lee, M.D.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Girkin, J.M.; Saunter, C.D.; McCarron, J.G. Mitochondrial ATP production provides long-range control of endothelial inositol trisphosphate-evoked calcium signaling. J. Biol. Chem. 2019, 294, 737–758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lee, M.D.; Wilson, C.; McCarron, J.G. Hydrogen peroxide depolarizes mitochondria and inhibits IP3-evoked Ca(2+) release in the endothelium of intact arteries. Cell Calcium 2019, 84, 102108. [Google Scholar] [CrossRef]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef]
- Kroller-Schon, S.; Jansen, T.; Schuler, A.; Oelze, M.; Wenzel, P.; Hausding, M.; Kerahrodi, J.G.; Beisele, M.; Lackner, K.J.; Daiber, A.; et al. Peroxisome proliferator-activated receptor gamma, coactivator 1alpha deletion induces angiotensin II-associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arter. Thromb. Vasc. Biol. 2013, 33, 1928–1935. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2(high) Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef] [Green Version]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Smith, S. The animal fatty acid synthase: One gene, one polypeptide, seven enzymes. FASEB J. 1994, 8, 1248–1259. [Google Scholar] [CrossRef]
- Browne, C.D.; Hindmarsh, E.J.; Smith, J.W. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. FASEB J. 2006, 20, 2027–2035. [Google Scholar] [CrossRef]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e815. [Google Scholar] [CrossRef] [Green Version]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Bruning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e813. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Ung, L.; Pattamatta, U.; Carnt, N.; Wilkinson-Berka, J.L.; Liew, G.; White, A.J.R. Oxidative stress and reactive oxygen species: A review of their role in ocular disease. Clin. Sci. (Lond.) 2017, 131, 2865–2883. [Google Scholar] [CrossRef]
- Muller, F. The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging. J. Am. Aging Assoc. 2000, 23, 227–253. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, P.; Pozos, R.S.; Somanathan, R.; Shangari, N.; O’Brien, P.J. Mechanism of mitochondrial uncouplers, inhibitors, and toxins: Focus on electron transfer, free radicals, and structure-activity relationships. Curr. Med. Chem. 2005, 12, 2601–2623. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Kauffman, K.J.; Sun, L.; Sun, H.; et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef]
- Moore, A.M.; Mahoney, E.; Dumitrescu, L.; De Jager, P.L.; Koran, M.E.I.; Petyuk, V.A.; Robinson, R.A.; Ruderfer, D.M.; Cox, N.J.; Schneider, J.A.; et al. APOE epsilon4-specific associations of VEGF gene family expression with cognitive aging and Alzheimer’s disease. NeuroBiol. Aging 2019. [Google Scholar] [CrossRef]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 5379–5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wang, Y.; Zhang, H.; Chen, R.; Lv, F.; Li, Z.; Jiang, T.; Lin, D.; Zhang, H.; Yang, L.; et al. The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. Int. J. Biol. Sci. 2019, 15, 1440–1451. [Google Scholar] [CrossRef]
- Colavitti, R.; Pani, G.; Bedogni, B.; Anzevino, R.; Borrello, S.; Waltenberger, J.; Galeotti, T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 2002, 277, 3101–3108. [Google Scholar] [CrossRef] [Green Version]
