ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry


Abstract
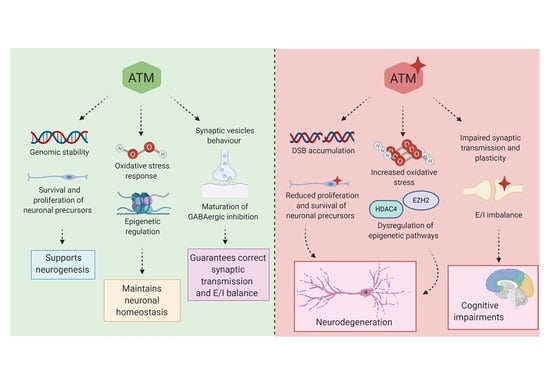
1. Introduction
ATM Protein
2. ATM Governs Genomic Integrity
2.1. ATM Mediates DDR
2.2. ATM Regulates Cell Cycle and Apoptosis
2.3. ATM and Cancer Susceptibility
3. Multiple Roles of ATM Protein Kinase: Beyond the DNA Damage Response
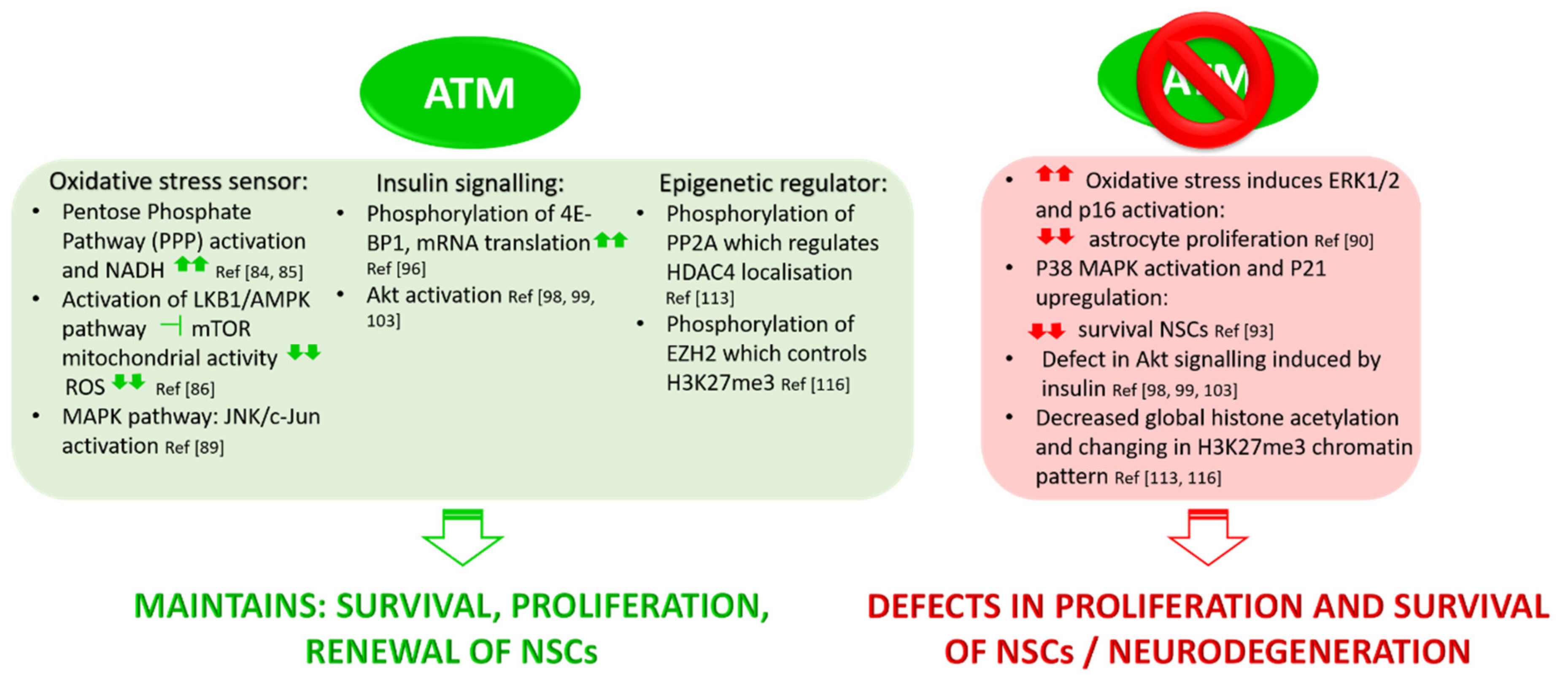
3.1. ATM Functions in Oxidative Stress
3.2. Role of ATM in Insulin Signalling Pathways
3.3. ATM Mediates Epigenetic Regulation
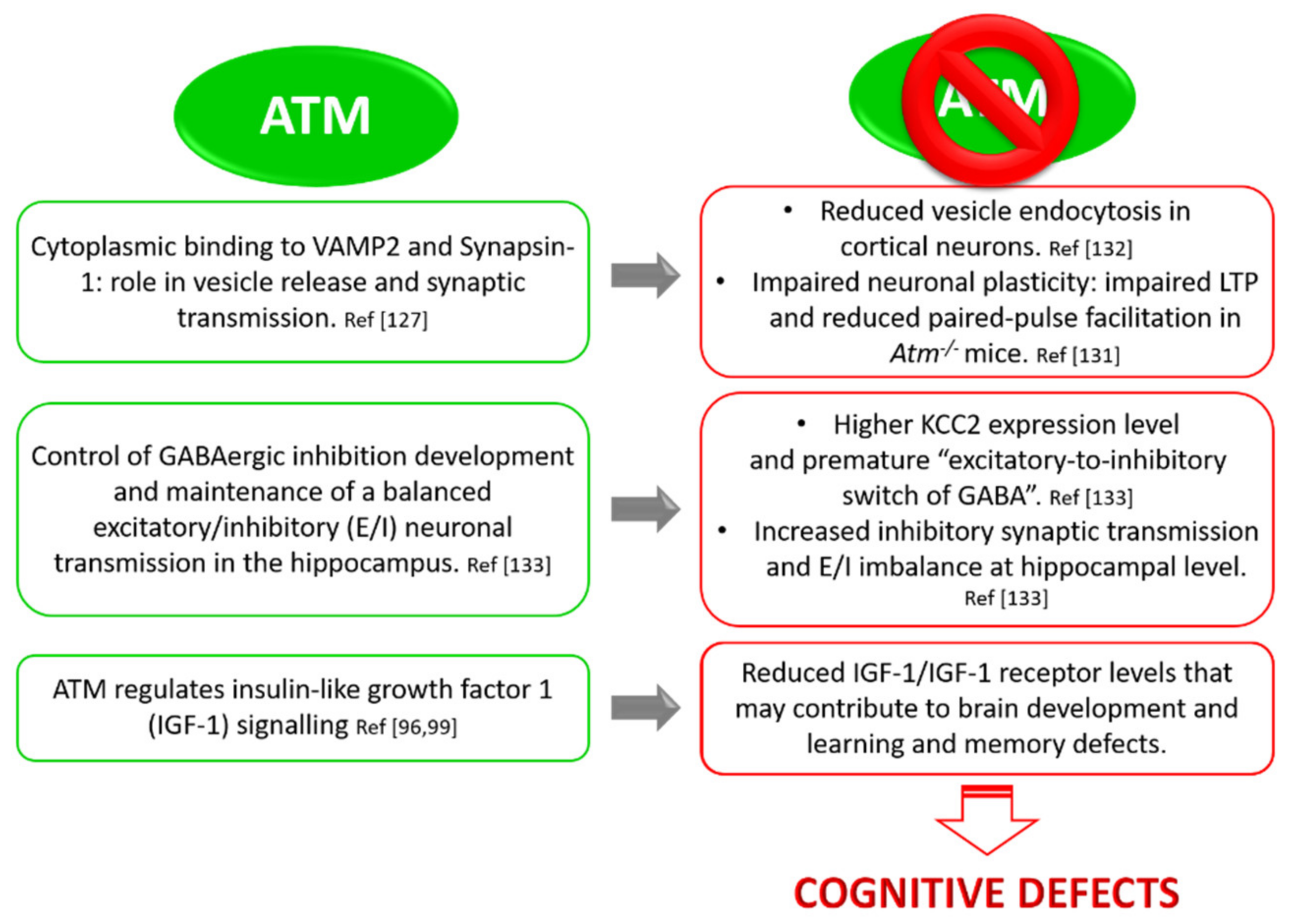
3.4. ATM and Neuronal Function: Implication in Synaptic Vesicles’ Behaviour in Neurons
3.5. ATM and Neuronal Function: Implication in GABAergic Development and Excitatory/Inhibitory Equilibrium
4. ATM Dysregulation in Neurological Diseases
4.1. ATM Involvement in Neurodegenerative Disorders and Brain Senescence
4.2. ATM Involvement in Cognition
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McKinnon, J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.J.; Lange, K.; et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature 1988, 336, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Savitsky, K.; Platzer, M.; Ziv, Y.; Helbitz, T.; Nehls, M.; Boehm, T.; Rosenthal, A.; Shiloh, Y.; Rotman, G. Genomic Organization of the ATM gene. Genomics 1996, 33, 317–320. [Google Scholar] [CrossRef]
- Sandoval, N.; Platzer, M.; Rosenthal, A.; Dörk, T.; Bendix, R.; Skawran, B.; Stuhrmann, M.; Wegner, R.-D.; Sperling, K.; Banin, S.; et al. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum. Mol. Genet. 1999, 8, 69–79. [Google Scholar] [CrossRef]
- Chun, H.H.; Gatti, R.A. Ataxia-telangiectasia, an evolving phenotype. DNA Repair 2004, 3, 1187–1196. [Google Scholar] [CrossRef]
- Boder, E.; Sedgwick, R. Ataxia-telangiectasia: A familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 1958, 21, 526–554. [Google Scholar] [PubMed]
- Biton, S.; Barzilai, A.; Shiloh, Y. The neurological phenotype of ataxia-telangiectasia: Solving a persistent puzzle. DNA Repair 2008, 7, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Becker-Catania, S.G.; Chen, G.; Hwang, M.J.; Wang, Z.; Sun, X.; Sanal, O.; Bernatowska-Matuszkiewicz, E.; Chessa, L.; Lee, E.Y.-H.P.; Gatti, R.A. Ataxia-telangiectasia: Phenotype/genotype studies of ATM protein expression, mutations, and radiosensitivity. Mol. Genet. Metab. 2000, 70, 122–133. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol. 2012, 7, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Becker-Catania, S.; Chun, H.H.; Sun, X.; Mitui, M.; Lai, C.H.; Khanlou, N.; Babaei, M.; Cheng, R.; Clark, C.; et al. The pathogenesis of ataxia-telangiectasia. Learning from a Rosetta Stone. Clin. Rev. Allergy Immunol. 2001, 20, 87–108. [Google Scholar] [CrossRef]
- Farina, L.; Uggetti, C.; Ottolini, A.; Martelli, A.; Bergamaschi, R.; Sibilla, L.; Zappoli, F.; Egitto, M.G.; Lanzi, G. Ataxia-telangiectasia: MR and CT findings. J. Comput. Assist. Tomogr. 1994, 18, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Zimmerman, R.A.; Berry, G.T.; Sullivan, K.; Gatti, R.; Bingham, P. Ataxia-telangiectasia: The pattern of cerebellar atrophy on MRI. Neuroradiology 2003, 45, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Vinters, H.V. Cerebellar pathology in ataxia-telangiectasia: The significance of basket cells. Kroc Found. Ser. 1985, 19, 225–332. [Google Scholar]
- Goldowitz, D.; Hamre, K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998, 21, 375–382. [Google Scholar] [CrossRef]
- Choy, K.R.; Watters, D.J. Neurodegeneration in ataxia-telangiectasia: Multiple roles of ATM kinase in cellular homeostasis. Dev. Dyn. 2018, 247, 33–46. [Google Scholar] [CrossRef]
- Crawford, T.O.; Skolasky, R.L.; Fernandez, R.; Rosquist, K.J.; Lederman, H.M. Survival probability in ataxia telangiectasia. Arch. Dis Child. 2006, 91, 610–611. [Google Scholar] [CrossRef]
