Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA Data Analysis
2.2. Cell Cultures
2.3. Cell Morphology Analysis
2.4. RT-qPCR
2.5. Western Blot
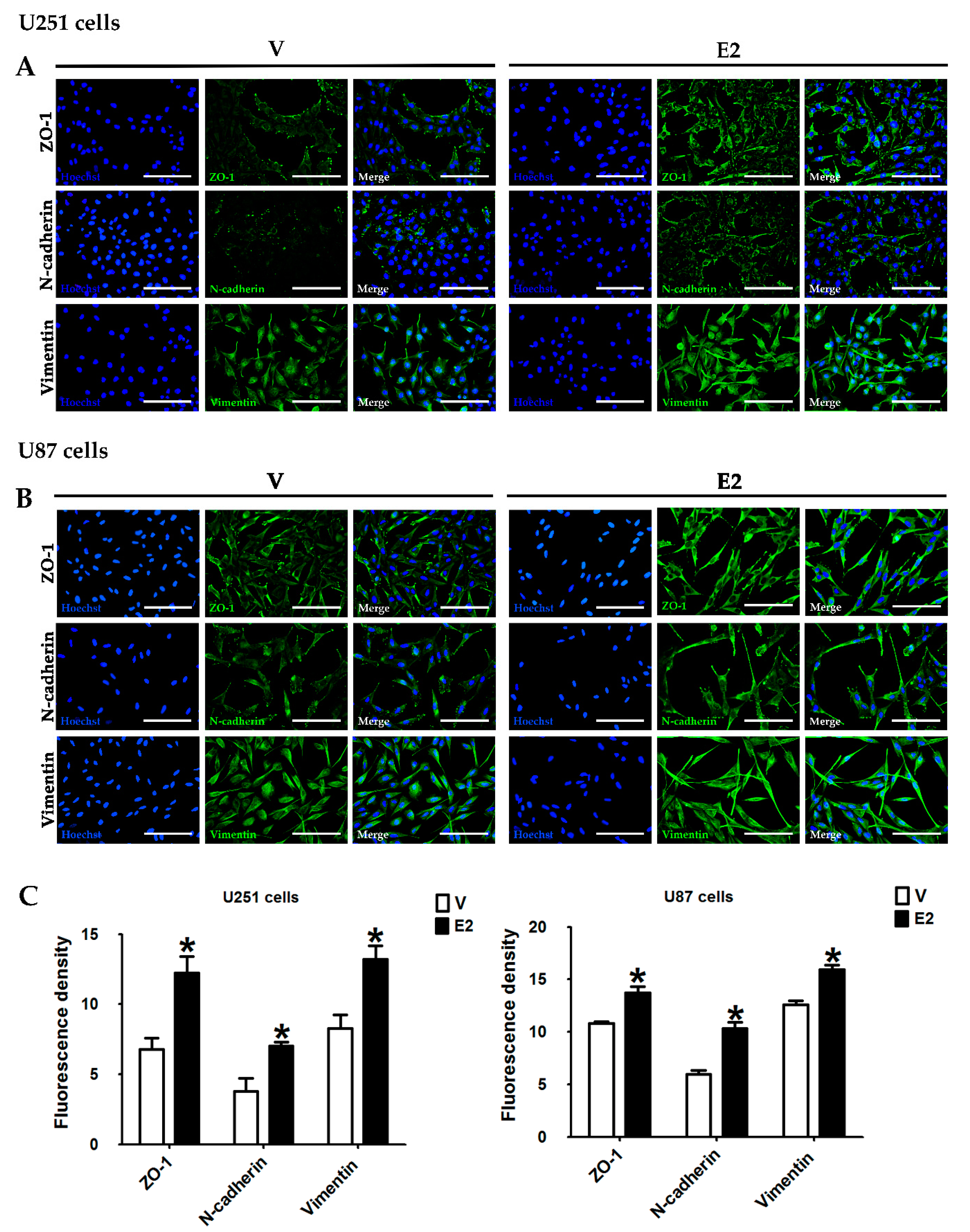
2.6. Immunofluorescence
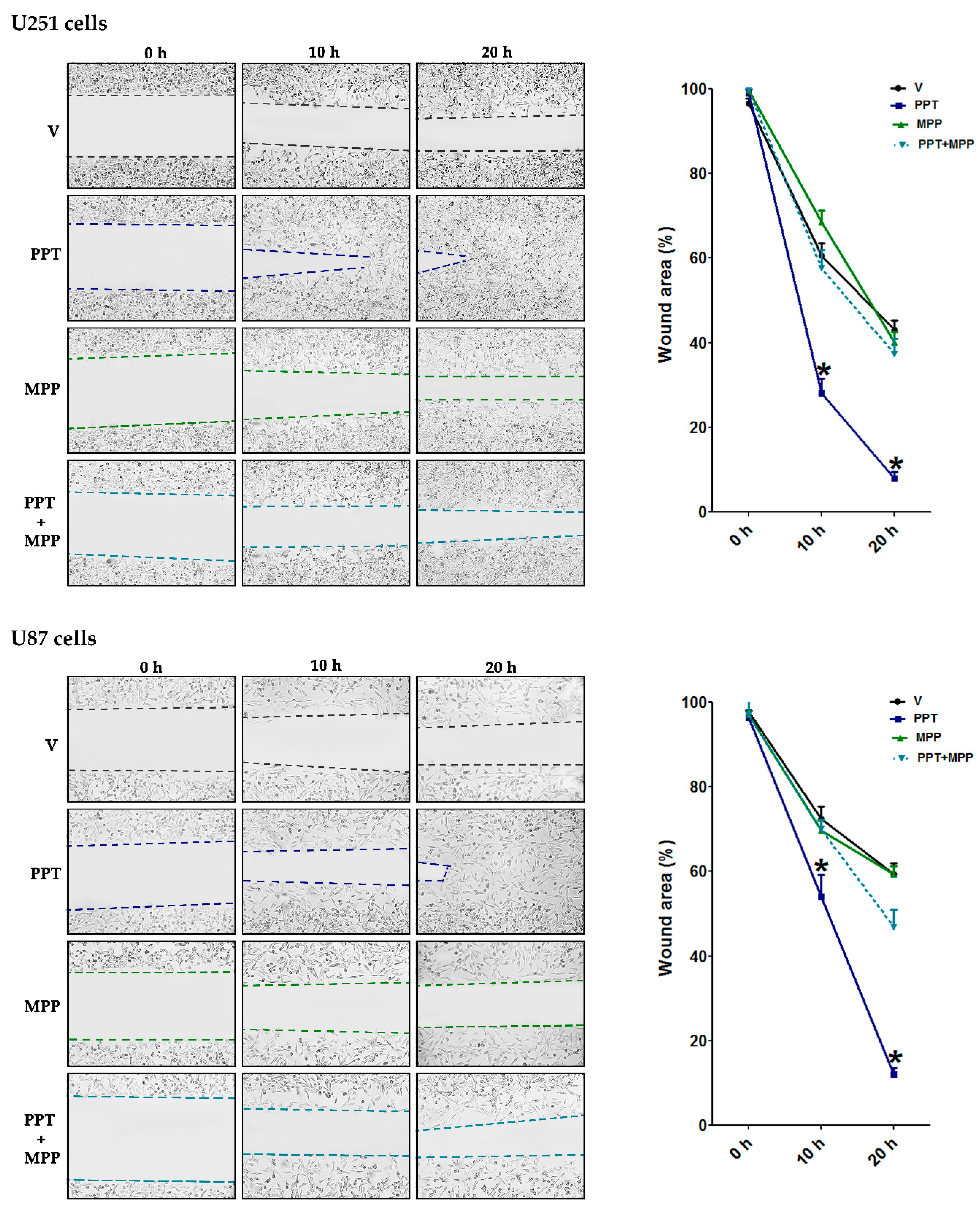
2.7. Migration Assay
2.8. Invasion Assay
2.9. Statistical Analysis
3. Results
3.1. Differential Expression of ERα and ERβ Subtypes in Human GBM-Derived Cells
3.2. Changes in Cell Morphology During E2-Induced EMT
3.3. E2-Induced Reorganization of Actin Filaments
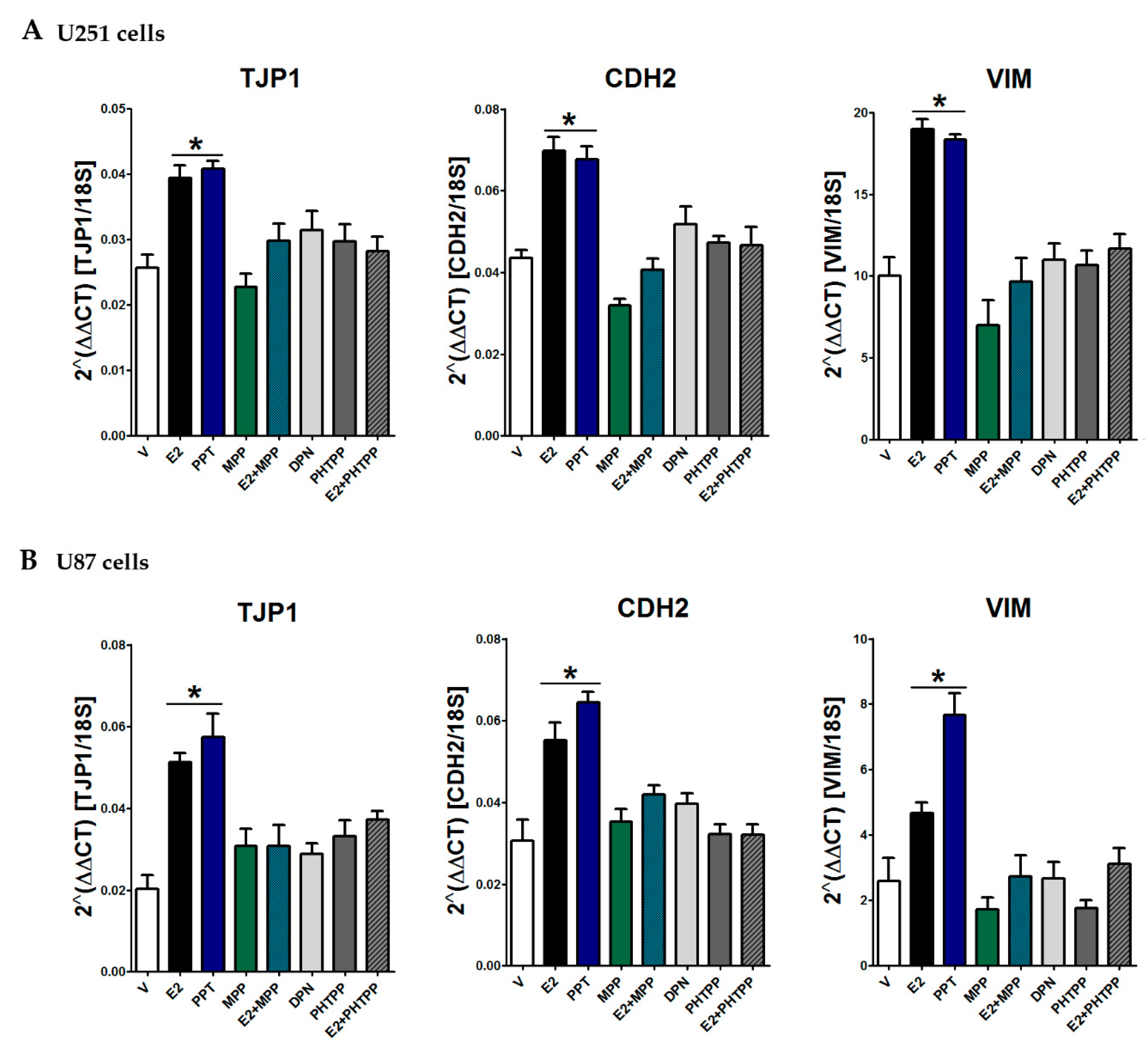
3.4. E2 Regulated EMT Marker Expression
3.5. E2 Promoted Migration and Invasion of Human GBM-Derived Cells
3.6. ER-α Mediated E2 Effects on EMT
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-κB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Thiery, J.P.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Trelstad, R.L.; Hay, E.D.; Revel, J.D. Cell contact during early morphogenesis in the chick embryo. Dev. Biol. 1967, 16, 78–106. [Google Scholar] [CrossRef]
- Nieto, M.A.; Sargent, M.; Wilkinson, D.; Cooke, J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 1994, 6, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A Dynamic Epicardial Injury Response Supports Progenitor Cell Activity during Zebrafish Heart Regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in non-epithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Nikkhah, G.; Maciaczyk, J. Epithelial-to-mesenchymal (-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett. 2013, 331, 131–138. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene Expression Profiling of Gliomas Strongly Predicts Survival. Cancer Res. 2004, 15, 6503–6510. [Google Scholar] [CrossRef] [PubMed]
- Tso, C.L.; Shintaku, P.; Chen, J.; Liu, Q.; Liu, J.; Chen, Z.; Yoshimoto, K.; Mischel, P.S.; Cloughesy, T.F.; Liau, L.M.; et al. Primary Glioblastomas Express Mesenchymal Stem-Like Properties. Mol. Cancer Res. 2006, 4, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Zarkoob, H.; Taube, J.H.; Singh, S.K.; Mani, S.A.; Kohandel, M. Investigating the Link between Molecular Subtypes of Glioblastoma, Epithelial-Mesenchymal Transition, and CD133 Cell Surface Protein. PLoS ONE 2013, 8, e64169. [Google Scholar] [CrossRef] [PubMed]
- Azcoitia, I.; Yague, J.G.; Garcia-Segura, L.M. Estradiol synthesis within the human brain. Neuroscience 2011, 191, 139–147. [Google Scholar] [CrossRef]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.M.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef]
- Toran-allerand, C.D. Sex steroids and the development of the newborn mouse hypothalamus and preoptic area In Vitro: Implications for sexual differentiation. Brain Res. 1976, 106, 407–412. [Google Scholar] [CrossRef]