- Kaisar, M.A.; Prasad, S.; Cucullo, L. Protecting the BBB endothelium against cigarette smoke-induced oxidative stress using popular antioxidants: Are they really beneficial? Brain Res. 2015, 1627, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Stephens, J.M.; Vidal-Puig, A.J. An update on visfatin/pre-B cell colony-enhancing factor, an ubiquitously expressed, illusive cytokine that is regulated in obesity. Curr. Opin. Lipidol. 2006, 17, 128–131. [Google Scholar] [CrossRef]
- Berndt, J.; Kloting, N.; Kralisch, S.; Kovacs, P.; Fasshauer, M.; Schon, M.R.; Stumvoll, M.; Bluher, M. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes 2005, 54, 2911–2916. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Chen, L.K.; Jian, D.Y.; Hsu, T.C.; Huang, W.C.; Kuan, T.T.; Wu, S.Y.; Kwok, C.F.; Ho, L.T.; Juan, C.C. Visfatin Promotes Monocyte Adhesion by Upregulating ICAM-1 and VCAM-1 Expression in Endothelial Cells via Activation of p38-PI3K-Akt Signaling and Subsequent ROS Production and IKK/NF-kappaB Activation. Cell Physiol. Biochem. 2019, 52, 1398–1411. [Google Scholar] [CrossRef]
- Cheng, J.; Yang, H.L.; Gu, C.J.; Liu, Y.K.; Shao, J.; Zhu, R.; He, Y.Y.; Zhu, X.Y.; Li, M.Q. Melatonin restricts the viability and angiogenesis of vascular endothelial cells by suppressing HIF-1alpha/ROS/VEGF. Int. J. Mol. Med. 2019, 43, 945–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Zhang, Y.; Zhong, S.; Gao, F.; Chen, Y.; Li, W.; Zheng, F.; Shi, G. N-n-butyl Haloperidol Iodide Protects against Hypoxia/Reoxygenation Injury in Cardiac Microvascular Endothelial Cells by Regulating the ROS/MAPK/Egr-1 Pathway. Front. Pharmacol. 2016, 7, 520. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongiardi, M.P.; Radice, G.; Piras, M.; Stagni, V.; Pacioni, S.; Re, A.; Putti, S.; Ferre, F.; Farsetti, A.; Pallini, R.; et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene 2019, 38, 5413–5424. [Google Scholar] [CrossRef]
- Yin, Y.; Zhou, Z.; Liu, W.; Chang, Q.; Sun, G.; Dai, Y. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int. J. Biochem. Cell Biol. 2017, 84, 22–34. [Google Scholar] [CrossRef]
- Foreman, K.E.; Tang, J. Molecular mechanisms of replicative senescence in endothelial cells. Exp. Gerontol. 2003, 38, 1251–1257. [Google Scholar] [CrossRef]
- Pearlstein, D.P.; Ali, M.H.; Mungai, P.T.; Hynes, K.L.; Gewertz, B.L.; Schumacker, P.T. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb. Vasc. Biol. 2002, 22, 566–573. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Liaudet, L.; Vassalli, G.; Pacher, P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front. Biosci. (Landmark Ed.) 2009, 14, 4809–4814. [Google Scholar] [CrossRef] [Green Version]
- Mathews, M.T.; Berk, B.C. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler Thromb. Vasc. Biol. 2008, 28, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Lassegue, B.; Griendling, K.K. Reactive oxygen species in hypertension; An update. Am. J. Hypertens 2004, 17, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Duchen, M.R. Endothelial mitochondria: Contributing to vascular function and disease. Circ. Res. 2007, 100, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Holland, I.B.; Cole, S.P.; Kuchler, K.; Higgins, C.F. ABC Proteins: From Bacteria to Man; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Linton, K.J.; Higgins, C.F. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. MicroBiol. 1998, 28, 5–13. [Google Scholar] [CrossRef]
