TonEBP Promotes β-Cell Survival under ER Stress by Enhancing Autophagy

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Transfection
2.3. Cell Viability Assay
2.4. Immunofluorescence Staining
2.5. Immunoblotting
2.6. Immunoprecipitation
2.7. Mice
2.8. Statistical Analysis
3. Results
3.1. TonEBP Suppresses the ER Stress-Induced Accumulation of Unfolded Proteins and β-Cell Death
3.2. TonEBP Is Required for ER Stress-Induced Autophagosome Formation
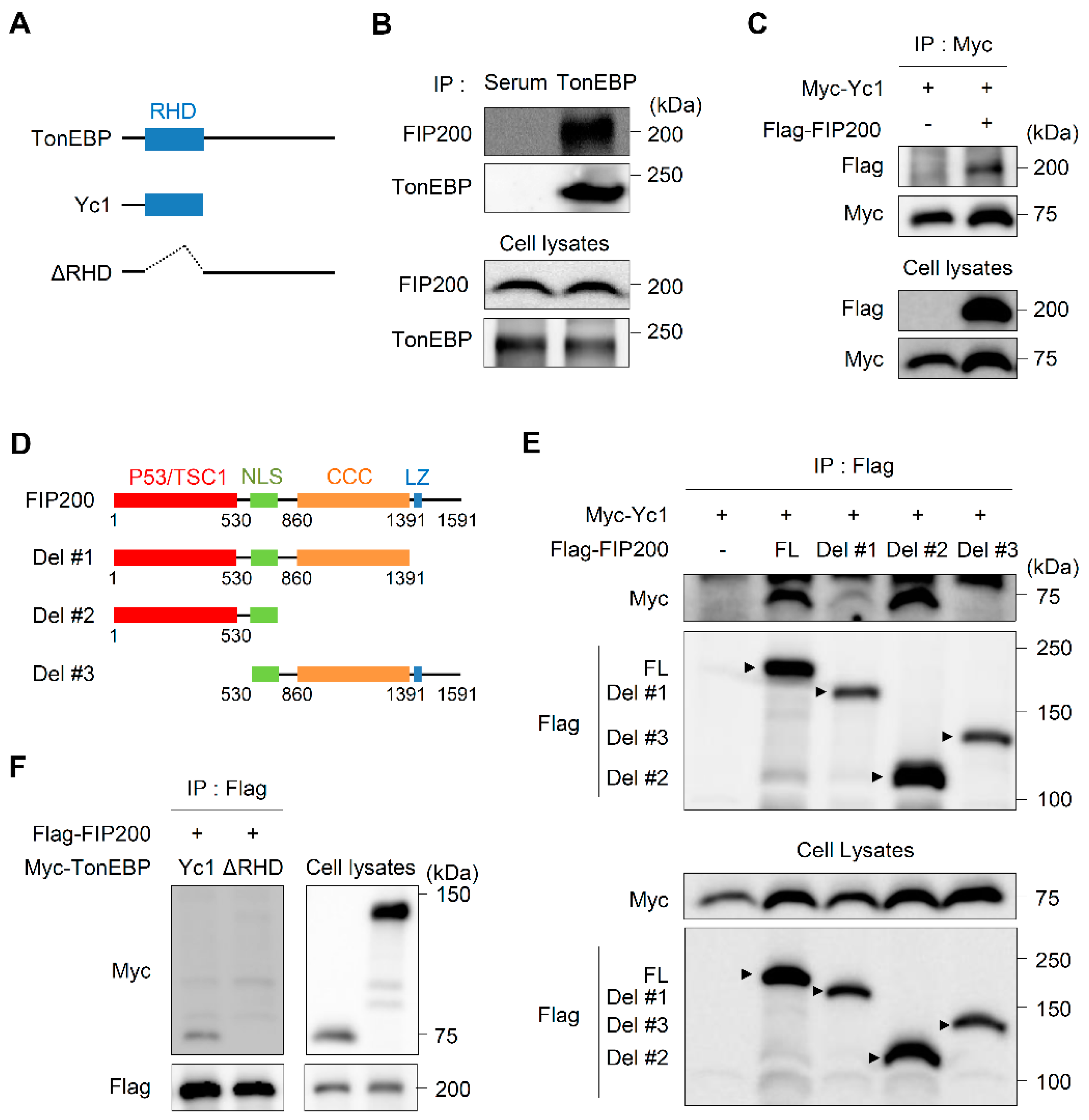
3.3. TonEBP Interacts with FIP200 through Its Rel-Homology Domain (RHD)
3.4. ER Stress Enhances the Stability of TonEBP Proteins
3.5. Deletion of TonEBP in Pancreatic Endocrine Progenitor Cells Perturbs Glucose Homeostasis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Schönthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef] [Green Version]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.S.; Lee, M.S. Macroautophagy in homeostasis of pancreatic beta-cell. Autophagy 2009, 5, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, H.; Woo, S.K.; Dahl, S.C.; Handler, J.S.; Kwon, H.M. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc. Natl. Acad. Sci. USA 1999, 96, 2538–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, W.Y.; Liu, X.; Roti, M.A.; Liu, F.; Ho, S.N. NFAT5/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proc. Natl. Acad. Sci. USA 2004, 101, 10673–10678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.D.; Choi, S.Y.; Lim, S.W.; Lamitina, S.T.; Ho, S.N.; Go, W.Y.; Kwon, H.M. TonEBP stimulates multiple cellular pathways for adaptation to hypertonic stress: Organic osmolyte-dependent and -independent pathways. Am. J. Physiol. Ren. Physiol. 2011, 300, F707–F715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.Y.; Lee-Kwon, W.; Kwon, H.M. The evolving role of TonEBP as an immunometabolic stress protein. Nat. Rev. Nephrol. 2020, 16, 352–364. [Google Scholar] [CrossRef]
- Choi, S.; You, S.; Kim, D.; Choi, S.Y.; Kwon, H.M.; Kim, H.S.; Hwang, D.; Park, Y.J.; Cho, C.S.; Kim, W.U. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J. Clin. Investig. 2017, 127, 954–969. [Google Scholar] [CrossRef]
- Ye, B.J.; Lee, H.H.; Yoo, E.J.; Lee, C.Y.; Lee, J.H.; Kang, H.J.; Jung, G.W.; Park, H.; Lee-Kwon, W.; Choi, S.Y.; et al. TonEBP in dendritic cells mediates pro-inflammatory maturation and Th1/Th17 responses. Cell Death Dis. 2020, 11, 421. [Google Scholar] [CrossRef]
- Halterman, J.A.; Kwon, H.M.; Leitinger, N.; Wamhoff, B.R. NFAT5 expression in bone marrow-derived cells enhances atherosclerosis and drives macrophage migration. Front. Physiol. 2012, 3, 313. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Suh, J.H.; Choi, S.Y.; Kang, H.J.; Lee, H.H.; Ye, B.J.; Lee, G.R.; Jung, S.W.; Kim, C.J.; Lee-Kwon, W.; et al. Tonicity-responsive enhancer-binding protein promotes hepatocellular carcinogenesis, recurrence and metastasis. Gut 2019, 68, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; An, S.M.; Ye, B.J.; Lee, J.H.; Yoo, E.J.; Jeong, G.W.; Kang, H.J.; Alfadda, A.A.; Lim, S.W.; Park, J.; et al. TonEBP/NFAT5 promotes obesity and insulin resistance by epigenetic suppression of white adipose tissue beiging. Nat. Commun. 2019, 10, 3536. [Google Scholar] [CrossRef] [PubMed]
