MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment


Abstract
1. Introduction
2. Prognostic Scoring Systems
3. Driver Mutations
4. JAK2 (Janus Kinase 2)
5. CALR (Calreticulin)
6. MPL (Myeloproliferative Leukaemia)
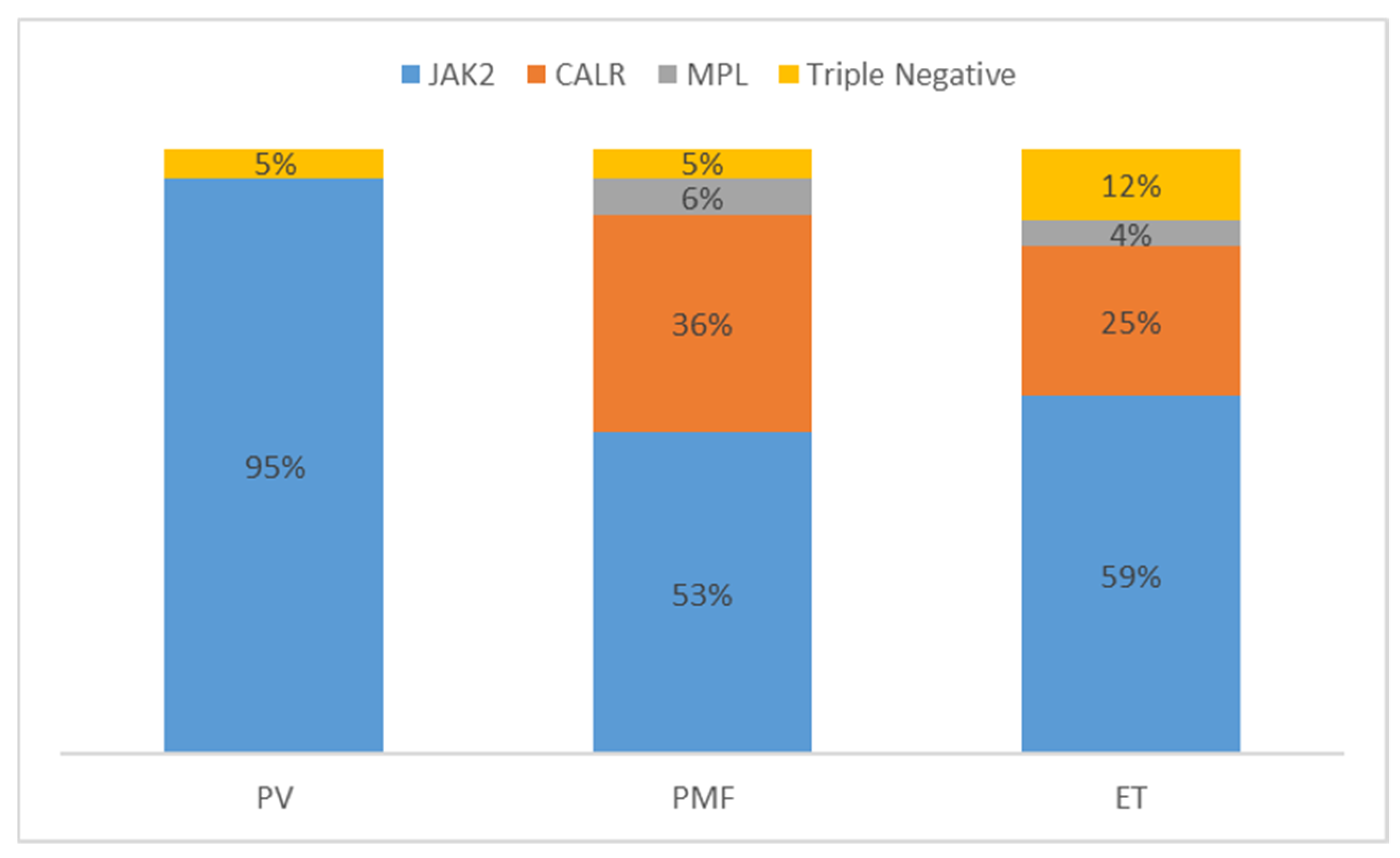
7. Triple Negative MPN
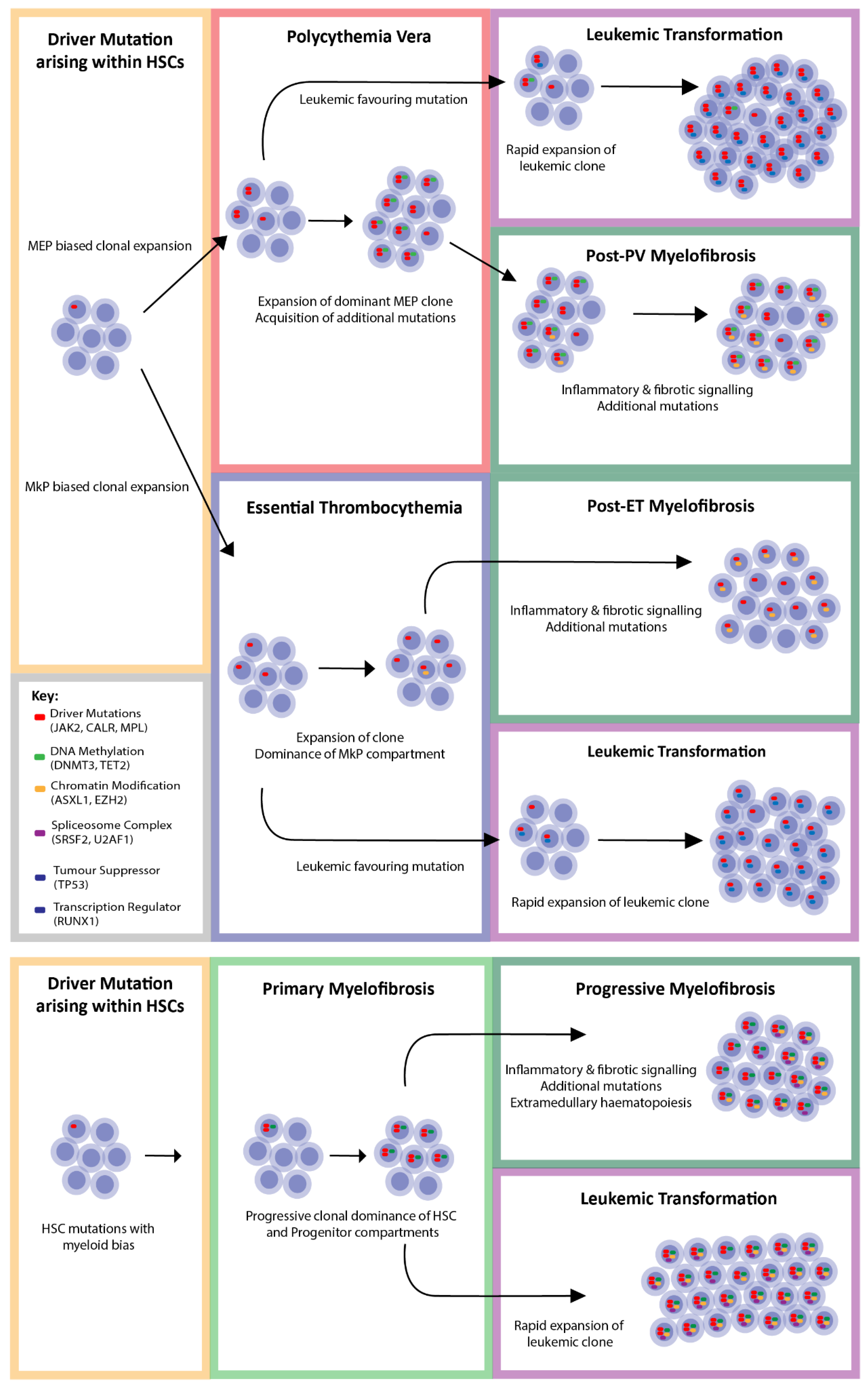
8. Coexisting Mutations
9. Chromatin Modifiers
9.1. ASXL1 (Additional Sex Comb Like-1)
9.2. EZH2 (Enhancer of Zeste 2)
10. Spliceosome Complex Components
10.1. SRSF2 (Splicing Factor, Serine/Arginine-Rich, 2)
10.2. U2AF1 (U2 Small Nuclear RNA Auxiliary Factor 1)
10.3. SF3B1 (Splicing Factor 3B, Subunit 1)
11. DNA Methylation Modifiers
11.1. DNMT3A (DNA Methyltransferase 3A)
11.2. TET2 (Ten-Eleven Translocation 2)
11.3. IDH1 and IDH2 (Isocitrate Dehydrogenase 1 and 2)
12. Tumour Suppressors
TP53 (Tumour Protein p53)
13. Transcriptional Regulators
RUNX1 (Runt-Related Transcription Factor 1)
14. Treatment of MPN—Existing and Emerging
14.1. Chemotherapy
14.2. JAK Inhibitors
14.3. Ruxolitinib
14.4. Fedratinib
14.5. Momelotinib
14.6. Pacritinib
14.7. Pegylated Interferon Alpha (IFNα)
14.8. Allogeneic Bone Marrow Transplantation
15. Future Directions
15.1. Checkpoint Inhibitors and Cellular Therapies
15.2. Ongoing Clinical Trials
16. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Heppner, J.; Nguyen, L.T.; Guo, M.; Naugler, C.; Rashid-Kolvear, F. Incidence of myeloproliferative neoplasms in Calgary, Alberta, Canada. BMC Res. Notes 2019, 12, 286. [Google Scholar] [CrossRef]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef]