- Díaz, N.F.; Guerra-Arraiza, C.; Díaz-Martínez, N.E.; Salazar, P.; Molina-Hernández, A.; Camacho-Arroyo, I.; Velasco, I. Changes in the content of estrogen α and progesterone receptors during differentiation of mouse embryonic stem cells to dopamine neurons. Brain Res. Bull. 2007, 73, 75–80. [Google Scholar] [CrossRef]
- Denley, M.C.S.; Gatford, N.J.F.; Sellers, K.J.; Srivastava, D.P. Estradiol and the development of the cerebral cortex: An unexpected role? Front. Neurosci. 2018, 12, 245. [Google Scholar] [CrossRef]
- Díaz, H.; Lorenzo, A.; Carrer, H.F.; Cáceres, A. Time lapse study of neurite growth in hypothalamic dissociated neurons in culture: Sex differences and estrogen effects. J. Neurosci. Res. 1992, 33, 266–281. [Google Scholar] [CrossRef]
- Pérez, J.; Naftolin, F.; García-Segura, L.M. Sexual differentiation of synaptic connectivity and neuronal plasma membrane in the arcuate nucleus of the rat hypothalamus. Brain Res. 1990, 527, 116–122. [Google Scholar] [CrossRef]
- Sugiyama, N.; Andersson, S.; Lathe, R.; Fan, X.; Schwend, T.; Nalvarte, I. Spatiotemporal dynamics of the expression of estrogen receptors in the postnatal mouse brain. Mol. Psychiatry 2009, 14, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Barth, C.; Villringer, A.; Sacher, J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front. Neurosci. 2015, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Hansberg-Pastor, V.; González-Arenas, A.; Piña-Medina, A.G.; Camacho-Arroyo, I. Sex hormones regulate cytoskeletal proteins involved in brain plasticity. Front. Psychiatry 2015, 6, 165. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors α and β. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. The role of estrogen in the initiation of breast cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 89–96. [Google Scholar] [CrossRef]
- Cunat, S.; Hoffmann, P.; Pujol, P. Estrogens and epithelial ovarian cancer. Gynecol. Oncol. 2004, 94, 25–32. [Google Scholar] [CrossRef]
- Kumar, M.M.; Davuluri, S.; Poojar, S.; Mukherjee, G.; Bajpai, A.K.; Bafna, U.D.; Devi, U.K.; Kallur, P.P.R.; Kshitish, A.K.; Jayshree, R.S. Role of estrogen receptor alpha in human cervical cancer-associated fibroblasts: A transcriptomic study. Tumor Biol. 2016, 37, 4409–4420. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di-Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors α and β in cancer. Crit. Rev. Oncol. Hematol. 2004, 50, 3–22. [Google Scholar] [CrossRef]
- Vaquero, J.; Marcos, M.L.; Martínez, R.; Bravo, G. Estrogen- and progesterone-receptor proteins in intracranial tumors. Surg. Neurol. 1983, 19, 11–13. [Google Scholar] [CrossRef]