- Higgins, C.F.; Gallagher, M.P.; Hyde, S.C.; Mimmack, M.L.; Pearce, S.R. Periplasmic binding protein-dependent transport systems: The membrane-associated components. Philos. Trans. R Soc. Lond. B Biol. Sci. 1990, 326, 353–364. [Google Scholar] [CrossRef]
- Liu, P.Q.; Liu, C.E.; Ames, G.F. Modulation of ATPase activity by physical disengagement of the ATP-binding domains of an ABC transporter, the histidine permease. J. Biol. Chem. 1999, 274, 18310–18318. [Google Scholar] [CrossRef] [Green Version]
- Hunke, S.; Mourez, M.; Jehanno, M.; Dassa, E.; Schneider, E. ATP modulates subunit-subunit interactions in an ATP-binding cassette transporter (MalFGK2) determined by site-directed chemical cross-linking. J. Biol. Chem. 2000, 275, 15526–15534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zutz, A.; Gompf, S.; Schagger, H.; Tampe, R. Mitochondrial ABC proteins in health and disease. Biochim. Biophys. Acta 2009, 1787, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, A.; Takahashi-Makise, N.; Yien, Y.Y.; Huston, N.C.; Whitman, J.C.; Musso, G.; Wallace, J.A.; Bradley, T.; Bergonia, H.A.; Kafina, M.D.; et al. Reductions in the mitochondrial ABC transporter Abcb10 affect the transcriptional profile of heme biosynthesis genes. J. Biol. Chem. 2017, 292, 16284–16299. [Google Scholar] [CrossRef] [Green Version]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 transporter: Gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 2000, 96, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef]
- Schaedler, T.A.; Thornton, J.D.; Kruse, I.; Schwarzlander, M.; Meyer, A.J.; van Veen, H.W.; Balk, J. A conserved mitochondrial ATP-binding cassette transporter exports glutathione polysulfide for cytosolic metal cofactor assembly. J. Biol. Chem. 2014, 289, 23264–23274. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cowan, J.A. Glutathione-coordinated [2Fe-2S] cluster: A viable physiological substrate for mitochondrial ABCB7 transport. Chem. Commun. (Camb.) 2015, 51, 2253–2255. [Google Scholar] [CrossRef]
- Krishnamurthy, P.C.; Du, G.; Fukuda, Y.; Sun, D.; Sampath, J.; Mercer, K.E.; Wang, J.; Sosa-Pineda, B.; Murti, K.G.; Schuetz, J.D. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006, 443, 586–589. [Google Scholar] [CrossRef]
- Ulrich, D.L.; Lynch, J.; Wang, Y.; Fukuda, Y.; Nachagari, D.; Du, G.; Sun, D.; Fan, Y.; Tsurkan, L.; Potter, P.M.; et al. ATP-dependent mitochondrial porphyrin importer ABCB6 protects against phenylhydrazine toxicity. J. Biol. Chem. 2012, 287, 12679–12690. [Google Scholar] [CrossRef] [Green Version]
- Hogue, D.L.; Liu, L.; Ling, V. Identification and characterization of a mammalian mitochondrial ATP-binding cassette membrane protein. J. Mol. Biol. 1999, 285, 379–389. [Google Scholar] [CrossRef]
- Faust, B.; Pike, A.C.W.; Shintre, C.A.; Quiqley, A.M.; Chu, A.; Barr, A.; Shrestha, L.; Mukhopadhyay, S.; Borkowska, O.; Chalk, R.; et al. The Crystal Structure of Human ABCB8 in an Outward-Facing State. Available online: https://www.rcsb.org/structure/5OCH (accessed on 8 September 2020).