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.M.; Rodeghiero, F.; Randi, M.L.; Vaidya, R.; Cazzola, M.; Rambaldi, A.; et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective identification of high-risk polycythemia vera patients based on JAK2V617F allele burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef]
- Barbui, T.; Vannucchi, A.M.; Carobbio, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Ruggeri, M.; Randi, M.L.; Cazzola, M.; Rambaldi, A.; et al. The effect of arterial hypertension on thrombosis in low-risk polycythemia vera. Am. J. Hematol. 2016, 92, E5–E6. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Guglielmelli, P.; Tefferi, A. Polycythemia vera and essential thrombocythemia. Curr. Opin. Hematol. 2018, 25, 112–119. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization–essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef]
- Gangat, N.; Tefferi, A. Myelofibrosis biology and contemporary management. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Ciboddo, M.; Mullally, A. JAK2 (and other genes) be nimble with MPN diagnosis, prognosis, and therapy. Hematol. 2018, 2018, 110–117. [Google Scholar] [CrossRef]
- Gowin, K.; Coakley, M.; Kosiorek, H.; Mesa, R.A. Discrepancies of applying primary myelofibrosis prognostic scores for patients with post polycythemia vera/essential thrombocytosis myelofibrosis. Haematol. 2016, 101, e405–e406. [Google Scholar] [CrossRef]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Prins, D.; Arias, C.G.; Klampfl, T.; Grinfeld, J.; Green, A.R. Mutant Calreticulin in the Myeloproliferative Neoplasms. HemaSphere 2020, 4, e333. [Google Scholar] [CrossRef]
- Online Mendelian Iinheritance in Man: An Online Catalog of Human Genes and Genetic Disorders [Internet]. Johns Hopkins University. 2020. Available online: https://omim.org/ (accessed on 27 May 2020).