- Carroll, R.S.; Zhang, J.; Dashner, K.; Sar, M.; Black, P.M. Steroid Hormone Receptors in Astrocytic Neoplasms. Neurosurgery 1995, 37, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Khalid, H.; Yasunaga, A.; Kishikawa, M.; Shibata, S. Immunohistochemical expression of the estrogen receptor-related antigen (ER-D5) in human intracranial tumors. Cancer 1995, 75, 2571–2578. [Google Scholar] [CrossRef]
- Batistatou, A.; Stefanou, D.; Goussia, A.; Arkoumani, E.; Papavassiliou, A.; Agnantis, N. Estrogen receptor beta (ERβ) is expressed in brain astrocytic tumors and declines with dedifferentiation of the neoplasm. J. Cancer Res. Clin. Oncol. 2004, 130, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Nair, B.C.; Gonugunta, V.K.; Zhang, Q.g.; Brenner, A.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. Therapeutic Significance of Estrogen Receptor Agonists in Gliomas. Mol. Cancer 2012, 11, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- González-Arenas, A.; Hansberg-Pastor, V.; Hernández-Hernández, O.T.; González-García, T.K.; Henderson-Villalpando, J.; Lemus-Hernández, D.; Cruz-Barrios, A.; Rivas-Suárez, M.; Camacho-Arroyo, I. Estradiol increases cell growth in human astrocytoma cell lines through ERa activation and its interaction with SRC-1 and SRC-3 coactivators. Biochim. Biophys. Acta 2012, 1823, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Winters, A.; Poteet, E.; Ryou, M.G.; Lin, S.; Hao, S.; Wu, Z.; Yuan, F.; Hatanpaa, K.J.; Simpkins, J.W.; et al. Involvement of estrogen receptor β5 in the progression of glioma. Brain Res. 2013, 1503, 97–107. [Google Scholar] [CrossRef]
- Dueñas, J.M.; Candanedo, A.; Santerre, A.; Orozco, S.; Sandoval, H.; Feria, I.; López-Elizalde, R.; Venegas, M.A.; Netel, B.; de la Torre-Valdovinos, B.; et al. Aromatase and estrogen receptor alpha mRNA expression as prognostic biomarkers in patients with astrocytomas. J. Neurooncol. 2014, 119, 275–284. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Zhang, K.; Bian, C.; Zhao, Y.; Zhang, J. Expression of estrogen receptors, androgen receptor and steroid receptor coactivator-3 is negatively correlated to the differentiation of astrocytic tumors. Cancer Epidemiol. 2014, 38, 291–297. [Google Scholar] [CrossRef]
- Tavares, C.B.; Gomes-Braga, F.; Sousa, E.B.; Borges, U.S.; Escórcio-Dourado, C.S.; Da Silva-Sampaio, J.P.; Borges da Silva, B. Evaluation of estrogen receptor expression in low-grade and high-grade astrocytomas. Rev. Assoc. Med. Bras. 2018, 64, 1129–1133. [Google Scholar] [CrossRef]