- Serr, I.; Scherm, M.G.; Zahm, A.M.; Schug, J.; Flynn, V.K.; Hippich, M.; Kälin, S.; Becker, M.; Achenbach, P.; Nikolaev, A.; et al. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Rodríguez, C.; Antos, C.L.; Shelton, J.M.; Richardson, J.A.; Lin, F.; Novobrantseva, T.I.; Bronson, R.T.; Igarashi, P.; Rao, A.; Olson, E.N. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 2392–2397. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Peng, T.; Sands, J.M.; Bagnasco, S.M. The TonE/TonEBP pathway mediates tonicity-responsive regulation of UT-A urea transporter expression. J. Biol. Chem. 2000, 275, 38275–38280. [Google Scholar] [CrossRef] [Green Version]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.K.; Beck, F.X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Jantsch, J.; Schatz, V.; Friedrich, D.; Schröder, A.; Kopp, C.; Siegert, I.; Maronna, A.; Wendelborn, D.; Linz, P.; Binger, K.J.; et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015, 21, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Buxadé, M.; Huerga Encabo, H.; Riera-Borrull, M.; Quintana-Gallardo, L.; López-Cotarelo, P.; Tellechea, M.; Martínez-Martínez, S.; Redondo, J.M.; Martín-Caballero, J.; Flores, J.M.; et al. Macrophage-specific MHCII expression is regulated by a remote Ciita enhancer controlled by NFAT5. J. Exp. Med. 2018, 215, 2901–2918. [Google Scholar] [CrossRef] [Green Version]
- Yoo, E.J.; Lee, H.H.; Ye, B.J.; Lee, J.H.; Lee, C.Y.; Kang, H.J.; Jeong, G.W.; Park, H.; Lim, S.W.; Lee-Kwon, W.; et al. TonEBP Suppresses the HO-1 Gene by Blocking Recruitment of Nrf2 to Its Promoter. Front. Immunol. 2019, 10, 850. [Google Scholar] [CrossRef]
- Buxadé, M.; Lunazzi, G.; Minguillón, J.; Iborra, S.; Berga-Bolaños, R.; Del Val, M.; Aramburu, J.; López-Rodríguez, C. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J. Exp. Med. 2012, 209, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Park, H.; Yoo, E.J.; Lee, J.H.; Choi, S.Y.; Lee-Kwon, W.; Lee, K.Y.; Hur, J.H.; Seo, J.K.; Ra, J.S.; et al. TonEBP Regulates PCNA Polyubiquitination in Response to DNA Damage through Interaction with SHPRH and USP1. Iscience 2019, 19, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Küper, C.; Beck, F.X.; Neuhofer, W. Generation of a conditional knockout allele for the NFAT5 gene in mice. Front. Physiol. 2014, 5, 507. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berke, S.J.; Paulson, H.L. Protein aggregation and the ubiquitin proteasome pathway: Gaining the UPPer hand on neurodegeneration. Curr. Opin. Genet. Dev. 2003, 13, 253–261. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy 2013, 9, 626–628. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. Cmls 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Aggrephagy: Selective Disposal of Protein Aggregates by Macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy: A potential link between obesity and insulin resistance. Cell Metab. 2010, 11, 449–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Virgin, H.W.; Levine, B. Autophagy genes in immunity. Nat. Immunol. 2009, 10, 461–470. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.Y.; Ryan, K.M. Autophagy and cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Chen, L. Endoplasmic Reticulum Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 167–177. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wang, C.; Yeo, S.; Liang, C.C.; Okamoto, T.; Sun, S.; Wen, J.; Guan, J.L. Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 2016, 30, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melkoumian, Z.K.; Peng, X.; Gan, B.; Wu, X.; Guan, J.L. Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res. 2005, 65, 6676–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [Green Version]