- Ardehali, H.; Chen, Z.; Ko, Y.; Mejia-Alvarez, R.; Marban, E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc. Natl. Acad. Sci. USA 2004, 101, 11880–11885. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Qiu, W.; Shirihai, O.S. Mitochondrial ABC transporters function: The role of ABCB10 (ABC-me) as a novel player in cellular handling of reactive oxygen species. Biochim. Biophys. Acta 2012, 1823, 1945–1957. [Google Scholar] [CrossRef] [Green Version]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabo, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Jairaj Naik, T.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [Green Version]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.I.; Liu, J.; Dutta, P. Iron transport kinetics through blood-brain barrier endothelial cells. Biochim. Biophys Acta Gen. Subj. 2018, 1862, 1168–1179. [Google Scholar] [CrossRef]
- Duck, K.A.; Simpson, I.A.; Connor, J.R. Regulatory mechanisms for iron transport across the blood-brain barrier. Biochem. Biophys. Res. Commun. 2017, 494, 70–75. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Chaston, T.; Chung, B.; Mascarenhas, M.; Marks, J.; Patel, B.; Srai, S.K.; Sharp, P. Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells. Gut 2008, 57, 374–382. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A. Hereditary hemochromatosis. Biochim. Biophys. Acta 2006, 1763, 700–710. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, G.; Wilkinson, N.; Pantopoulos, K. Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front. Pharmacol. 2016, 7, 160. [Google Scholar] [CrossRef]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam. Horm. 2019, 110, 71–99. [Google Scholar] [CrossRef]
- Canali, S.; Wang, C.Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 2017, 92, 1204–1213. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Dev, S.; Canali, S.; Bayer, A.; Xu, Y.; Agarwal, A.; Wang, C.Y.; Babitt, J.L. Endothelial Bone Morphogenetic Protein 2 (Bmp2) Knockout Exacerbates Hemochromatosis in Homeostatic Iron Regulator (Hfe) Knockout Mice but not Bmp6 Knockout Mice. Hepatology 2019. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Robb, E.L.; Gawel, J.M.; Aksentijevic, D.; Cocheme, H.M.; Stewart, T.S.; Shchepinova, M.M.; Qiang, H.; Prime, T.A.; Bright, T.P.; James, A.M.; et al. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 2015, 89, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Gaenzer, H.; Marschang, P.; Sturm, W.; Neumayr, G.; Vogel, W.; Patsch, J.; Weiss, G. Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J. Am. Coll. Cardiol. 2002, 40, 2189–2194. [Google Scholar] [CrossRef] [Green Version]
- Lekakis, J.; Papamicheal, C.; Stamatelopoulos, K.; Cimponeriu, A.; Voutsas, A.; Vemmos, K.; Mavrikakis, M.; Stamatelopoulos, S. Hemochromatosis associated with endothelial dysfunction: Evidence for the role of iron stores in early atherogenesis. Vasc. Med. 1999, 4, 147–148. [Google Scholar] [CrossRef]
- Vinchi, F.; Muckenthaler, M.U.; Da Silva, M.C.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis and iron: From epidemiology to cellular level. Front. Pharmacol. 2014, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Mollet, I.G.; Patel, D.; Govani, F.S.; Giess, A.; Paschalaki, K.; Periyasamy, M.; Lidington, E.C.; Mason, J.C.; Jones, M.D.; Game, L.; et al. Low Dose Iron Treatments Induce a DNA Damage Response in Human Endothelial Cells within Minutes. PLoS ONE 2016, 11, e0147990. [Google Scholar] [CrossRef]