- Wan, S.; Jiang, J.; Zheng, C.; Wang, N.; Zhai, X.; Fei, X.; Wu, R.; Jiang, X. Estrogen nuclear receptors affect cell migration by altering sublocalization of AQP2 in glioma cell lines. Cell Death Discov. 2018, 4, 49. [Google Scholar] [CrossRef]
- Park, S.H.; Cheung, L.W.T.; Wong, A.S.T.; Leung, P.C.K. Estrogen Regulates Snail and Slug in the Down-Regulation of E-Cadherin and Induces Metastatic Potential of Ovarian Cancer Cells through Estrogen Receptor α. Mol. Endocrinol. 2008, 22, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Tai, Q.; Gu, X.; Schmitz, J.; Poullard, A.; Fajardo, R.J.; Mahalingam, D.; Chen, X.; Zhu, X.; Sun, L.Z. Estrogen and Estrogen Receptor Alpha Promotes Malignancy and Osteoblastic Tumorigenesis in Prostate Cancer. Oncotarget 2015, 6, 44388–44402. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Peng, Y.; Du, X.; Liu, H.; Klocker, H.; Lin, Q.; Shi, J.; Zhang, J. Estradiol promotes epithelial-to-mesenchymal transition in human benign prostatic epithelial cells. Prostate 2017, 77, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Kajita, M.; Wade, P.A. The Transcription Factor Snail Mediates Epithelial to Mesenchymal Transitions by Repression of Estrogen Receptor-α. Mol. Endocrinol. 2007, 21, 2907–2918. [Google Scholar] [CrossRef]
- Al Saleh, S.; Al Mulla, F.; Luqmani, Y.A. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS ONE 2011, 6, e20610. [Google Scholar] [CrossRef]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen receptor alpha mediates epithelial to mesenchymal transition, expression of specific matrix effectors and functional properties of breast cancer cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Andreeva, O.E.; Shatskaya, V.A.; Krasil’Nikov, M.A. The relationships between snail1 and estrogen receptor signaling in breast cancer cells. J. Cell Biochem. 2012, 113, 2147–2155. [Google Scholar] [CrossRef]
- Wik, E.; Ræder, M.B.; Krakstad, C.; Trovik, J.; Birkeland, E.; Hoivik, E.A.; Mjos, S.; Werner, H.M.J.; Mannelqvist, M.; Stefansson, I.M.; et al. Lack of estrogen receptor-α is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin. Cancer Res. 2013, 19, 1094–1105. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef]
- Bakal, C.; Church, G.; Perrimon, N. Quantitative Morphological Signatures Define Local Signaling Networks Regulating Cell Morphology. Science 2007, 316, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, M.; Wu, G.; Yao, C.; Zhang, J. Recent Advances in Morphological Cell Image Analysis. Comput. Math. Methods Med. 2012, 2012, 101536. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.X.; Yu, H.B.; Li, J.S.; Shen, J.L.; Du, W.S. Suitable parameter choice on quantitative morphology of A549 cell in epithelial—mesenchymal transition. Biosci. Rep. 2015, 35, e00202. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Lin, N.; Yan, W.; Gao, K.; Wang, Y.; Zhang, J.; You, Y. Prevalence and clinicopathologic characteristics of the molecular subtypes in malignant glioma: A multi-institutional analysis of 941 cases. PLoS ONE 2014, 9, e94871. [Google Scholar] [CrossRef]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef]