- Lefton-Greif, M.A.; Crawford, T.O.; Winkelstein, J.A.; Loughlin, G.M.; Koerner, C.B.; Zahurak, M.; Lederman, H.M. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. J. Pediatr. 2000, 136, 225–231. [Google Scholar] [CrossRef]
- Donath, H.; Woelke, S.; Theis, M.; Heß, U.; Knop, V.; Herrmann, E.; Krauskopf, D.; Kieslich, M.; Schubert, R.; Zielen, S. Progressive Liver Disease in Patients With Ataxia Telangiectasia. Front Pediatr. 2019, 7, 458. [Google Scholar] [CrossRef]
- Paulino, T.L.; Rafael, M.N.; Hix, S.; Shigueoka, D.C.; Ajzen, S.A.; Kochi, C.; Suano-Souza, F.L.; da Silva, R.; Costa-Carvalho, B.T.; Sarni, R.O. Is age a risk factor for liver disease and metabolic alterations in ataxia Telangiectasia patients? Orphanet J. Rare Dis. 2017, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Aloj, G.; Giardino, G.; Valentino, L.; Maio, F.; Gallo, V.; Esposito, T.; Naddei, R.; Cirillo, E.; Pignata, C. Severe combined immunodeficiences: New and old scenarios. Int. Rev. Immunol. 2012, 31, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Boder, E. Ataxia-telangiectasia: An overview. Kroc Found. Ser. 1985, 19, 1–63. [Google Scholar] [PubMed]
- Hoche, F.; Frankenberg, E.; Rambow, J.; Theis, M.; Harding, J.A.; Qirshi, M.; Seidel, K.; Barbosa-Sicard, E.; Porto, L.; Schmahmann, J.D.; et al. Cognitive phenotype in ataxia-telangiectasia. Pediatr. Neurol. 2014, 51, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Hoche, F.; Daly, M.P.; Chutake, Y.K.; Valera, E.; Sherman, J.C.; Schmahmann, J.D. The Cerebellar Cognitive Affective Syndrome in Ataxia-Telangiectasia. Cerebellum 2019, 18, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Mandir, A.S.; Lefton-Greif, M.A.; Goodman, S.N.; Goodman, B.K.; Sengul, H.; Lederman, H.M. Quantitative neurologic assessment of ataxia-telangiectasia. Neurology 2000, 54, 1505–1509. [Google Scholar] [CrossRef]
- Jackson, T.J.; Chow, G.; Suri, M.; Byrd, P.; Taylor, M.R.; Whitehouse, W.P. Longitudinal analysis of the neurological features of ataxia-telangiectasia. Dev. Med. Child. Neurol. 2016, 58, 690–697. [Google Scholar] [CrossRef]
- Brown, K.D.; Barlow, C.; Wynshaw-Boris, A. Multiple ATM-dependent pathways: An explanation for pleiotropy. Am. J. Hum. Genet. 1999, 64, 46–50. [Google Scholar] [CrossRef][Green Version]
- Kanner, S.; Goldin, M.; Galron, R.; Jacob, E.B.; Bonifazi, P.; Barzilai, A. Astrocytes restore connectivity and synchronization in dysfunctional cerebellar networks. Proc. Natl. Acad. Sci. USA 2018, 115, 8025–8030. [Google Scholar] [CrossRef]
- Lau, W.C.; Li, Y.; Liu, Z.; Gao, Y.; Zhang, Q.; Huen, M.S. Structure of the human dimeric ATM kinase. Cell Cycle 2016, 15, 1117–1124. [Google Scholar] [CrossRef]
- Shafman, T.; Khanna, K.K.; Kedar, P.; Spring, K.; Kozlov, S.; Yen, T.; Hobson, K.; Gatei, M.; Zhang, N.; Watters, D.; et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature 1997, 387, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Llorca, O.; Rivera-Calzada, A.; Grantham, J.; Willison, K.R. Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene 2003, 22, 3867–3874. [Google Scholar] [CrossRef]
- Banin, S.; Moyal, L.; Shieh, S.Y.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Chen, P.C.; Lavin, M.F.; Kidson, C.; Moss, D. Identification of ataxia telangiectasia heterozygotes, a cancer prone population. Nature 1978, 274, 484–486. [Google Scholar] [CrossRef]
- Chen, P.C.; Lavin, M.F.; Kidson, C.; Moss, D. Ataxia telangiectasia: A human mutation with abnormal radiation sensitivity. Nature 1975, 258, 427–429. [Google Scholar]
- Gotoff, S.; Amirmokri, E.; Liebner, E.J. Ataxia telangiectasia. Neoplasia, untoward response to x-irradiation, and tuberous sclerosis. Am. J. Dis. Child. 1967, 114, 617–625. [Google Scholar] [CrossRef]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R., III; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.J.; Taylor, A.M.R. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; BekkShiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef]
