A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Spheroid Formation
2.3. Dynarray Preparation
2.4. Antibodies and Dyes
2.5. Data Acquisition
2.6. Data Analysis
3. Results
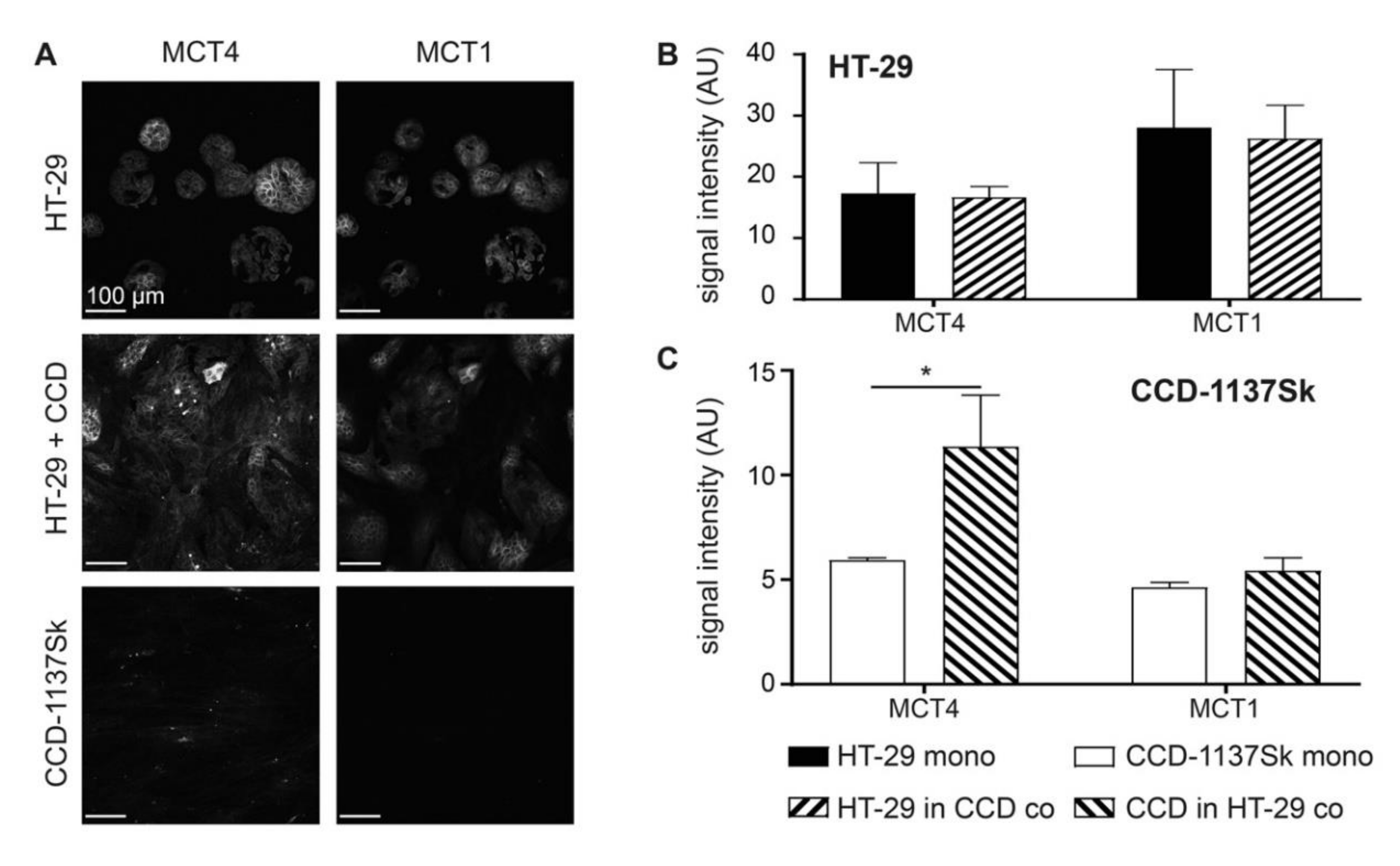
3.1. MCT4 as well as Markers for Glycolysis and Autophagy are Upregulated in CCD-1337Sk Fibroblasts