- Ozawa, T.; Riester, M.; Cheng, Y.; Huse, J.T.; Squatrito., M.; Helmy., K.; Charles, N.; Michor, F.; Holland, E.C. Most Human Non-GCIMP Glioblastoma Subtypes Evolve from a Common Proneural-like Precursor Glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-mesenchymal transition: Phenotypic plasticity to acquire multitherapy resistance in glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Stanzani, E.; Martínez-Soler, F.; Mateos, T.M.; Vidal, N.; Villanueva, A.; Pujana, M.A.; Serra-Musach, J.; Iglesia, N.; Giménez-Bonafé, P.; Tortosa, A. Radioresistance of mesenchymal glioblastoma initiating cells correlates with patient outcome and is associated with activation of inflammatory program. Oncotarget 2017, 8, 73640–73653. [Google Scholar] [CrossRef]
- Kaffes, I.; Szulzewsky, F.; Chen, Z.; Herting, C.J.; Gabanic, B.; Velazquez, J.E.; Shelton, J.; Switchenko, J.M.; Ross, J.L.; McSwain, L.F.; et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors presence compared to Proneural and Classical tumors. Oncoimmunology 2019, 8, e1655360. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef]
- Baker, A.E.; Brautigam, V.M.; Watters, J.J. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor β. Endocrinology 2004, 145, 5021–5032. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Sanchez-Gonzalez, R.; Azcoitia, I.; Arevalo, M.A.; Garcia-Segura, L.M. Role of Astrocytes in the Neuroprotective Actions of 17β-estradiol and Selective Estrogen Receptor Modulators. Mol. Cell Endocrinol. 2014, 389, 48–57. [Google Scholar] [CrossRef]
- Pinacho-Garcia, L.M.; Valdez, R.A.; Navarrete, A.; Cabeza, M.; Segovia, J.; Romano, M.C. The Effect of Finasteride and Dutasteride on the Synthesis of Neurosteroids by Glioblastoma Cells. Steroids 2020, 155, 108556. [Google Scholar] [CrossRef] [PubMed]
- Reijm, E.; Jansen, M.; Ruigrok-Ritstier, K.; van Staveren, I.L.; Look, M.; van Gelder, M.E.M.; Sieuwerts, A.M.; Sleijfer, S.; Foekens, J.A.; Berns, E.M.J.J. Decreased expression of EZH2 is associated with upregulation of ER and favorable outcome to tamoxifen in advanced breast cancer. Breast Cancer Res Treat 2011, 125, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Weaver, A.; Pradhan, M.; Dai, Y.; Miller, L.D.; Lin, C.Y.; Stanculescu, A. Positive cross-talk between estrogen receptor and NF-κB in breast cancer. Cancer Res. 2009, 14, 8918–8925. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; El-Shennawy, L.; Stender, J.; Kastrati, I. NFκB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Mol. Cell Endocrinol. 2015, 418, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Hongying, Z.; Qingbin, K.; Yangfu, J. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.; Mayne, C.; Katzenellenbogen, B.; Greene, G.; Chandarlapaty, S. Structural Underpinnings of Estrogen Receptor Mutations in Endocrine Therapy Resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- Christoforos, T.; Gustafsson, J. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol Metab 2015, 26, 467–476. [Google Scholar] [CrossRef]
- Thompson, S.; Petti, F.; Sujka-Kwok, I.; Mercado, P.; Bean, J.; Monaghan, M.; Seymour, S.L.; Argast, G.M.; Epstein, D.M.; Haley, J.D. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis 2011, 28, 137–155. [Google Scholar] [CrossRef]
- D´Souza, R.; Knitte, A.; Nagaraj, N.; van Dinther, M.; Choudhary, C.; Dijke, P.; Mann, M.; Sharma, K. Time-resolved dissection of early phosphoproteome and ensuing proteome changes in response to TGF-β. Sci Signal 2014, 7, 5. [Google Scholar] [CrossRef]
- Rojas-Puente, L.; Cardona, A.; Carranza, H.; Vargas, C.; Jaramillo, L.; Zea, D.; Cetina, L.; Wills, B.; Ruiz-Garcia, E.; Arrieta, O. Epithelial-mesenchymal transition, proliferation, and angiogenesis in locally advanced cervical cancer treated with chemoradiotherapy. Cancer Med. 2016, 5, 1989–1999. [Google Scholar] [CrossRef]
- Qun-Ying, L.; Zhang, H.; Zhao, B.; Zheng-Yu, Z.; Bai, F.; Xin-Hai, P.; Zhao, S.; Xiong, Y.; Kun-Liang, G. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef]
- Castruccio, C.; Longhitano, L.; Distefano, A.; Anfuso, D.; Kalampoka, S.; Spina, E.L.; Astuto, M.; Avola, R.; Caruso, M.; Nicolosi, D.; et al. Role of 17β-Estradiol on Cell Proliferation and Mitochondrial Fitness in Glioblastoma Cells. J. Oncol. 2020, 2020, 2314693. [Google Scholar] [CrossRef]
- DePasquale, J.A. Rearrangement of the F-actin cytoskeleton in estradiol-treated MCF-7 breast carcinoma cells. Histochem. Cell Biol. 1999, 112, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Briz, V.; Baudry, M. Estrogen regulates protein synthesis and actin polymerization in hippocampal neurons through different molecular mechanisms. Front. Endocrinol 2014, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, J.; Malbouyres., M.; De Mets, R.; Balland, M.; Beauchef, G.; Vié, K.; Chamot, C.; Lionnet, C.; Ruggiero, F.; Vanacker, J.M. Estrogens Induce Rapid Cytoskeleton Re-Organization in Human Dermal Fibroblasts via the Non-Classical Receptor GPR30. PLoS ONE 2015, 10, e0120672. [Google Scholar] [CrossRef]