- Gnanasambandan, K.; Sayeski, P.P. A structure-function perspective of Jak2 mutations and implications for alternate drug design strategies: The road not taken. Curr. Med. Chem. 2011, 18, 4659–4673. [Google Scholar] [CrossRef]
- Saharinen, P.; Takaluoma, K.; Silvennoinen, O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol. Cell. Biol. 2000, 20, 3387–3395. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet Lond. Engl. J. 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.; Teo, S.-S.; Tiedt, R.; Passweg, J.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- Lu, X.; Levine, R.; Tong, W.; Wernig, G.; Pikman, Y.; Zarnegar, S.; Gilliland, D.G.; Lodish, H.F. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc. Natl. Acad. Sci. USA 2005, 102, 18962–18967. [Google Scholar] [CrossRef]
- Garçon, L.; Rivat, C.; James, C.; Lacout, C.; Camara-Clayette, V.; Ugo, V.; Lécluse, Y.; Bennaceur-Griscelli, A.; Vainchenker, W. Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood 2006, 108, 1551–1554. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Cordua, S.; Kjær, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef]
- Mansier, O.; Kilani, B.; Guitart, A.V.; Guy, A.; Gourdou-Latyszenok, V.; Marty, C.; Parrens, M.; Plo, I.; Vainchencker, W.; James, C. Description of a knock-in mouse model of JAK2V617F MPN emerging from a minority of mutated hematopoietic stem cells. Blood 2019, 134, 2383–2387. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Kralovics, R. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp. Hematol. 2002, 30, 229–236. [Google Scholar] [CrossRef]
- Xing, S.-W.; Wanting, T.H.; Zhao, W.; Ma, J.; Wang, S.; Xu, X.; Li, Q.; Fu, X.; Xu, M.; Zhao, Z.J. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood 2008, 111, 5109–5117. [Google Scholar] [CrossRef]
- Saliba, J.; Hamidi, S.; Lenglet, G.; Langlois, T.; Yin, J.; Cabagnols, X.; Secardin, L.; Legrand, C.; Galy, A.; Opolon, P.; et al. Heterozygous and Homozygous JAK2V617F States Modeled by Induced Pluripotent Stem Cells from Myeloproliferative Neoplasm Patients. PLoS ONE 2013, 8, e74257. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Ortmann, C.A.; Silber, Y.; Bellosillo, B.; Guglielmelli, P.; Harrison, C.N.; Reilly, J.T.; Stegelmann, F.; et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygous subclone. Blood 2012, 120, 2704–2707. [Google Scholar] [CrossRef]
- Chen, E.; Beer, P.A.; Godfrey, A.L.; Ortmann, C.A.; Li, J.; Costa-Pereira, A.P.; Ingle, C.E.; Dermitzakis, E.T.; Campbell, P.J.; Green, A.R. Distinct Clinical Phenotypes Associated with JAK2V617F Reflect Differential STAT1 Signaling. Cancer Cell 2010, 18, 524–535. [Google Scholar] [CrossRef]
- Duek, A.; Lundberg, P.; Shimizu, T.; Grisouard, J.; Karow, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Loss of Stat1 decreases megakaryopoiesis and favors erythropoiesis in a JAK2-V617F–driven mouse model of MPNs. Blood 2014, 123, 3943–3950. [Google Scholar] [CrossRef]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of myeloproliferative neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef]
- Grisouard, J.; Shimizu, T.; Duek, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Deletion of Stat3 in hematopoietic cells enhances thrombocytosis and shortens survival in a JAK2-V617F mouse model of MPN. Blood 2015, 125, 2131–2140. [Google Scholar] [CrossRef]
- Yan, N.; Hutchison, R.E.; Mohi, G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood 2012, 119, 3539–3549. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef]
- Gilles, L.; Arslan, A.D.; Marinaccio, C.; Wen, Q.J.; Arya, P.; McNulty, M.; Yang, Q.; Zhao, J.C.; Konstantinoff, K.; Lasho, T.L.; et al. Downregulation of GATA1 drives impaired hematopoiesis in primary myelofibrosis. J. Clin. Investig. 2017, 127, 1316–1320. [Google Scholar] [CrossRef]
- Abbonante, V.; Di Buduo, C.A.; Gruppi, C.; Malara, A.; Gianelli, U.; Celesti, G.; Anselmo, A.; Laghi, L.; Vercellino, M.; Visai, L.; et al. Thrombopoietin/TGF-β1 Loop Regulates Megakaryocyte Extracellular Matrix Component Synthesis. Stem Cells 2016, 34, 1123–1133. [Google Scholar] [CrossRef]
- Malara, A.; Currao, M.; Gruppi, C.; Celesti, G.; Viarengo, G.; Buracchi, C.; Laghi, L.; Kaplan, D.L.; Balduini, A. Megakaryocytes contribute to the bone marrow-matrix environment by expressing fibronectin, type IV collagen, and laminin. Stem Cells 2014, 32, 926–937. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Klampfl, T.; Them, N.C.C.; Berg, T.; Vladimer, G.I.; Bagienski, K.; Milanesi, C.; Casetti, I.C.; Sant’Antonio, E.; Ferretti, V.V.; Schischlik, F.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.-M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2009, 24, 665–683. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, N.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through trans-Activation of LRP on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.-I.; Defour, J.-P.; Nivarthi, H.; Hug, E.; et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef]
- Masubuchi, N.; Araki, M.; Yang, Y.; Hayashi, E.; Imai, M.; Edahiro, Y.; Hironaka, Y.; Mizukami, Y.; Kihara, Y.; Takei, H.; et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 2019, 34, 499–509. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Imai, M.; Mizukami, Y.; Kihara, Y.; Sunami, Y.; Masubuchi, N.; Edahiro, Y.; Hironaka, Y.; Osaga, S.; et al. Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia 2018, 33, 122–131. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- How, J.; Hobbs, G.S.; Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood 2019, 134, 2242–2248. [Google Scholar] [CrossRef]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Faculty Opinions recommendation of Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Fac. Opin. Post-Publ. Peer Rev. Biomed. Lit. 2016, 30, 431–438. [Google Scholar] [CrossRef]