- Das, B.B.; Antony, S.; Gupta, S.; Dexheimer, T.S.; Redon, C.E.; Garfield, S.; Shiloh, Y.; Pommier, Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009, 28, 3667–3680. [Google Scholar] [CrossRef]
- Frappart, O.; McKinnon, J. Mouse models of DNA double-strand break repair and neurological disease. DNA Repair 2008, 7, 1051–1060. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Jeggo, A. The role of double-strand break repair-insights from human genetics. Nat. Rev. Genet. 2006, 7, 45–54. [Google Scholar] [CrossRef]
- Spencer, S. Hypothesis: Etiologic and Molecular Mechanistic Leads for Sporadic Neurodegenerative Diseases Based on Experience With Western Pacific ALS/PDC. Front Neurol. 2019, 10, 754. [Google Scholar] [CrossRef]
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathol. 2018, 38, 98–107. [Google Scholar] [CrossRef]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992, 71, 543–546. [Google Scholar] [CrossRef]
- Beamish, H.; Lavin, M.F. Radiosensitivity in ataxia-telangiectasia: Anomalies in radiation-induced cell cycle delay. Int. J. Radiat. Biol. 1994, 65, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, J.; Lavin, M.F. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980, 8, 3709–3720. [Google Scholar] [CrossRef]
- Kastan, M.B.; Zhan, Q.; El-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef]
- Painter, R.B.; Young, B.R. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc. Natl. Acad. Sci. USA 1980, 77, 7315–7317. [Google Scholar] [CrossRef]
- Herzog, K.H.; Chong, M.J.; Kapsetaki, M.; Morgan, J.I.; McKinnon, P.J. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 1998, 280, 1089–1091. [Google Scholar] [CrossRef]
- Lee, Y.; Barnes, D.E.; Lindahl, T.; McKinnon, P.J. Defective neurogenesis resulting from DNA ligase IV deficiency requires Atm. Genes Dev. 2000, 14, 2576–2580. [Google Scholar] [CrossRef]
- Lee, Y.; Chong, M.J.; McKinnon, J. Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. J. Neurosci. 2001, 21, 6687–6693. [Google Scholar] [CrossRef] [PubMed]
- Orii, K.E.; Lee, Y.; Kondo, N.; McKinnon, P.J. Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc. Natl. Acad. Sci. USA 2006, 103, 10017–10022. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001, 21, 2661–2668. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Brückner, M.K.; Mosch, B.; Lösche, A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 2010, 177, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Frade, J.M.; Lopez-Sanchez, N. A novel hypothesis for Alzheimer disease based on neuronal tetraploidy induced by p75 (NTR). Cell Cycle 2010, 9, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Schaffner, C.; Litterst, A.; Piebisch, P.; Gilad, S.; Bar-Shira, A.; James, M.R.; Lichter, P.; Döhner, H. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nat. Med. 1997, 3, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Vořechovský, I.; Luo, L.; Dyer, M.J.; Catovsky, D.; Amlot, P.L.; Yaxley, J.C.; Foroni, L.; Hammarström, L.; Webster, D.B.; Yuille, M.A. Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukaemia. Nat. Genet. 1997, 17, 96–99. [Google Scholar] [CrossRef]
- Bullrich, F.; Rasio, D.; Kitada, S.; Starostik, P.; Kipps, T.; Keating, M.; Albitar, M.; Reed, J.C.; Croce, C.M. ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res. 1999, 59, 24–27. [Google Scholar]
- Stankovic, T.; Weber, P.; Stewart, G.; Bedenham, T.; Murray, J.; Byrd, P.J.; Moss, P.A.; Taylor, A.M.R. Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lance 1999, 353, 26–29. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkäs, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM rare variants and cancer risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef]