- Flamini, M.I.; Sanchez, A.M.; Goglia, L.; Tosi, V.; Genazzani, A.R.; Simoncini, T. Differential actions of estrogen and SERMs in regulation of the actin cytoskeleton of endometrial cells. Mol. Hum. Reprod. 2009, 15, 675–685. [Google Scholar] [CrossRef]
- O’Neill, G.M. The coordination between actin filaments and adhesion in mesenchymal migration. Cell Adhes. Migr. 2009, 3, 355–357. [Google Scholar] [CrossRef]
- Hotulainen, P.; Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Ballestrem, C.; Kam, Z.; Geiger, B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 2003, 116, 4605–4613. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Martínez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef]
- Ebnet, K.; Schulz, C.U.; Meyer Zu Brickwedde, M.K.; Pendl, G.G.; Vestweber, D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J. Biol. Chem. 2000, 275, 27979–27988. [Google Scholar] [CrossRef]
- McNeil, E.; Capaldo, C.T.; Macara, I.G. Zonula Occludens-1 Function in the Assembly of Tight Junctions in Madin-Darby Canine Kidney Epithelial Cells. Mol. Biol. Cell 2006, 17, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Anderson, J.M. Zonula Occludens-1 and -2 Are Cytosolic Scaffolds That Regulate the Assembly of Cellular Junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef]
- Patil, V.; Pal, J.; Somasundaram, K. Elucidating the cancer-specific genetic alteration spectrum of glioblastoma derived cell lines from whole exome and RNA sequencing. Oncotarget 2015, 6, 43452–43471. [Google Scholar] [CrossRef]
- Li, H.; Lei, B.; Xiang, W.; Wang, H.; Feng, W.; Liu, Y.; Qi, S. Differences in protein expression between the U251 and U87 cell lines. Turk. Neurosurg. 2017, 27, 894–903. [Google Scholar] [CrossRef]
- Derycke, L.D.M.; Bracke, M.E. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 2004, 48, 463–476. [Google Scholar] [CrossRef]
- Shih, W.; Yamada, S. N-cadherin-mediated cell-cell adhesion promotes cell migration in a three-dimensional matrix. J. Cell Sci. 2012, 125, 3661–3670. [Google Scholar] [CrossRef]
- Shih, W.; Yamada, S. N-cadherin as a key regulator of collective cell migration in a 3D environment. Cell Adhes. Migr. 2012, 6, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, T.; Mitsushima, A.; Kurimura, T. The role of the vimentin intermediate filaments in rat 3Y1 cells elucidated by immunoelectron microscopy and computer-graphic reconstruction. Biol. Cell 1990, 68, 139–146. [Google Scholar] [CrossRef]
- Chang, L.; Goldman, R.D. Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 2004, 5, 601–613. [Google Scholar] [CrossRef]