- Angona, A.; Alvarez-Larrán, A.; Bellosillo, B.; Longarón, R.; Camacho, L.; Fernández-Rodríguez, M.C.; Pairet, S.; Besses, C. Characterization of CD34+ hematopoietic progenitor cells in JAK2 V617F and CALR -mutated myeloproliferative neoplasms. Leuk. Res. 2016, 48, 11–15. [Google Scholar] [CrossRef]
- Nam, A.S.; Kim, K.-T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef]
- McNamara, C.J.; Panzarella, T.; Kennedy, J.A.; Arruda, A.; Claudio, J.O.; Daher-Reyes, G.; Ho, J.; Siddiq, N.; Devlin, R.; Tsui, H.; et al. The mutational landscape of accelerated- and blast-phase myeloproliferative neoplasms impacts patient outcomes. Blood Adv. 2018, 2, 2658–2671. [Google Scholar] [CrossRef] [PubMed]
- Venton, G.; Courtier, F.; Charbonnier, A.; D’Incan, E.; Saillard, C.; Mohty, B.; Mozziconacci, M.-J.; Birnbaum, D.; Murati, A.; Vey, N.; et al. Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am. J. Hematol. 2017, 93, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adélaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Geneschromosom. Cancer 2019, 59, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef]
- De Laval, B.; Pawlikowska, P.; Petit-Cocault, L.; Bilhou-Nabera, C.; Aubin-Houzelstein, G.; Souyri, M.; Pouzoulet, F.; Gaudry, M.; Porteu, F. Thrombopoietin-Increased DNA-PK-Dependent DNA Repair Limits Hematopoietic Stem and Progenitor Cell Mutagenesis in Response to DNA Damage. Cell Stem Cell 2013, 12, 37–48. [Google Scholar] [CrossRef]
- Guenther, K.L.; Cheruku, P.S.; Cash, A.; Smith, R.H.; Alvarado, L.J.; Burkett, S.; Townsley, D.M.; Winkler, T.; LaRochelle, A. Eltrombopag promotes DNA repair in human hematopoietic stem and progenitor cells. Exp. Hematol. 2019, 73, 1–6.e6. [Google Scholar] [CrossRef]
- Barbieri, D.; Elvira-Matelot, E.; Pelinski, Y.; Genève, L.; De Laval, B.; Yogarajah, G.; Pecquet, C.; Constantinescu, S.N.; Porteu, F. Thrombopoietin protects hematopoietic stem cells from retrotransposon-mediated damage by promoting an antiviral response. J. Exp. Med. 2018, 215, 1463–1480. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Defour, J.-P.; Chachoua, I.; Pecquet, C.; Constantinescu, S.N. Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: Implications for myeloproliferative neoplasms. Leukemia 2015, 30, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Guglielmelli, P.; Bordoni, R.; Casetti, I.; Milanesi, C.; Sant’Antonio, E.; Ferretti, V.V.; Pancrazzi, A.; Rotunno, G.; et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013, 121, 4388–4395. [Google Scholar] [CrossRef] [PubMed]
- Pecquet, C.; Diaconu, C.C.; Staerk, J.; Girardot, M.; Marty, C.; Royer, Y.; Defour, J.-P.; Dusa, A.; Besancenot, R.; Giraudier, S.; et al. Thrombopoietin receptor down-modulation by JAK2 V617F: Restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood 2012, 119, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Maifrede, S.; Dasgupta, Y.; Sullivan, K.; Flis, S.; Le, B.V.; Solecka, M.; Belyaeva, E.A.; Kubovcakova, L.; Nawrocki, M.; et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017, 130, 2848–2859. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.; Roberts, A.W.; Nicola, N.A.; Li, R.; Metcalf, D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood 1996, 87, 2162–2170. [Google Scholar] [CrossRef]
- Alexander, W.S.; Roberts, A.W.; Maurer, A.B.; Nicola, N.A.; Dunn, A.R.; Metcalf, D. Studies of the c‐Mpl Thrombopoietin Receptor through Gene Disruption and Activation. Stem Cells 1996, 14, 124–132. [Google Scholar] [CrossRef]
- Tiedt, R.; Coers, J.; Ziegler, S.; Wiestner, A.; Hao-Shen, H.; Bornmann, C.; Schenkel, J.; Karakhanova, S.; De Sauvage, F.J.; Jackson, C.W.; et al. Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood 2009, 113, 1768–1777. [Google Scholar] [CrossRef]
- Lannutti, B.J.; Epp, A.; Roy, J.; Chen, J.; Josephson, N.C. Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood 2009, 113, 1778–1785. [Google Scholar] [CrossRef]
- Feenstra, J.D.M.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; Milanesi, C.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.-C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Favier, R.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Pardanani, A.; Vannucchi, A.M. Myelofibrosis Treatment Algorithm 2018. Blood Cancer J. 2018, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of polycomb-associated geneASXL1in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, J.C.; Alonso, A.G.D.A.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Müller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef]
- Balasubramani, A.; Larjo, A.; Bassein, J.A.; Chang, X.; Hastie, R.B.; Togher, S.M.; Lähdesmäki, H.; Rao, A. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1–BAP1 complex. Nat. Commun. 2015, 6, 7307. [Google Scholar] [CrossRef]
- Asada, S.; Goyama, S.; Inoue, D.; Shikata, S.; Takeda, R.; Fukushima, T.; Yonezawa, T.; Fujino, T.; Hayashi, Y.; Kawabata, K.C.; et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat. Commun. 2018, 9, 2733. [Google Scholar] [CrossRef]
- Kitamura, T. ASXL1 mutations gain a function. Blood 2018, 131, 274–275. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Score, J.; Hidalgo-Curtis, C.; Jones, A.V.; Winkelmann, N.; Skinner, A.; Ward, D.; Zoi, K.; Ernst, T.; Stegelmann, F.; Döhner, K.; et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012, 119, 1208–1213. [Google Scholar] [CrossRef]