- Alvarez-Quilón, A.; Serrano-Benítez, A.; Lieberman, J.A.; Quintero, C.; Sánchez-Gutiérrez, D.; Escudero, L.M.; Cortés-Ledesma, F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014, 5, 3347. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Guleria, A.; Chandna, S. ATM kinase: Much more than a DNA damage responsive protein. DNA Repair 2016, 39, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Huang, X.; Tanaka, T.; Kurose, A.; Traganos, F.; Darzynkiewicz, Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int. J. Oncol. 2006, 29, 495–501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Dennery, P.A.; Shigenaga, M.K.; Smith, M.A.; Morrow, J.D.; Roberts, L.J.; Wynshaw-Boris, A.; Levine, R.L. Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc. Natl. Acad. Sci. USA 1999, 96, 9915–9919. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, S.; Kozlov, S.; Farooqi, A.A.; Naqi, A.; Lavin, M.; Khanna, K.K. ATM protein kinase: The linchpin of cellular defenses to stress. Cell Mol. Life Sci. 2011, 68, 2977–3006. [Google Scholar] [CrossRef]
- Takao, N.; Li, Y.; Yamamoto, K. Protective roles for ATM in cellular response to oxidative stress. FEBS Lett. 2000, 472, 133–136. [Google Scholar] [CrossRef]
- Reliene, R.; Fischer, E.; Schiestl, R.H. Effect of N-acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in atm-deficient mice. Cancer Res. 2004, 64, 5148–5153. [Google Scholar] [CrossRef]
- Kamsler, A.; Daily, D.; Hochman, A.; Stern, N.; Shiloh, Y.; Rotman, G.; Barzilai, A. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Res. 2001, 61, 1849–1854. [Google Scholar] [PubMed]
- Watters, D.J. Oxidative stress in ataxia telangiectasia. Redox Rep. 2003, 8, 23–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Ralser, M. ATM is a redox sensor linking genome stability and carbon metabolism. Sci. Signal. 2011, 4, pe17. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.-L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. The axis of mTOR-mitochondria-ROS and stemness of the hematopoietic stem cells. Cell Cycle 2009, 8, 1158–1160. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Lee, S.A.; Dritschilo, A.; Jung, M. Role of ATM in oxidative stress-mediated c-Jun phosphorylation in response to ionizing radiation and CdCl2. J. Biol. Chem. 2001, 276, 11783–11790. [Google Scholar] [CrossRef]
- Liu, N.; Stoica, G.; Yan, M.; Scofield, V.L.; Qiang, W.; Lynn, W.S.; Wong, P.K. ATM deficiency induces oxidative stress and endoplasmic reticulum stress in astrocytes. Lab. Investig. 2005, 85, 1471–1480. [Google Scholar] [CrossRef]
- Carlessi, L.; De Filippis, L.; Lecis, D.; Vescovi, A.; Delia, D. DNA-damage response, survival and differentiation in vitro of a human neural stem cell line in relation to ATM expression. Cell Death Differ. 2009, 16, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wong, K. Loss of ATM impairs proliferation of neural stem cells through oxidative stress-mediated p38 MAPK signaling. Stem Cells 2009, 27, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwangbo, J.; Wong, K. p38 MAPK-Mediated Bmi-1 down-regulation and defective proliferation in ATM-deficient neural stem cells can be restored by Akt activation. PLoS ONE 2011, 6, e16615. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; LeRoith, D. Insulin and insulin-like growth factor receptors in the brain: Physiological and pathological aspects. Eur. Neuropsychopharmacol. 2014, 24, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Yang, D.Q.; Kastan, M.B. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat. Cell Biol. 2000, 2, 893–898. [Google Scholar] [CrossRef]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef]
- Halaby, M.J.; Hibma, J.C.; He, J.; Yang, D.Q. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal. 2008, 20, 1555–1563. [Google Scholar] [CrossRef]
- Viniegra, J.G.; Martínez, N.; Modirassari, P.; Losa, J.H.; Cobo, C.P.; Lobo, V.J.S.A.; Luquero, C.I.A.; Álvarez-Vallina, L.; Cajal, S.R.; Sánchez-Prieto, R. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J. Biol. Chem. 2005, 280, 4029–4036. [Google Scholar] [CrossRef]