upon Co-Culture with HT-29 Cells
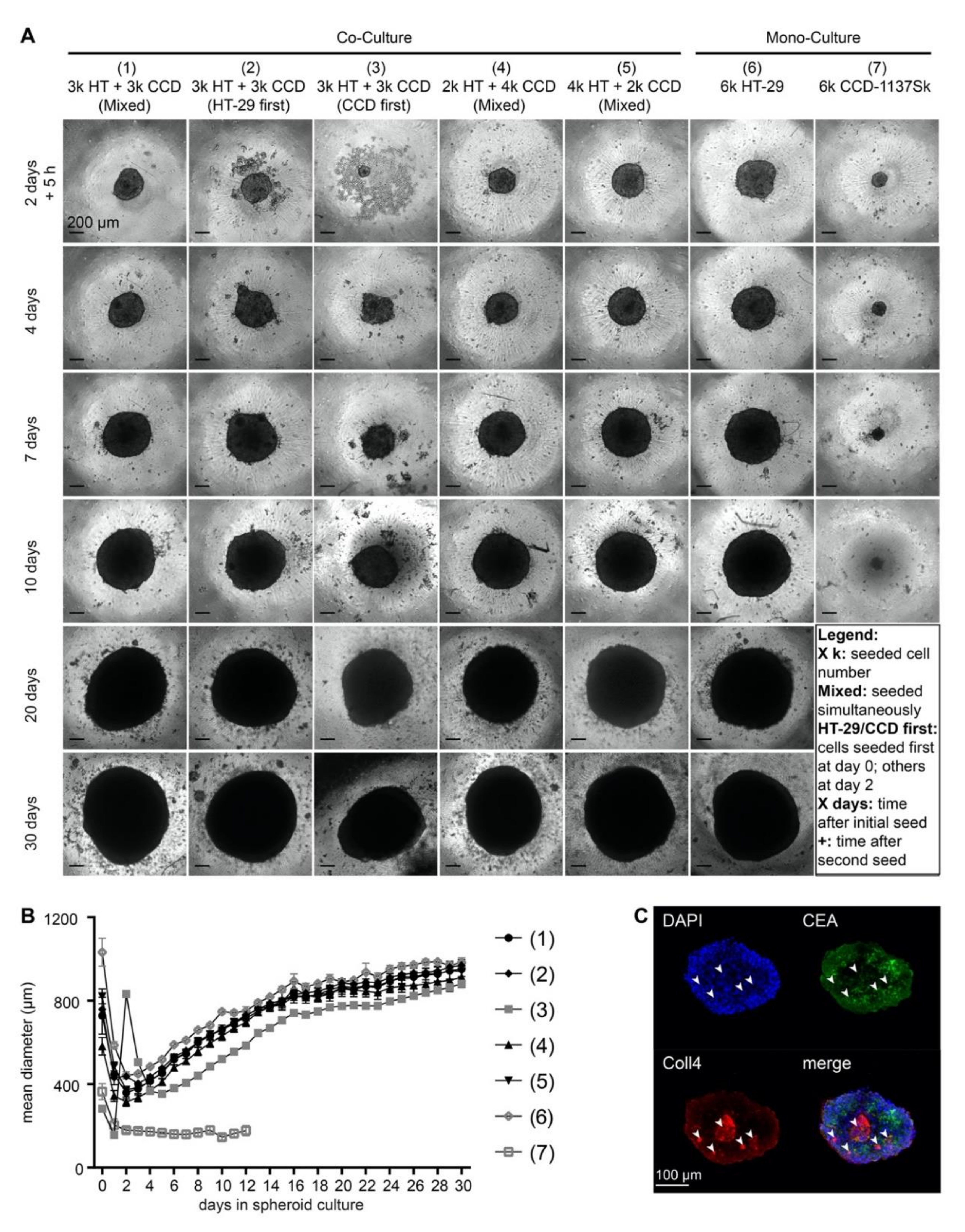
3.2. Addition of CCD-1137Sk Fibroblasts does not Alter Growth of HT-29 Spheroids