- van Tits, L.J.; Jacobs, E.M.; Swinkels, D.W.; Lemmers, H.L.; van der Vleuten, G.M.; de Graaf, J.; Stalenhoef, A.F. Non-transferrin-bound iron is associated with plasma level of soluble intercellular adhesion molecule-1 but not with in vivo low-density lipoprotein oxidation. Atherosclerosis 2007, 194, 272–278. [Google Scholar] [CrossRef]
- Kell, D.B. Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genom. 2009, 2, 2. [Google Scholar] [CrossRef]
- Silva-Gomes, S.; Vale-Costa, S.; Appelberg, R.; Gomes, M.S. Iron in intracellular infection: To provide or to deprive? Front. Cell Infect. Microbiol. 2013, 3, 96. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Chen, M.P.; Cao, J.M.; Chan, G.C.; Cheung, Y.F. Carvedilol protects against iron-induced microparticle generation and apoptosis of endothelial cells. Acta Haematol. 2014, 132, 200–210. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Georgiou, N.A.; Visseren, F.L.; van Kats-Renaud, H.; van Asbeck, B.S.; Marx, J.J. Endothelial activation and induction of monocyte adhesion by nontransferrin-bound iron present in human sera. FASEB J. 2006, 20, 353–355. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Georgiou, N.A.; Visseren, F.L.; van Kats-Renaud, H.; van Asbeck, B.S.; Marx, J.J. Intracellular labile iron modulates adhesion of human monocytes to human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2257–2262. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Qiao, Y.; Zhou, Q.; Wang, Z.; Chen, X.; Liu, D.; Yin, D.; He, M. Iron Overload Damages the Endothelial Mitochondria via the ROS/ADMA/DDAHII/eNOS/NO Pathway. Oxid. Med. Cell Longev. 2019, 2019, 2340392. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Sullivan, J.L. Iron in arterial plaque: Modifiable risk factor for atherosclerosis. Biochim. Biophys. Acta 2009, 1790, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Falk, E.; Shah, P.K.; Fuster, V. Coronary plaque disruption. Circulation 1995, 92, 657–671. [Google Scholar] [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef]
- Partida, R.A.; Libby, P.; Crea, F.; Jang, I.K. Plaque erosion: A new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur. Heart J. 2018, 39, 2070–2076. [Google Scholar] [CrossRef]
- Shah, P.K. Molecular mechanisms of plaque instability. Curr. Opin. Lipidol. 2007, 18, 492–499. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, atherosclerosis, and coronary heart disease. Eur. Heart J. 2017, 38, 3195–3201. [Google Scholar] [CrossRef]
- Shah, P.K.; Lecis, D. Inflammation in atherosclerotic cardiovascular disease. F1000Res 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Zacharski, L.R.; Chow, B.; Lavori, P.W.; Howes, P.S.; Bell, M.R.; DiTommaso, M.A.; Carnegie, N.M.; Bech, F.; Amidi, M.; Muluk, S. The iron (Fe) and atherosclerosis study (FeAST): A pilot study of reduction of body iron stores in atherosclerotic peripheral vascular disease. Am. Heart J. 2000, 139, 337–345. [Google Scholar] [CrossRef]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Reduction of iron stores and cardiovascular outcomes in patients with peripheral arterial disease: A randomized controlled trial. JAMA 2007, 297, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Kiechl, S.; Willeit, J.; Egger, G.; Poewe, W.; Oberhollenzer, F. Body iron stores and the risk of carotid atherosclerosis: Prospective results from the Bruneck study. Circulation 1997, 96, 3300–3307. [Google Scholar] [CrossRef]