- Bar, R.S.; Levis, W.R.; Rechler, M.M.; Harrison, L.C.; Siebert, C.; Podskalny, J.; Royh, J.; Muggeo, M. Extreme insulin resistance in ataxia telangiectasia: Defect in affinity of insulin receptors. N. Engl. J. Med. 1978, 298, 1164–1171. [Google Scholar] [CrossRef]
- López-Carballo, G.; Moreno, L.; Masiá, S.; Pérez, P.; Barettino, D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J. Biol. Chem. 2002, 277, 25297–25304. [Google Scholar] [CrossRef] [PubMed]
- Boehrs, J.K.; He, J.; Halaby, M.J.; Yang, D.Q. Constitutive expression and cytoplasmic compartmentalization of ATM protein in differentiated human neuron-like SH-SY5Y cells. J. Neurochem. 2007, 100, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiong, H.; Yang, D.Q. Functional switching of ATM: Sensor of DNA damage in proliferating cells and mediator of Akt survival signal in post-mitotic human neuron-like cells. Chin. J. Cancer 2012, 31, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Maya-Vetencourt, J.F.; Baroncelli, L.; Viegi, A.; Tiraboschi, E.; Castren, E.; Cattaneo, A.; Maffei, L. IGF-1 restores visual cortex plasticity in adult life by reducing local GABA levels. Neural Plast. 2012, 2012, 250421. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Aleman, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Trejo, J.L.; Piriz, J.; Llorens-Martin, M.V.; Fernandez, A.M.; Bolos, M.; LeRoith, D.; Nuñez, A.; Torres-Aleman, I. Central actions of liver-derived insulin-like growth factor I underlying its pro-cognitive effects. Mol. Psychiatry 2007, 12, 1118–1128. [Google Scholar] [CrossRef]
- Baroncelli, L.; Cenni, M.C.; Melani, R.; Deidda, G.; Landi, S.; Narducci, R.; Cancessa, L.; Maffei, L.; Berardi, N. Early IGF-1 primes visual cortex maturation and accelerates developmental switch between NKCC1 and KCC2 chloride transporters in enriched animals. Neuropharmacology 2017, 113, 167–177. [Google Scholar] [CrossRef]
- Cotman, C.W.; Berchtold, N.C. Physical activity and the maintenance of cognition: Learning from animal models. Alzheimers Dement. 2007, 3, S30–S37. [Google Scholar] [CrossRef]
- Ching, J.K.; Luebbert, S.H.; Collins, R.L., IV; Zhang, Z.; Marupudi, N.; Banerjee, S.; Hurd, R.D.; Ralston, L.; Fisher, J.S. Ataxia telangiectasia mutated impacts insulin-like growth factor 1 signalling in skeletal muscle. Exp. Physiol. 2013, 98, 526–535. [Google Scholar] [CrossRef]
- D’Mello, S.R. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009, 22, 513–524. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Darcy, M.J.; Calvin, K.; Cavnar, K.; Ouimet, C.C. Regional and subcellular distribution of HDAC4 in mouse brain. J. Comp. Neurol. 2010, 518, 722–740. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Ricupero, C.L.; Hart, R.P.; Schwartz, M.S.; Kusnecov, A.; Herrup, K. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med. 2012, 18, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Bolger, T.A.; Yao, T. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 2005, 25, 9544–9553. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Cepko, C.L. HDAC4 regulates neuronal survival in normal and diseased retinas. Science 2009, 323, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hart, R.P.; Mallimo, E.M.; Swerdel, M.R.; Kusnecov, A.W.; Herrup, K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat. Neurosci. 2013, 16, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Weber, P.; Stankovic, T.; Rottinghaus, S.T.; Taylor, A.M.; Jackson, S.P. Analysis of the ATM protein in wild-type and ataxia telangiectasia cells. Oncogene 1996, 13, 2707–2716. [Google Scholar] [PubMed]
- Watters, D.; Khanna, K.K.; Beamish, H.; Birrell, G.; Spring, K.; Kedar, P.; Gatei, M.; Stenzel, D.; Hobson, K.; Kozlov, S.; et al. Cellular localisation of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogene 1997, 14, 1911–1921. [Google Scholar] [CrossRef]