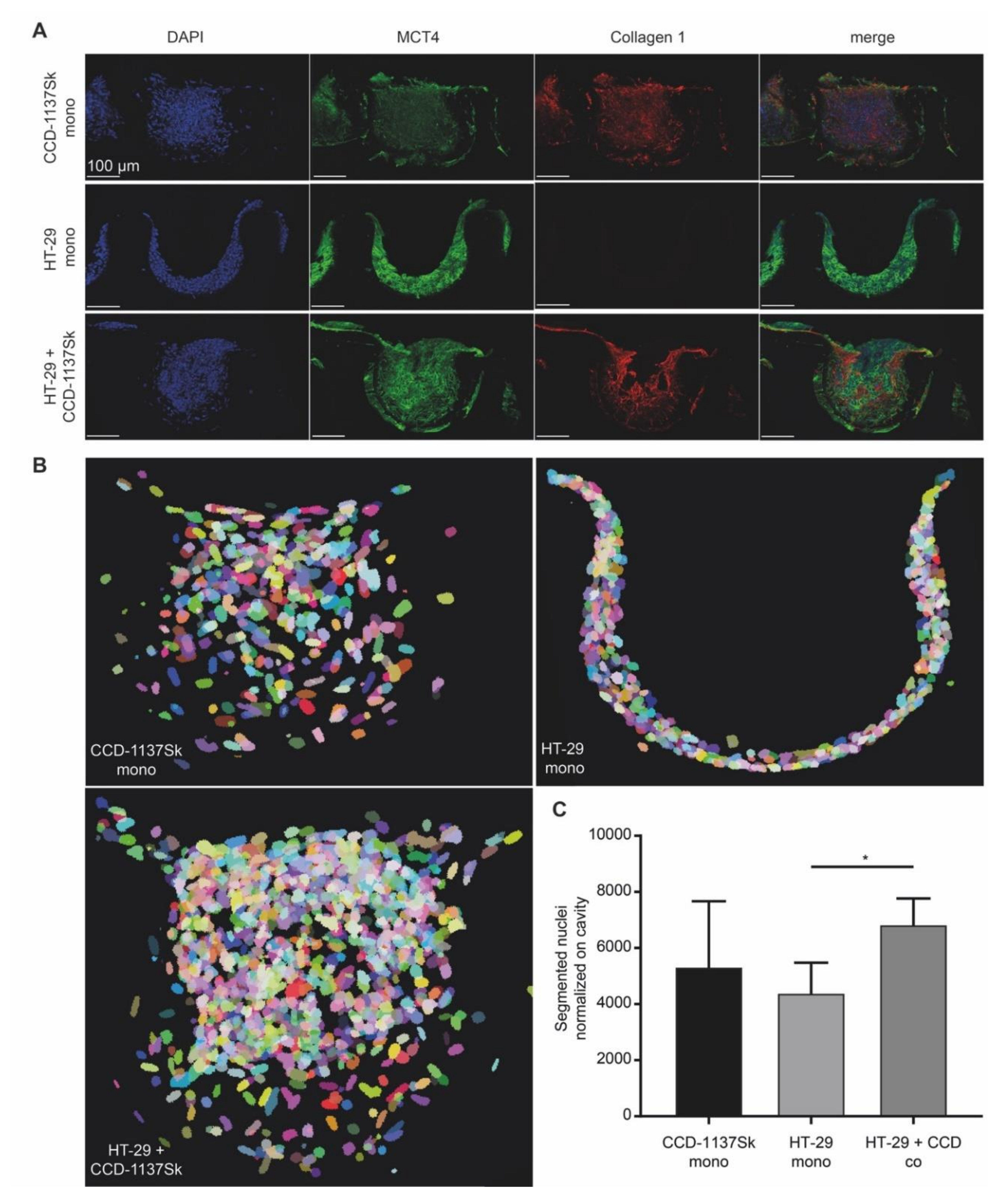
3.3. Growth of CCD-1137Sk and HT-29 Co-Cultures is Enhanced in Dynarray Chips
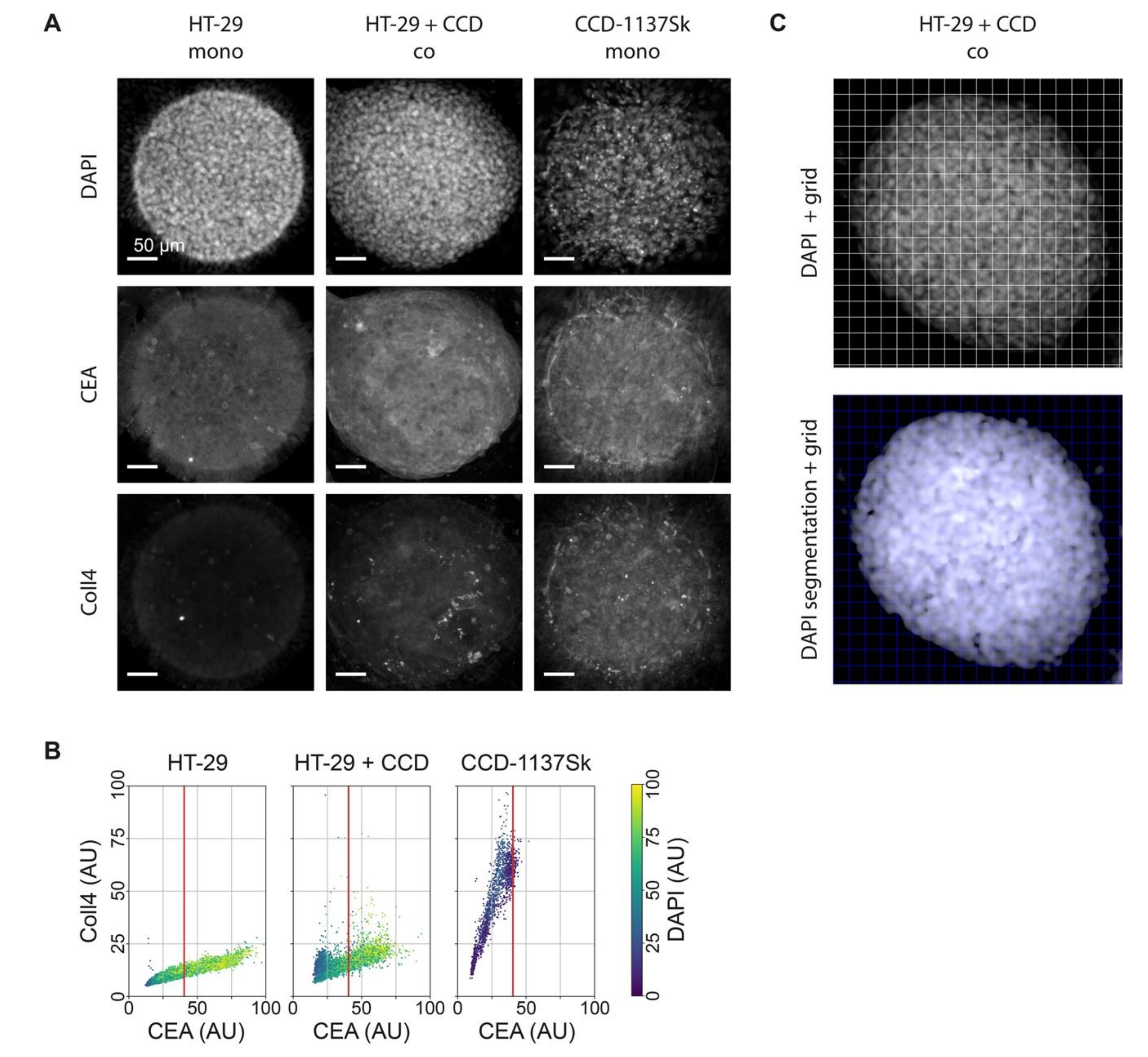