- Li, B.; Chng, W.-J. EZH2 abnormalities in lymphoid malignancies: Underlying mechanisms and therapeutic implications. J. Hematol. Oncol. 2019, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosén, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Saunthararajah, Y.; Maciejewski, J. Polycomb segment myeloid malignancies. Blood 2012, 119, 1097–1098. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Velcheti, V.; Wong, K.-K.; Saunthararajah, Y. EZH2 Inhibitors: Take It EZy, It Is All About Context. Cancer Discov. 2019, 9, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Ennishi, D.; Takata, K.; Beguelin, W.; Duns, G.; Mottok, A.; Farinha, P.; Bashashati, A.; Saberi, S.; Boyle, M.; Meissner, B.; et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019, 9, 546–563. [Google Scholar] [CrossRef]
- Yang, Y.; Akada, H.; Nath, D.; Hutchison, R.E.; Mohi, G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood 2016, 127, 3410–3423. [Google Scholar] [CrossRef]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Komeno, Y.; Huang, Y.-J.; Qiu, J.; Lin, L.; Xu, Y.; Zhou, Y.; Chen, L.; Monterroza, D.D.; Li, H.; DeKelver, R.C.; et al. SRSF2 Is Essential for Hematopoiesis, and Its Myelodysplastic Syndrome-Related Mutations Dysregulate Alternative Pre-mRNA Splicing. Mol. Cell. Boil. 2015, 35, 3071–3082. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Smeets, M.F.; Tan, S.Y.; Xu, J.J.; Anande, G.; Unnikrishnan, A.; Chalk, A.M.; Taylor, S.R.; Pimanda, J.E.; Wall, M.; Purton, L.E.; et al. Srsf2 P95H initiates myeloid bias and myelodysplastic/myeloproliferative syndrome from hemopoietic stem cells. Blood 2018, 132, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Illar, G.M.; Bailey, N.G. Clinicopathologic characterisation of myeloid neoplasms with concurrent spliceosome mutations and myeloproliferative-neoplasm-associated mutations. J. Clin. Pathol. 2020, 1–9. [Google Scholar] [CrossRef]
- Soares, L.M.; Zanier, K.; Mackereth, C.; Sattler, M.; Valcárcel, J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science 2006, 312, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef]
- Isono, K.; Mizutani-Koseki, Y.; Komori, T.; Schmidt-Zachmann, M.S.; Koseki, H. Mammalian Polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 2005, 19, 536–541. [Google Scholar] [CrossRef]
- Cazzola, M.; Rossi, M.; Malcovati, L.; Mieloproliferative, A.I.P.L.R.S.C.G.I.M. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013, 121, 260–269. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 mutations in primary myelofibrosis: Clinical, histopathology and genetic correlates among 155 patients. Leukemia 2011, 26, 1135–1137. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.; Hellström-Lindberg, E.; Gambacorti-Passerini, C.; et al. SomaticSF3B1Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Feenstra, J.D.M.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Anteneh, H.; Fang, J.; Song, J. Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat. Commun. 2020, 11, 2294. [Google Scholar] [CrossRef] [PubMed]
- Izzo, F.; Lee, S.C.; Poran, A.; Chaligne, R.; Gaiti, F.; Gross, B.; Murali, R.R.; Deochand, S.D.; Ang, C.; Jones, P.W.; et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 2020, 52, 378–387. [Google Scholar] [CrossRef]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couédic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Ostrander, E.L.; Kramer, A.C.; Mallaney, C.; Celik, H.; Koh, W.K.; Fairchild, J.; Haussler, E.; Zhang, C.R.; Challen, G.A. Divergent Effects of Dnmt3a and Tet2 Mutations on Hematopoietic Progenitor Cell Fitness. Stem Cell Rep. 2020, 14, 551–560. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Hillis, D.M.; Patton, J.C.; Bradley, R.D.; Baker, R.J. Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol. Boil. Evol. 1998, 15, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, S.-M.; Shin, S.W.; Park, J.-W. Inactivation of mitochondrial NADP+-dependent isocitrate dehydrogenase by hypochlorous acid. Free. Radic. Res. 2008, 42, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, Ş.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef]
- Delic, S.; Rose, M.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Somasundara, A.V.H.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.T.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.-W.; Micol, J.-B.; Zhang, X.J.; De Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Boil. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Kubesova, B.; Pavlova, S.; Malcikova, J.; Kabathova, J.; Radova, L.; Tom, N.; Tichy, B.; Plevova, K.; Kantorova, B.; Fiedorova, K.; et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: Association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia 2017, 32, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Marcellino, B.K.; Hoffman, R.; Tripodi, J.; Lu, M.; Kosiorek, H.; Mascarenhas, J.; Rampal, R.K.; Dueck, A.; Najfeld, V. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018, 2, 3581–3589. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 Lesions in Leukemic Transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef]
- Tsuruta-Kishino, T.; Koya, J.; Kataoka, K.; Narukawa, K.; Sumitomo, Y.; Kobayashi, H.; Sato, T.; Kurokawa, M. Loss of p53 induces leukemic transformation in a murine model of Jak2 V617F-driven polycythemia vera. Oncogene 2017, 36, 3300–3311. [Google Scholar] [CrossRef]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, R.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef]