- Watters, D.; Kedar, P.; Spring, K.; Bjorkman, J.; Chen, P.; Gatei, M.; Birrell, G.; Garrone, B.; Srinivasa, P.; Crane, D.I.; et al. Localization of a portion of extranuclear ATM to peroxisomes. J. Biol. Chem. 1999, 274, 34277–34282. [Google Scholar] [CrossRef]
- Blignaut, M.; Loos, B.; Botchway, S.W.; Parker, A.W.; Huisamen, B. Ataxia-Telangiectasia Mutated is located in cardiac mitochondria and impacts oxidative phosphorylation. Sci. Rep. 2019, 9, 4782. [Google Scholar] [CrossRef]
- Valentin-Vega, Y.A.; Kastan, M.B. A new role for ATM: Regulating mitochondrial function and mitophagy. Autophagy 2012, 8, 840–841. [Google Scholar] [CrossRef][Green Version]
- Valentin-Vega, Y.A.; MacLean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.D.; Ziv, Y.; Sadanandan, S.N.; Chessa, L.; Collins, F.S.; Shiloh, Y.; Tagle, D.A. The ataxia-telangiectasia gene product, a constitutively expressed nuclear protein that is not up-regulated following genome damage. Proc. Natl. Acad. Sci. USA 1997, 94, 1840–1845. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Kirsch, D.G.; Canman, C.E.; Ahn, J.H.; Ziv, Y.; Newman, L.S.; Darnell, R.B.; Shiloh, Y.; Kastan, M.B. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc. Natl. Acad. Sci. USA 1998, 95, 10146–10151. [Google Scholar] [CrossRef] [PubMed]
- De Camilli, P.; Emr, S.D.; McPherson, P.S.; Novick, P. Phosphoinositides as regulators in membrane traffic. Science 1996, 271, 1533–1539. [Google Scholar] [CrossRef]
- Oka, A.; Takashima, S. Expression of the ataxia-telangiectasia gene (ATM) product in human cerebellar neurons during development. Neurosci. Lett. 1998, 252, 195–198. [Google Scholar] [CrossRef]
- Li, J.; Han, Y.R.; Plummer, M.R.; Herrup, K. Cytoplasmic ATM in neurons modulates synaptic function. Curr. Biol. 2009, 19, 2091–2096. [Google Scholar] [CrossRef]
- Greengard, P.; Benfenati, F.; Valtorta, F. Synapsin I, an actin-binding protein regulating synaptic vesicle traffic in the nerve terminal. Adv. Second Messenger Phosphoprot. Res. 1994, 29, 31–45. [Google Scholar]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef]
- Schiavo, G.G.; Benfenati, F.; Poulain, B.; Rossetto, O.; de Laureto, P.P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar] [CrossRef]
- Vail, G.; Cheng, A.; Han, Y.R.; Zhao, T.; Du, S.; Loy, M.M.; Herrup, K.; Plummer, M.R. ATM protein is located on presynaptic vesicles and its deficit leads to failures in synaptic plasticity. J. Neurophysiol. 2016, 116, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Zhao, T.; Tse, K.H.; Chow, H.M.; Cui, Y.; Jiang, L.; Du, S.; Loy, M.M.T.; Herrup, K. ATM and ATR play complementary roles in the behavior of excitatory and inhibitory vesicle populations. Proc. Natl. Acad. Sci. USA 2018, 115, E292–E301. [Google Scholar] [CrossRef] [PubMed]
- Pizzamiglio, L.; Focchi, E.; Murru, L.; Tamborini, M.; Passafaro, M.; Menna, E.; Matteoli, M.; Antonucci, F. New Role of ATM in Controlling GABAergic Tone During Development. Cereb Cortex 2016, 26, 3879–3888. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.F.; Boyce, L.H.; Davis, M.B.; Kriegstein, A.R. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 1996, 16, 6414–6423. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, E.; Gaiarsa, J.L.; Ben-Ari, Y. GABA: An excitatory transmitter in early postnatal life. Trends Neurosci. 1991, 14, 515–519. [Google Scholar] [CrossRef]
- Ben-Ari, Y. The GABA excitatory/inhibitory developmental sequence: A personal journey. Neuroscience 2014, 279, 187–219. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Chudotvorova, I.; Ivanov, A.; Rama, S.; Hübner, C.A.; Pellegrino, C.; Ben-Ari, Y.; Medina, I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 2005, 566, 671–679. [Google Scholar] [CrossRef]
- Ganguly, K.; Schinder, A.F.; Wong, S.T.; Poo, M.M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 2001, 105, 521–532. [Google Scholar] [CrossRef]