- Kiechl, S.; Willeit, J. In a Nutshell: Findings from the Bruneck Study. Gerontology 2019, 65, 9–19. [Google Scholar] [CrossRef]
- Tuomainen, T.P.; Punnonen, K.; Nyyssonen, K.; Salonen, J.T. Association between body iron stores and the risk of acute myocardial infarction in men. Circulation 1998, 97, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Wish, J.B. Assessing iron status: Beyond serum ferritin and transferrin saturation. Clin. J. Am. Soc. Nephrol. 2006, 1, S4–S8. [Google Scholar] [CrossRef]
- Pang, J.H.; Jiang, M.J.; Chen, Y.L.; Wang, F.W.; Wang, D.L.; Chu, S.H.; Chau, L.Y. Increased ferritin gene expression in atherosclerotic lesions. J. Clin. Invest. 1996, 97, 2204–2212. [Google Scholar] [CrossRef]
- Marques, V.B.; Leal, M.A.S.; Mageski, J.G.A.; Fidelis, H.G.; Nogueira, B.V.; Vasquez, E.C.; Meyrelles, S.D.S.; Simoes, M.R.; Dos Santos, L. Chronic iron overload intensifies atherosclerosis in apolipoprotein E deficient mice: Role of oxidative stress and endothelial dysfunction. Life Sci. 2019, 233, 116702. [Google Scholar] [CrossRef]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2019. [Google Scholar] [CrossRef]
- Stadler, N.; Lindner, R.A.; Davies, M.J. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: Evidence for the presence of elevated levels of iron and copper. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Kumar, A.; Tsao, J.W. Alzheimer Disease; StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Roth, L.; Prahst, C.; Ruckdeschel, T.; Savant, S.; Westrom, S.; Fantin, A.; Riedel, M.; Heroult, M.; Ruhrberg, C.; Augustin, H.G. Neuropilin-1 mediates vascular permeability independently of vascular endothelial growth factor receptor-2 activation. Sci. Signal. 2016, 9, ra42. [Google Scholar] [CrossRef] [Green Version]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-beta and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- Holsinger, R.M.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimers. Dis. 2019. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Perez, J.M.; Evans, A.C.; Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Shin, Y.; Choi, S.H.; Kim, E.; Bylykbashi, E.; Kim, J.A.; Chung, S.; Kim, D.Y.; Kamm, R.D.; Tanzi, R.E. Blood-Brain Barrier Dysfunction in a 3D In Vitro Model of Alzheimer’s Disease. Adv. Sci. 2019, 6, 1900962. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, L.T.; Thal, D.R. Vascular pathology in the aged human brain. Acta Neuropathol. 2010, 119, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Toledo, J.B.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef]
- Ghiso, J.; Fossati, S.; Rostagno, A. Amyloidosis associated with cerebral amyloid angiopathy: Cell signaling pathways elicited in cerebral endothelial cells. J. Alzheimers. Dis. 2014, 42, S167–S176. [Google Scholar] [CrossRef] [Green Version]
- Marco, S.; Skaper, S.D. Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 2006, 401, 219–224. [Google Scholar] [CrossRef]