- Sood, R.; Kamikubo, Y.; Liu, P.P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Bartalucci, N.; Contini, E.; Rotunno, G.; Pacilli, A.; Romagnoli, S.; Mannelli, L.; Mannelli, F.; Coltro, G.; Pancrazzi, A.; et al. Involvement of RUNX1 Pathway Is a Common Event in the Leukemic Transformation of Chronic Myeloproliferative Neoplasms (MPNs). Blood 2019, 134, 2968. [Google Scholar] [CrossRef]
- Šaban, N.; Bujak, M. Hydroxyurea and hydroxamic acid derivatives as antitumor drugs. Cancer Chemother. Pharmacol. 2009, 64, 213–221. [Google Scholar] [CrossRef]
- Fruchtman, S.M.; Mack, K.; Kaplan, M.E.; Peterson, P.; Berk, P.D.; Wasserman, L.R. From efficacy to safety: A Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin. Hematol. 1997, 34, 17–23. [Google Scholar] [PubMed]
- Cortelazzo, S.; Finazzi, G.; Ruggeri, M.; Vestri, O.; Galli, M.; Rodeghiero, F.; Barbui, T. Hydroxyurea for Patients with Essential Thrombocythemia and a High Risk of Thrombosis. N. Engl. J. Med. 1995, 332, 1132–1137. [Google Scholar] [CrossRef]
- Martínez-Trillos, A.; Gaya, A.; Maffioli, M.; Arellano-Rodrigo, E.; Calvo, X.; Díaz-Beyá, M.; Cervantes, F.; Calvet, X. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: Results in 40 patients. Ann. Hematol. 2010, 89, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, A.; Mascarenhas, J.; Mesa, R.A.; Kosiorek, H.E.; Rampal, R.K.; Silver, R.T.; Salama, M.E.; Siwoski, O.; Dueck, A.C.; Sandy, L.; et al. Final Results of Prospective Treatment with Pegylated Interferon Alfa-2a for Patients with Polycythemia Vera and Essential Thrombocythemia in First and Second-Line Settings. Blood 2019, 134, 2943. [Google Scholar] [CrossRef]
- Finazzi, G.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef]
- Nand, S.; Stock, W.; Godwin, J.; Fisher, S.G. Leukemogenic risk of hydroxyurea therapy in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Am. J. Hematol. 1996, 52, 42–46. [Google Scholar] [CrossRef]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Martínez-Avilés, L.; Hernández-Boluda, J.C.; Ferrer-Marín, F.; Antelo, M.L.; Burgaleta, C.; Mata, M.I.; Xicoy, B.; Martínez-Trillos, A.; Gómez-Casares, M.T.; et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann. Hematol. 2014, 93, 2037–2043. [Google Scholar] [CrossRef]
- Shvidel, L.; Sigler, E.; Haran, M.; Klepfish, A.; Duek, A.; Berrebi, A.; Shtalrid, M. Busulphan is safe and efficient treatment in elderly patients with essential thrombocythemia. Leukemia 2007, 21, 2071–2072. [Google Scholar] [CrossRef]
- Fang, X.; Yin, H.; Zhang, H.; Wu, F.; Liu, Y.; Fu, Y.; Yu, D.; Zong, L. p53 mediates hydroxyurea resistance in aneuploid cells of colon cancer. Exp. Cell Res. 2019, 376, 39–48. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.; Al-Ali, H.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Andraos, R.; Qian, Z.; Bonenfant, D.; Rubert, J.; Vangrevelinghe, E.; Scheufler, C.; Marque, F.; Régnier, C.H.; De Pover, A.; Ryckelynck, H.; et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012, 2, 512–523. [Google Scholar] [CrossRef]
- Tvorogov, D.; Thomas, D.; Liau, N.P.D.; Dottore, M.; Barry, E.F.; Lathi, M.; Kan, W.L.; Hercus, T.R.; Stomski, F.; Hughes, T.P.; et al. Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci. Adv. 2018, 4, eaat3834. [Google Scholar] [CrossRef]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef]
- Meyer, S.C.; Levine, R.L. Molecular pathways: Molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef]
- Singer, J.W.; Al-Fayoumi, S.; Taylor, J.; Velichko, S.; O’Mahony, A. Comparative phenotypic profiling of the JAK2 inhibitors ruxolitinib, fedratinib, momelotinib, and pacritinib reveals distinct mechanistic signatures. PLoS ONE 2019, 14, e0222944. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, C.; Han, L.; Jain, N.; Roberts, K.G.; Ma, H.; Cai, T.; Cavazos, A.; Tabe, Y.; Jacamo, R.O.; et al. Inhibition of mTORC1/C2 signaling improves anti-leukemia efficacy of JAK/STAT blockade in CRLF2 rearranged and/or JAK driven Philadelphia chromosome-like acute B-cell lymphoblastic leukemia. Oncotarget 2018, 9, 8027–8041. [Google Scholar] [CrossRef]
- Kong, X.; Sun, H.; Pan, P.; Li, D.; Zhu, F.; Chang, S.; Xu, L.; Li, Y.; Hou, T. How Does the L884P Mutation Confer Resistance to Type-II Inhibitors of JAK2 Kinase: A Comprehensive Molecular Modeling Study. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.; Cervantes, F.; Harrison, C.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Kiladjian, J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Vannucchi, A.M.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Kirito, K.; et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016, 101, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Griesshammer, M.; Saydam, G.; Palandri, F.; Benevolo, G.; Egyed, M.; Callum, J.; Devos, T.; Sivgin, S.; Guglielmelli, P.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80-week follow-up from the RESPONSE-2 trial. Ann. Hematol. 2018, 97, 1591–1600. [Google Scholar] [CrossRef]
- Harrison, C.; Mead, A.J.; Panchal, A.; Fox, S.; Yap, C.; Gbandi, E.; Houlton, A.; Alimam, S.; Ewing, J.; Wood, M.; et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamide. Blood 2017, 130, 1889–1897. [Google Scholar] [CrossRef]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rumi, E.; Gattoni, E.; Pieri, L.; Zhen, H.; Granier, M.; Assad, A.; et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: Long-term phase 2 study results. Blood 2017, 130, 1768–1771. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Rotunno, G.; Bogani, C.; Mannarelli, C.; Giunti, L.; Provenzano, A.; Giglio, S.; Squires, M.; Stalbovskaya, V.; Gopalakrishna, P.; et al. Ruxolitinib is an effective treatment forCALR-positive patients with myelofibrosis. Br. J. Haematol. 2015, 173, 938–940. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Verstovsek, S.; Guglielmelli, P.; Griesshammer, M.; Burn, T.C.; Naim, A.; Paranagama, D.; Marker, M.; Gadbaw, B.; Kiladjian, J.-J. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann. Hematol. 2017, 96, 1113–1120. [Google Scholar] [CrossRef]