- Ludwig, A.; Uvarov, P.; Soni, S.; Thomas-Crusells, J.; Airaksinen, M.S.; Rivera, C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 2011, 31, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Le Magueresse, C.; Monyer, H. GABAergic interneurons shape the functional maturation of the cortex. Neuron 2013, 77, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Hensch, T.K.; Fagiolini, M.; Mataga, N.; Stryker, M.P.; Baekkeskov, S.; Kash, S.F. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 1998, 282, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Represa, A.; Ben-Ari, Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005, 28, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Barbin, G.; Pollard, H.; Gaiarsa, J.L.; Ben-Ari, Y. Involvement of GABAA receptors in the outgrowth of cultured hippocampal neurons. Neurosci Lett. 1993, 152, 150–154. [Google Scholar] [CrossRef]
- Herrup, K.; Li, J.; Chen, J. The role of ATM and DNA damage in neurons: Upstream and downstream connections. DNA Repair 2013, 12, 600–604. [Google Scholar] [CrossRef]
- Perlman, S.L.; Boder Deceased, E.; Sedgewick, R.P.; Gatti, R.A. Ataxia-telangiectasia. Handb Clin Neurol 2012, 103, 307–332. [Google Scholar]
- Katyal, S.; McKinnon, J. DNA repair deficiency and neurodegeneration. Cell Cycle 2007, 6, 2360–2365. [Google Scholar] [CrossRef]
- Yang, Y.; Herrup, K. Loss of neuronal cell cycle control in ataxia-telangiectasia: A unified disease mechanism. J. Neurosci. 2005, 25, 2522–2529. [Google Scholar] [CrossRef]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef]
- Petersen, A.J.; Rimkus, S.A.; Wassarman, D.A. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc. Natl. Acad. Sci. USA 2012, 109, E656–E664. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in Vulnerable Regions of the Alzheimer’s Disease Brain Display Reduced ATM Signaling. Eneuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Illuzzi, J.; Yerkes, S.; Parekh-Olmedo, H.; Kmiec, E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J. Neurosci. Res. 2009, 87, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, P.; de Cristofaro, T.; Affaitati, A.; Pizzulo, G.M.; Feliciello, A.; Criscuolo, C.; De Michele, G.; Filla, A.; Avvedimento, E.V.; Varrone, S. DNA damage induced by polyglutamine-expanded proteins. Hum. Mol. Genet. 2003, 12, 2301–2309. [Google Scholar] [CrossRef]
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178. [Google Scholar] [CrossRef]
- Toledo-Sherman, L.; Breccia, P.; Cachope, R.; Bate, J.R.; Angulo-Herrera, I.; Wishart, G.; Matthews, K.L.; Martin, S.L.; Cox, H.C.; McAllister, G.; et al. Optimization of Potent and Selective Ataxia Telangiectasia-Mutated Inhibitors Suitable for a Proof-of-Concept Study in Huntington’s Disease Models. J. Med. Chem. 2019, 62, 2988–3008. [Google Scholar] [CrossRef]
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.; Truant, R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum. Mol. Genet. 2017, 26, 395–406. [Google Scholar] [CrossRef]
- Grimes, A.; Chandra, S.B. Significance of cellular senescence in aging and cancer. Cancer Res. Treat. 2009, 41, 187–195. [Google Scholar] [CrossRef]
- Kanu, N.; Penicud, K.; Hristova, M.; Wong, B.; Irvine, E.; Plattner, F.; Raivich, G.; Behrens, A. The ATM cofactor ATMIN protects against oxidative stress and accumulation of DNA damage in the aging brain. J. Biol. Chem. 2010, 285, 38534–38542. [Google Scholar] [CrossRef]
- Kreis, P.; Gallrein, C.; Rojas-Puente, E.; Mack, T.G.; Kroon, C.; Dinkel, V.; Willmes, C.; Dinkel, V.; Willmes, C.; Murk, K.; et al. ATM phosphorylation of the actin-binding protein drebrin controls oxidation stress-resistance in mammalian neurons and C. elegans. Nat. Commun. 2019, 10, 486. [Google Scholar] [CrossRef]
- Mikati, M.A.; Grintsevich, E.E.; Reisler, E. Drebrin-induced stabilization of actin filaments. J. Biol. Chem. 2013, 288, 19926–19938. [Google Scholar] [CrossRef]