- Abbi, S.; Ueda, H.; Zheng, C.; Cooper, L.A.; Zhao, J.; Christopher, R.; Guan, J.L. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell 2002, 13, 3178–3191. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Abbi, S.; Zheng, C.; Guan, J.L. Suppression of Pyk2 kinase and cellular activities by FIP200. J. Cell Biol. 2000, 149, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Gan, B.; Guan, J.L. FIP200, a key signaling node to coordinately regulate various cellular processes. Cell. Signal. 2008, 20, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.; Schwamborn, K.; Urlaub, H.; Gan, B.; Guan, J.L.; Dejean, A. Spatial interplay between PIASy and FIP200 in the regulation of signal transduction and transcriptional activity. Mol. Cell. Biol. 2008, 28, 2771–2781. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Lim, S.W.; Salimi, S.; Yoo, E.J.; Lee-Kwon, W.; Lee, H.H.; Lee, J.H.; Mitchell, B.D.; Sanada, S.; Parsa, A.; et al. Tonicity-Responsive Enhancer-Binding Protein Mediates Hyperglycemia-Induced Inflammation and Vascular and Renal Injury. J. Am. Soc. Nephrol. JASN 2018, 29, 492–504. [Google Scholar] [CrossRef] [Green Version]
- In’t Veld, P.; Marichal, M. Microscopic anatomy of the human islet of Langerhans. In The Islets of Langerhans; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2010; Volume 654, pp. 1–19. [Google Scholar] [CrossRef]
- Rojas, J.; Bermudez, V.; Palmar, J.; Martínez, M.S.; Olivar, L.C.; Nava, M.; Tomey, D.; Rojas, M.; Salazar, J.; Garicano, C.; et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J. Diabetes Res. 2018, 2018, 9601801. [Google Scholar] [CrossRef] [Green Version]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Jiménez, A.; Peón, A.N.; Terrazas, L.I. Alternatively activated macrophages in types 1 and 2 diabetes. Mediat. Inflamm. 2012, 2012, 815953. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Sanada, S.; An, S.M.; Ye, B.J.; Lee, J.H.; Seo, Y.K.; Lee, C.; Lee-Kwon, W.; Küper, C.; Neuhofer, W.; et al. LPS-induced NFκB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci. Rep. 2016, 6, 24921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.Y.; Lee, H.H.; Lee, J.H.; Ye, B.J.; Yoo, E.J.; Kang, H.J.; Jung, G.W.; An, S.M.; Lee-Kwon, W.; Chiong, M.; et al. TonEBP suppresses IL-10-mediated immunomodulation. Sci. Rep. 2016, 6, 25726. [Google Scholar] [CrossRef] [Green Version]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.J.; Yoo, E.J.; Lee, H.H.; An, S.M.; Park, H.; Lee-Kwon, W.; Choi, S.Y.; Kwon, H.M. TonEBP Promotes β-Cell Survival under ER Stress by Enhancing Autophagy. Cells 2020, 9, 1928. https://doi.org/10.3390/cells9091928
Kang HJ, Yoo EJ, Lee HH, An SM, Park H, Lee-Kwon W, Choi SY, Kwon HM. TonEBP Promotes β-Cell Survival under ER Stress by Enhancing Autophagy. Cells. 2020; 9(9):1928. https://doi.org/10.3390/cells9091928
Chicago/Turabian StyleKang, Hyun Je, Eun Jin Yoo, Hwan Hee Lee, Seung Min An, Hyun Park, Whaseon Lee-Kwon, Soo Youn Choi, and Hyug Moo Kwon. 2020. "TonEBP Promotes β-Cell Survival under ER Stress by Enhancing Autophagy" Cells 9, no. 9: 1928. https://doi.org/10.3390/cells9091928