- Austin, R.J.; Straube, J.; Bruedigam, C.; Pali, G.; Jacquelin, S.; Vu, T.; Green, J.; Gräsel, J.; Lansink, L.; Cooper, L.; et al. Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations. Leukemia 2019, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F Expression Causes a Lethal Myeloproliferative Neoplasm with Differential Effects on Hematopoietic Stem and Progenitor Cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.Y.; McNamara, C.; Kennedy, J.A.; Panzarella, T.; Arruda, A.; Stockley, T.; Sukhai, M.; Thomas, M.; Bartoszko, J.; Ho, J.; et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. 2017, 1, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Vannucchi, A.M.; Kiladjian, J.; Al-Ali, H.; Sirulnik, A.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.S.; Passamonti, F.; et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013, 122, 4047–4053. [Google Scholar] [CrossRef]
- Fedratinib Becomes New Option in Myelofibrosis. Cancer Discov. 2019, 9, 1332. [CrossRef]
- Mullally, A.; Hood, J.; Harrison, C.; Mesa, R. Fedratinib in myelofibrosis. Blood Adv. 2020, 4, 1792–1800. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Harrison, C.; Schaap, N.P.M.; Vannucchi, A.M.; Kiladjian, J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Harrison, C.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.-J.; Jourdan, E.; Silver, R.T.; Schouten, H.C.; Passamonti, F.; Zweegman, S.; Talpaz, M.; et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: An updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am. J. Hematol. 2020, 95, 594–603. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; McLornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor–Naïve Patients With Myelofibrosis. J. Clin. Oncol. 2017, 35, 3844–3850. [Google Scholar] [CrossRef]
- Harrison, C.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Barraco, D.; Lasho, T.L.; Shah, S.; Begna, K.H.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Hanson, C.A.; Ketterling, R.P.; et al. Momelotinib therapy for myelofibrosis: A 7-year follow-up. Blood Cancer J. 2018, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.W.; Al-Fayoumi, S.; Ma, H.; Komrokji, R.S.; Mesa, R.; Verstovsek, S. Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor. J. Exp. Pharmacol. 2016, 8, 11–19. [Google Scholar] [CrossRef]
- Mesa, R.A.; Vannucchi, A.M.; Mead, A.J.; Egyed, M.; Szőke, A.; Suvorov, A.; Jakucs, J.; Perkins, A.; Prasad, R.; Mayer, J.; et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): An international, randomised, phase 3 trial. Lancet Haematol. 2017, 4, e225–e236. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szőke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis. JAMA Oncol. 2018, 4, 652. [Google Scholar] [CrossRef]
- Diaz, A.E.; Mesa, R.A. Pacritinib and its use in the treatment of patients with myelofibrosis who have thrombocytopenia. Future Oncol. 2018, 14, 797–807. [Google Scholar] [CrossRef]
- Bellucci, S.; Harousseau, J.; Brice, P.; Tobelem, G. Treatment of essential thrombocythaemia by alpha 2a interferon. Lancet 1988, 332, 960–961. [Google Scholar] [CrossRef]
- Green, R.R.; Ireton, R.C.; Gale, J.M. Interferon-stimulated genes: New platforms and computational approaches. Mamm. Genome 2018, 29, 593–602. [Google Scholar] [CrossRef]
- Raftery, N.; Stevenson, N.J. Advances in anti-viral immune defence: Revealing the importance of the IFN JAK/STAT pathway. Cell. Mol. Life Sci. 2017, 74, 2525–2535. [Google Scholar] [CrossRef]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Onai, N.; Yoshihara, H.; Arai, F.; Suda, T.; Ohteki, T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nat. Med. 2009, 15, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef]
- Hasan, S.; Lacout, C.; Marty, C.; Cuingnet, M.; Solary, E.; Vainchenker, W.; Villeval, J.-L. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood 2013, 122, 1464–1477. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Estrov, Z.; Pierce, S.; Richie, M.A.; Borthakur, G.; Konopleva, M.; Cortes, J.; et al. Pegylated Interferon Alfa-2a Yields High Rates of Hematologic and Molecular Response in Patients With Advanced Essential Thrombocythemia and Polycythemia Vera. J. Clin. Oncol. 2009, 27, 5418–5424. [Google Scholar] [CrossRef]
- Ianotto, J.-C.; Chauveau, A.; Boyer-Perrard, F.; Gyan, E.; Laribi, K.; Cony-Makhoul, P.; Demory, J.-L.; De Renzis, B.; Dosquet, C.; Rey, J.; et al. Benefits and pitfalls of pegylated interferon-α2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: A French Intergroup of Myeloproliferative neoplasms (FIM) study. Haematologica 2017, 103, 438–446. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Abdel-Wahab, O.; Manshouri, T.; Kilpivaara, O.; Cortes, J.; Roupie, A.L.; Zhang, S.-J.; Harris, D.; Estrov, Z.; Kantarjian, H.; et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon α-2a. Blood 2013, 122, 893–901. [Google Scholar] [CrossRef]
- Verger, E.; Cassinat, B.; Chauveau, A.; Dosquet, C.; Giraudier, S.; Schlageter, M.-H.; Ianotto, J.-C.; Yassin, M.A.; Al-Dewik, N.; Carillo, S.; et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 2015, 126, 2585–2591. [Google Scholar] [CrossRef]