- Goldman, R.D.; Khuon, S.; Chou, Y.H.; Opal, P.; Steinert, P.M. The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell Biol. 1996, 134, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, S.R.; Coletta, C.J.; Tedesco, R.; Nishiguchi, G.; Carlson, K.; Sun, J.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Pyrazole ligands: Structure-Affinity/activity relationships and estrogen receptor-α-selective agonists. J. Med. Chem. 2000, 43, 4934–4947. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Huang, Y.R.; Harrington, W.R.; Sheng, S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Antagonists selective for estrogen receptor α. Endocrinology 2002, 143, 941–947. [Google Scholar] [CrossRef]
- Meyers, M.J.; Sun, J.; Carlson, K.E.; Marriner, G.A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor-β potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001, 44, 4230–4251. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Image |
|---|---|---|
| Area | The area included in the polygon that defines the figure contour |  |
| Axis major | Major axis length of an imaginary ellipse surrounding figure |  |
| Axis minor | Minor axis length of an imaginary ellipse surrounding figure |  |
| Aspect | The ratio between the major and minor axis of an ellipse |  |
| Bound box height | Bounding box height of the figure |  |
| Bound box width | Bounding box width of the figure |  |
| Box XY | The ratio of width to height of bounding box |  |
| Circularity | The ratio of figure area to the diameter of a circle around it |  |
| Perimeter | Length of the region surrounding the figure |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Vega, A.M.; Del Moral-Morales, A.; Zamora-Sánchez, C.J.; Piña-Medina, A.G.; González-Arenas, A.; Camacho-Arroyo, I. Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells. Cells 2020, 9, 1930. https://doi.org/10.3390/cells9091930
Hernández-Vega AM, Del Moral-Morales A, Zamora-Sánchez CJ, Piña-Medina AG, González-Arenas A, Camacho-Arroyo I. Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells. Cells. 2020; 9(9):1930. https://doi.org/10.3390/cells9091930
Chicago/Turabian StyleHernández-Vega, Ana M., Aylin Del Moral-Morales, Carmen J. Zamora-Sánchez, Ana G. Piña-Medina, Aliesha González-Arenas, and Ignacio Camacho-Arroyo. 2020. "Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells" Cells 9, no. 9: 1930. https://doi.org/10.3390/cells9091930
APA StyleHernández-Vega, A. M., Del Moral-Morales, A., Zamora-Sánchez, C. J., Piña-Medina, A. G., González-Arenas, A., & Camacho-Arroyo, I. (2020). Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells. Cells, 9(9), 1930. https://doi.org/10.3390/cells9091930