- Janota, C.; Lemere, C.A.; Brito, M.A. Dissecting the Contribution of Vascular Alterations and Aging to Alzheimer’s Disease. Mol. NeuroBiol. 2016, 53, 3793–3811. [Google Scholar] [CrossRef]
- Wan, W.; Chen, H.; Li, Y. The potential mechanisms of Abeta-receptor for advanced glycation end-products interaction disrupting tight junctions of the blood-brain barrier in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 75–81. [Google Scholar] [CrossRef]
- Chen, W.; Chan, Y.; Wan, W.; Li, Y.; Zhang, C. Abeta1-42 induces cell damage via RAGE-dependent endoplasmic reticulum stress in bEnd.3 cells. Exp. Cell Res. 2018, 362, 83–89. [Google Scholar] [CrossRef]
- Sole, M.; Esteban-Lopez, M.; Taltavull, B.; Fabregas, C.; Fado, R.; Casals, N.; Rodriguez-Alvarez, J.; Minano-Molina, A.J.; Unzeta, M. Blood-brain barrier dysfunction underlying Alzheimer’s disease is induced by an SSAO/VAP-1-dependent cerebrovascular activation with enhanced Abeta deposition. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2189–2202. [Google Scholar] [CrossRef]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Liu, D.X.; Liang, Y.; Xing, L.L.; Zhao, W.H.; Qin, X.X.; Shang, D.S.; Li, B.; Fang, W.G.; Cao, L.; et al. Cystatin C Shifts APP Processing from Amyloid-beta Production towards Non-Amyloidgenic Pathway in Brain Endothelial Cells. PLoS ONE 2016, 11, e0161093. [Google Scholar] [CrossRef]
- Meadowcroft, M.D.; Connor, J.R.; Smith, M.B.; Yang, Q.X. MRI and histological analysis of beta-amyloid plaques in both human Alzheimer’s disease and APP/PS1 transgenic mice. J. Magn. Reson. Imaging 2009, 29, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, R.; Wengenack, T.M.; Poduslo, J.F.; Garwood, M.; Jack, C.R., Jr. Magnetic resonance imaging of amyloid plaques in transgenic mouse models of Alzheimer’s disease. Curr. Med. Imaging Rev. 2011, 7, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Bonda, D.J.; Lee, H.G.; Blair, J.A.; Zhu, X.; Perry, G.; Smith, M.A. Role of metal dyshomeostasis in Alzheimer’s disease. Metallomics 2011, 3, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.G.; Connor, J.R.; Meadowcroft, M.D. The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer’s disease: Two sides of the same coin. NeuroBiol. Dis. 2015, 81, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, L.; Camaschella, C. A potential pathogenetic role of iron in Alzheimer’s disease. J. Cell Mol. Med. 2008, 12, 1548–1550. [Google Scholar] [CrossRef]
- Liao, H.K.; Wang, Y.; Noack Watt, K.E.; Wen, Q.; Breitbach, J.; Kemmet, C.K.; Clark, K.J.; Ekker, S.C.; Essner, J.J.; McGrail, M. Tol2 gene trap integrations in the zebrafish amyloid precursor protein genes appa and aplp2 reveal accumulation of secreted APP at the embryonic veins. Dev. Dyn. 2012, 241, 415–425. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, R.C.; Park, Y.H.; Kosman, D.J. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. 2014, 15, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. beta-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, R.C.; Kosman, D.J. Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Front. Mol. Neurosci. 2015, 8, 31. [Google Scholar] [CrossRef]