- Mikkelsen, S.U.; Kjær, L.; Bjørn, M.E.; Knudsen, T.A.; Sørensen, A.L.; Andersen, C.B.L.; Bjerrum, O.W.; Brochmann, N.; El Fassi, D.; Kruse, T.A.; et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018, 7, 3571–3581. [Google Scholar] [CrossRef] [PubMed]
- Carreras, E.; Dufour, C.; Mohty, M.; Kröger, N. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Kröger, N.M.; Deeg, J.H.; Olavarria, E.; Niederwieser, D.; Bacigalupo, A.; Barbui, T.; Rambaldi, A.; Mesa, R.; Tefferi, A.; Griesshammer, M.; et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: A consensus process by an EBMT/ELN international working group. Leukemia 2015, 29, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.; De Wreede, L.C.; Wolschke, C.; Schetelig, J.; Eikema, D.-J.; Van Lint, M.T.; Knelange, N.S.; Beelen, D.; Brecht, A.; Niederwieser, D.; et al. Long-term outcome after allogeneic hematopoietic cell transplantation for myelofibrosis. Haematologica 2019, 104, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Gowin, K.; Ballen, K.; Ahn, K.W.; Hu, Z.-H.; Ali, H.; Arcasoy, M.O.; Devlin, R.; Coakley, M.; Gerds, A.T.; Green, M.; et al. Survival following allogeneic transplant in patients with myelofibrosis. Blood Adv. 2020, 4, 1965–1973. [Google Scholar] [CrossRef]
- Tamari, R.; Rapaport, F.; Zhang, N.; McNamara, C.; Kuykendall, A.; Sallman, D.A.; Komrokji, R.; Arruda, A.; Najfeld, V.; Sandy, L.; et al. Impact of High-Molecular-Risk Mutations on Transplantation Outcomes in Patients with Myelofibrosis. Boil. Blood Marrow Transplant. 2019, 25, 1142–1151. [Google Scholar] [CrossRef]
- Devlin, R.; Gupta, V. Myelofibrosis: To transplant or not to transplant? Hematology 2016, 2016, 543–551. [Google Scholar] [CrossRef][Green Version]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617Fcauses PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef]
- Holmström, M.O.; Hjortsø, M.D.; Ahmad, S.M.; Met, Ö.; Martinenaite, E.; Riley, C.; Straten, P.T.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2016, 31, 495–498. [Google Scholar] [CrossRef]
- Holmström, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, Ö.; Friese, C.; Kjær, L.; Riley, C.H.; Straten, P.T.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2017, 32, 429–437. [Google Scholar] [CrossRef]
- Schieber, M.; Crispino, J.D.; Stein, B. Myelofibrosis in 2019: Moving beyond JAK2 inhibition. Blood Cancerj. 2019, 9, 74. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Harrison, C. Emerging treatments for classical myeloproliferative neoplasms. Blood 2017, 129, 693–703. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scoring System | Criteria | Low Risk | Int Risk | High Risk |
|---|---|---|---|---|
| MIPSS-PV | Age > 67 years (2 points) Leukocyte count ≥15 × 109/L (1 point) Adverse Mutation—SRSF2 (3 points) Thrombosis history (1 point) | 0–1 point Median OS 24 years | 2–3 points Median OS 13.1 years | ≥4 points Median OS 3.2 years |
| MIPSS-ET | Age > 60 years (4 points) Male gender (1 point) Leukocyte count ≥11 × 109/L (1 point) Adverse Mutations—SRSF2, SF3B1, U2AF1, TP53 (2 points) | 0–1 point Median OS 34.4 years | 2–5 points Median OS 14.1 years | ≥6 points Median OS 7.9 years |
| Scoring System | Criteria | Very Low Risk | Low Risk | Intermediate Risk | High Risk | Very High Risk |
|---|---|---|---|---|---|---|
| MIPSS70-Plus Version 2.0 (Any time point) | Severe Anaemia (2 points) (Hb < 80g/L female, < 90g/L male) Moderate Anaemia (1 point) (Hb 80–100g/L female, 90–110g/L male) Circulating blasts ≥ 2% (1 point) Constituitional symptoms (2 points) Very high risk karyotype (4 points) Unfavourable karyotype (3 points) ≥2 HMR mutations (3 points) 1 HMR mutation (2 points) Type 1/like CALR absent (2 points) | 0 points Median OS NR | 1–2 points Median OS 16.4 years | 3–4 points Median OS 7.7 years | 5–8 points Median OS 4.1 years | 9+ points Median OS 1.8 years |
| Scoring System | Criteria | Low Risk | Int-1 Risk | Int-2 Risk | High Risk |
|---|---|---|---|---|---|
| DIPSS (Any time point) | Age > 65 years (1 point) Constitutional Symptoms (1 point) Hb < 100g/L (2 points) Leukocyte count ≥25 × 109/L (1 point) Circulating blasts > 1% (1 point) | 0 points Median OS 14.6 years | 1–2 points Median OS 7.4 years | 3-4 points Median OS 4 years | 5–6 points Median OS 2.3 years |
| Mutation Group | Gene | PV [7] | ET [7] | PMF [64] | sMF [64] | LT [61] |
|---|---|---|---|---|---|---|
| Driver Mutations | JAK2 | 98% | 52% | 62% | 81% | 60% |
| CALR | 0% | 26% | 22% | 14% | 21% | |
| MPL | 0% | 4% | 5% | 3% | 13% | |
| DNA Methylation | TET2 | 22% | 16% | 15% | 39% | 19% |
| DNMT3A | 2% | 6% | 9% | 5% | 3% | |
| IDH1 | 0% | 0% | 2% * | 1% * | 12% | |
| IDH2 | 2% | 1% | NA | NA | 7% | |
| Chromatin Modification | ASXL1 | 12% | 11% | 48% | 27% | 47% |
| EZH2 | 0% | 3% | 6% | 14% | 15% | |
| Spliceosome Complex | SRSF2 | 3% | 2% | 14% | 3% | 13% |
| U2AF1 | 0% | 1% | 17% | 7% | 5% | |
| SF3B1 | 3% | 5% | 13% | 5% | 7% | |
| Tumour Suppressor | TP53 $ | 1% | 2% | 6% | 14% | 16% |
| Transcription Regulator | RUNX1 | 2% | 2% | 3% | 3% | 17% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells 2020, 9, 1901. https://doi.org/10.3390/cells9081901
Grabek J, Straube J, Bywater M, Lane SW. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells. 2020; 9(8):1901. https://doi.org/10.3390/cells9081901
Chicago/Turabian StyleGrabek, Julian, Jasmin Straube, Megan Bywater, and Steven W. Lane. 2020. "MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment" Cells 9, no. 8: 1901. https://doi.org/10.3390/cells9081901
APA StyleGrabek, J., Straube, J., Bywater, M., & Lane, S. W. (2020). MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells, 9(8), 1901. https://doi.org/10.3390/cells9081901