- Kojima, N.; Shirao, T. Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex: A study of neurological disorders accompanied by cognitive deficits. Neurosci. Res. 2007, 58, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Liu, Z.; Peng, L.; Tang, X.; Meng, F.; Ao, Y.; Zhou, M.; Wang, M.; Cao, X.; Qin, B.; et al. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife 2018, 7, e34836. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D.; Sherman, J.C. The cerebellar cognitive affective syndrome. Brain 1998, 121, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, C.M.; O’Hearn, E.; Leroi, I.; Gourley, L.; Ross, C.A.; Margolis, R.L. Cognitive impairment and psychiatric symptoms in 133 patients with diseases associated with cerebellar degeneration. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 109–112. [Google Scholar] [CrossRef]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Dager, S.R.; Dickson, P.E.; Estes, A.M.; et al. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef]
- Volkow, N.D.; Tomasi, D.; Wang, G.J.; Studentsova, Y.; Margus, B.; Crawford, T.O. Brain glucose metabolism in adults with ataxia-telangiectasia and their asymptomatic relatives. Brain 2014, 137, 1753–1761. [Google Scholar] [CrossRef]
- Mostofsky, S.H.; Kunze, J.C.; Cutting, L.E.; Lederman, H.M.; Denckla, M.B. Judgment of duration in individuals with ataxia-telangiectasia. Dev. Neuropsychol. 2000, 17, 63–74. [Google Scholar] [CrossRef]
- Vinck, A.; Verhagen, M.M.; Gerven, M.V.; de Groot, I.J.; Weemaes, C.M.; Maassen, B.A.; Willemsen, M.A. Cognitive and speech-language performance in children with ataxia telangiectasia. Dev. Neurorehabil. 2011, 14, 315–322. [Google Scholar] [CrossRef]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Robison, S.H.; Munzer, J.S.; Mrcp, R.T.; Bradley, W.G. Alzheimer’s disease cells exhibit defective repair of alkylating agent-induced DNA damage. Ann. Neurol. 1987, 21, 250–258. [Google Scholar] [CrossRef]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat. Res. 2007, 614, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef]





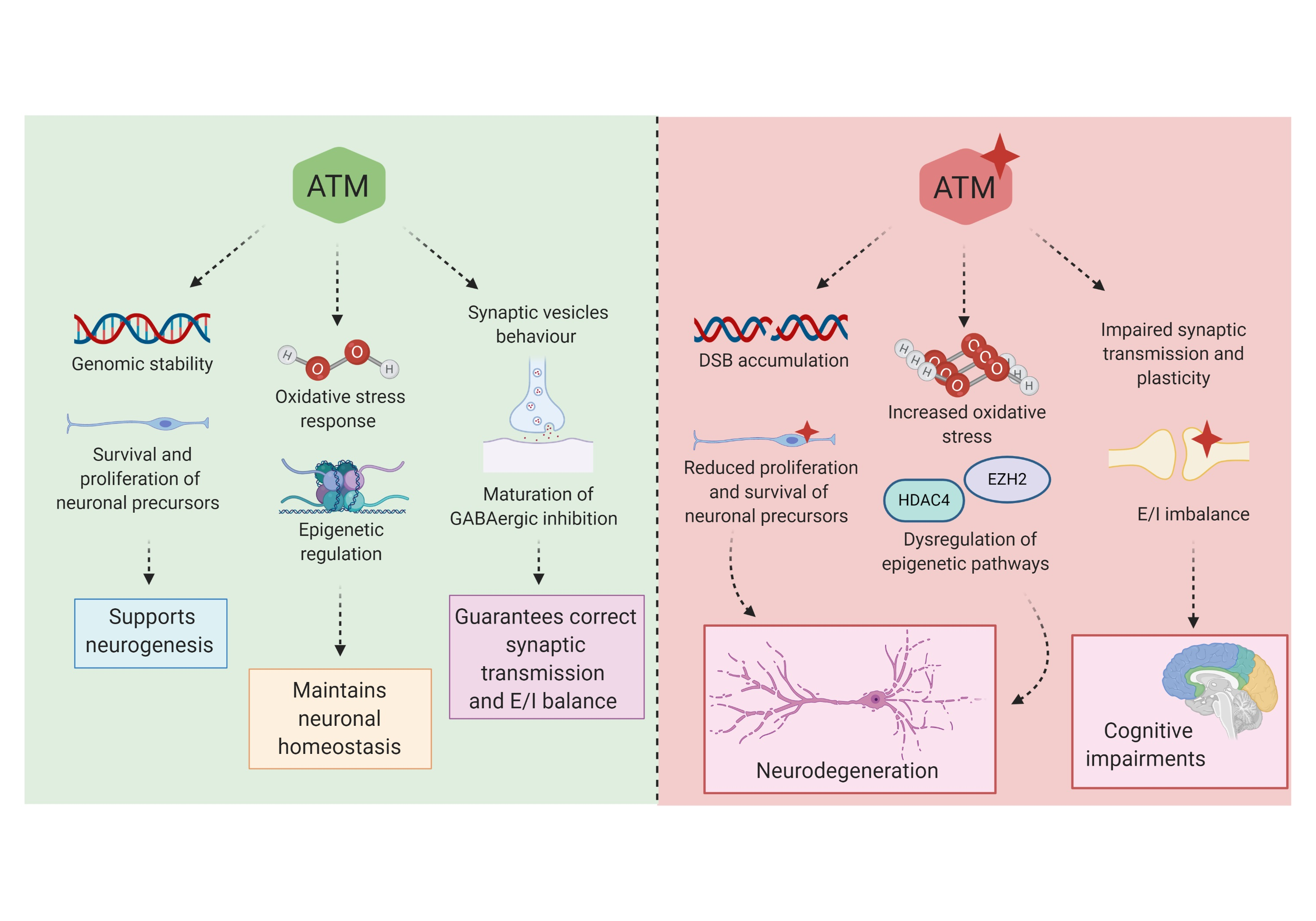{kind=link}
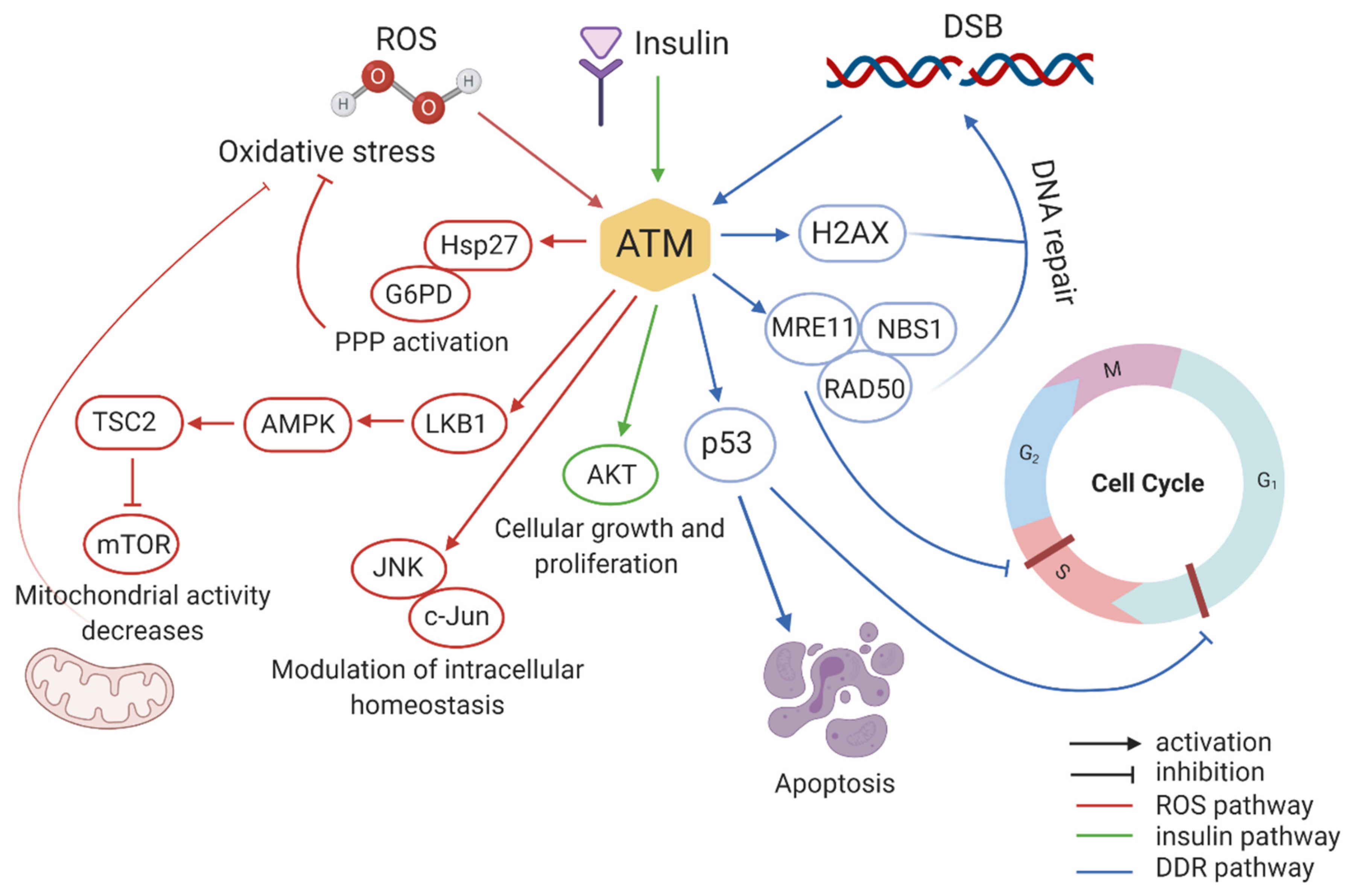{kind=link}
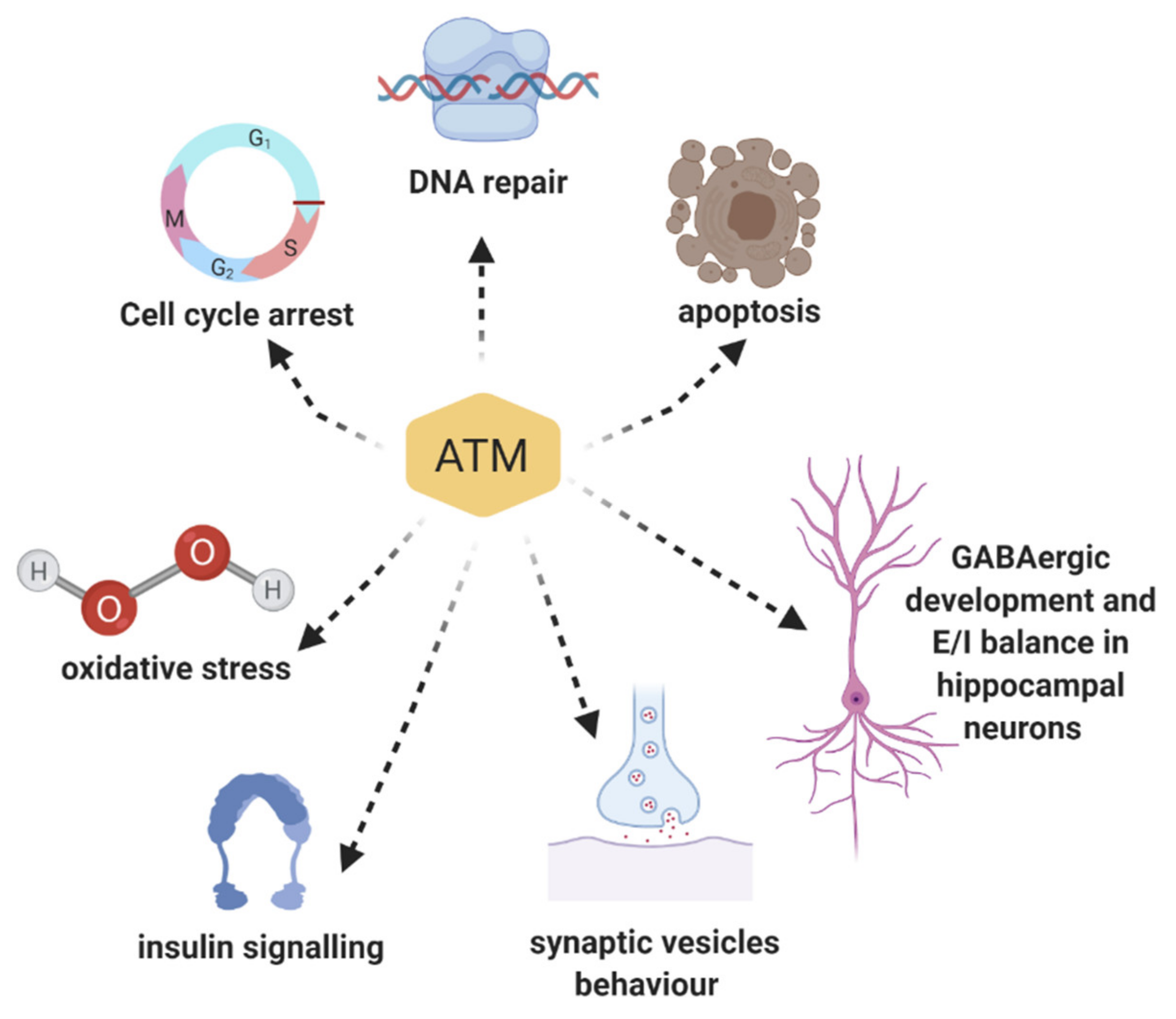{kind=link}
{kind=link}
{kind=link}
| References | Experimental Models | ATM Levels | Changes |
|---|---|---|---|
| Petersen et al. [151] | • Drosophila mutant for ATM | Low | • Neuron and glial cell death in the adult brain • High inflammation • Reduction in mobility and longevity |
| Shen et al. [152] | • R1.40, PS/APP, 3xTg mouse models of AD • Human AD brains | Low | • Nuclear translocation ofhistone deacetylase 4 • Trimethylation of histone H3 • The presence of cell cycle activity • Low ATM signaling in neurons in regions where degeneration is prevalent |
| Illuzzi et al. [153] Giuliano et al. [154] | • Cells transfected with mHTT | High (activation) | • Elevated DNA damage and oxidative stress |
| Lu et al. [155] | • BACHA mouse model of HD • Human HD brain | High | Reducing Atm ameliorates: • mHTT fragment toxicity in cells • spontaneous locomotion, motor coordination, depressive-like behaviours |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizzamiglio, L.; Focchi, E.; Antonucci, F. ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry. Cells 2020, 9, 1969. https://doi.org/10.3390/cells9091969
Pizzamiglio L, Focchi E, Antonucci F. ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry. Cells. 2020; 9(9):1969. https://doi.org/10.3390/cells9091969
Chicago/Turabian StylePizzamiglio, Lara, Elisa Focchi, and Flavia Antonucci. 2020. "ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry" Cells 9, no. 9: 1969. https://doi.org/10.3390/cells9091969
APA StylePizzamiglio, L., Focchi, E., & Antonucci, F. (2020). ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry. Cells, 9(9), 1969. https://doi.org/10.3390/cells9091969