- Holmes, C. Review: Systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. NeuroBiol. 2013, 39, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.R.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J. Neurol. Sci. 2008, 272, 164–170. [Google Scholar] [CrossRef]
- Huang, C.W.; Tsai, M.H.; Chen, N.C.; Chen, W.H.; Lu, Y.T.; Lui, C.C.; Chang, Y.T.; Chang, W.N.; Chang, A.Y.; Chang, C.C. Clinical significance of circulating vascular cell adhesion molecule-1 to white matter disintegrity in Alzheimer’s dementia. Thromb. Haemost. 2015, 114, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Hamel, E.; Nicolakakis, N.; Aboulkassim, T.; Ongali, B.; Tong, X.K. Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer’s disease. Exp. Physiol. 2008, 93, 116–120. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Grammas, P.; Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J. Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2011, 4, 616–627. [Google Scholar] [PubMed]
- Fonseca, A.C.; Ferreiro, E.; Oliveira, C.R.; Cardoso, S.M.; Pereira, C.F. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim. Biophys. Acta 2013, 1832, 2191–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Chen, S.; Ku, G.; Ahmed, S.H.; Xu, J.; Chen, H.; Hsu, C.Y. Amyloid beta peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J. Cereb. Blood Flow Metab. 2001, 21, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Leuner, K.; Schutt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox. Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; Trimmer, P.A.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; et al. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 2000, 48, 148–155. [Google Scholar] [CrossRef]
- Onyango, I.G.; Ahn, J.Y.; Tuttle, J.B.; Bennett, J.P., Jr.; Swerdlow, R.H. Nerve growth factor attenuates oxidant-induced beta-amyloid neurotoxicity in sporadic Alzheimer’s disease cybrids. J. Neurochem. 2010, 114, 1605–1618. [Google Scholar] [CrossRef]
- Kukreja, L.; Kujoth, G.C.; Prolla, T.A.; Van Leuven, F.; Vassar, R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, H.; Diaz, F.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 14163–14168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.; Pickrell, A.M.; Fukui, H.; Moraes, C.T. Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiol. Aging 2013, 34, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]



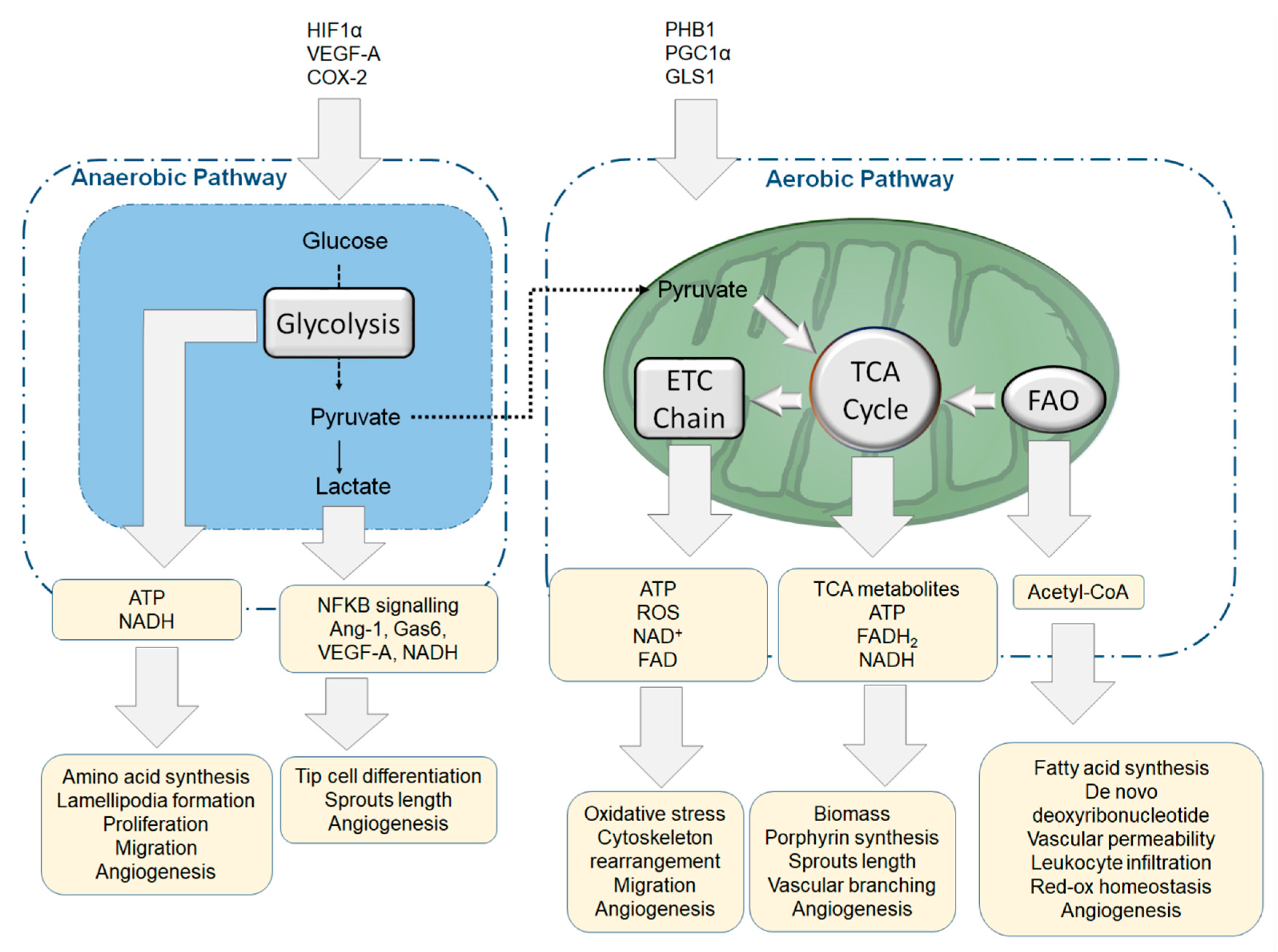

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosseboeuf, E.; Raimondi, C. Signalling, Metabolic Pathways and Iron Homeostasis in Endothelial Cells in Health, Atherosclerosis and Alzheimer’s Disease. Cells 2020, 9, 2055. https://doi.org/10.3390/cells9092055
Bosseboeuf E, Raimondi C. Signalling, Metabolic Pathways and Iron Homeostasis in Endothelial Cells in Health, Atherosclerosis and Alzheimer’s Disease. Cells. 2020; 9(9):2055. https://doi.org/10.3390/cells9092055
Chicago/Turabian StyleBosseboeuf, Emy, and Claudio Raimondi. 2020. "Signalling, Metabolic Pathways and Iron Homeostasis in Endothelial Cells in Health, Atherosclerosis and Alzheimer’s Disease" Cells 9, no. 9: 2055. https://doi.org/10.3390/cells9092055
APA StyleBosseboeuf, E., & Raimondi, C. (2020). Signalling, Metabolic Pathways and Iron Homeostasis in Endothelial Cells in Health, Atherosclerosis and Alzheimer’s Disease. Cells, 9(9), 2055. https://doi.org/10.3390/cells9092055