3.4. Fluorescence Staining of CEA, Coll4, and DAPI Shows Different Intensities in HT-29 and CCD-1137Sk Cells
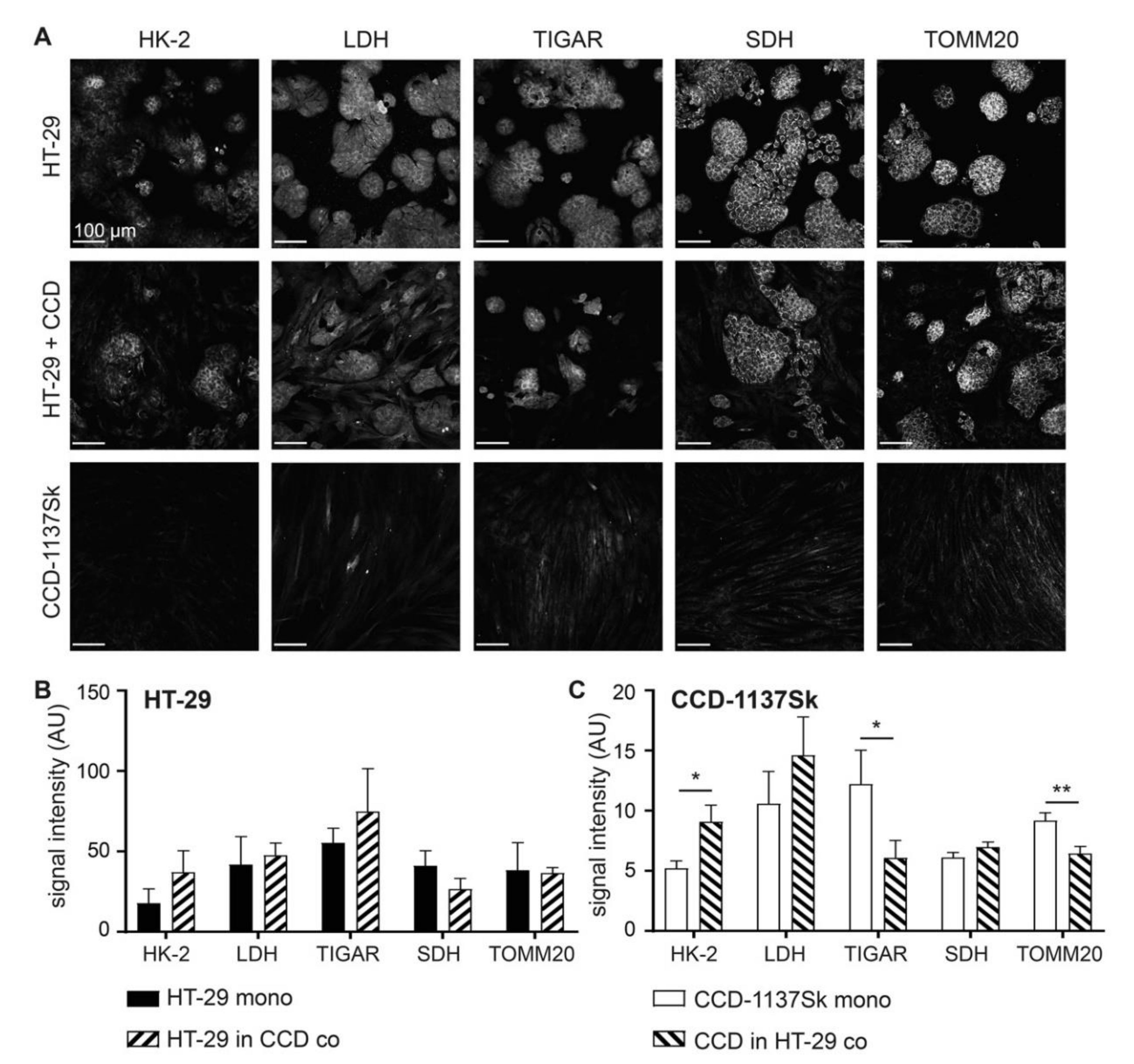
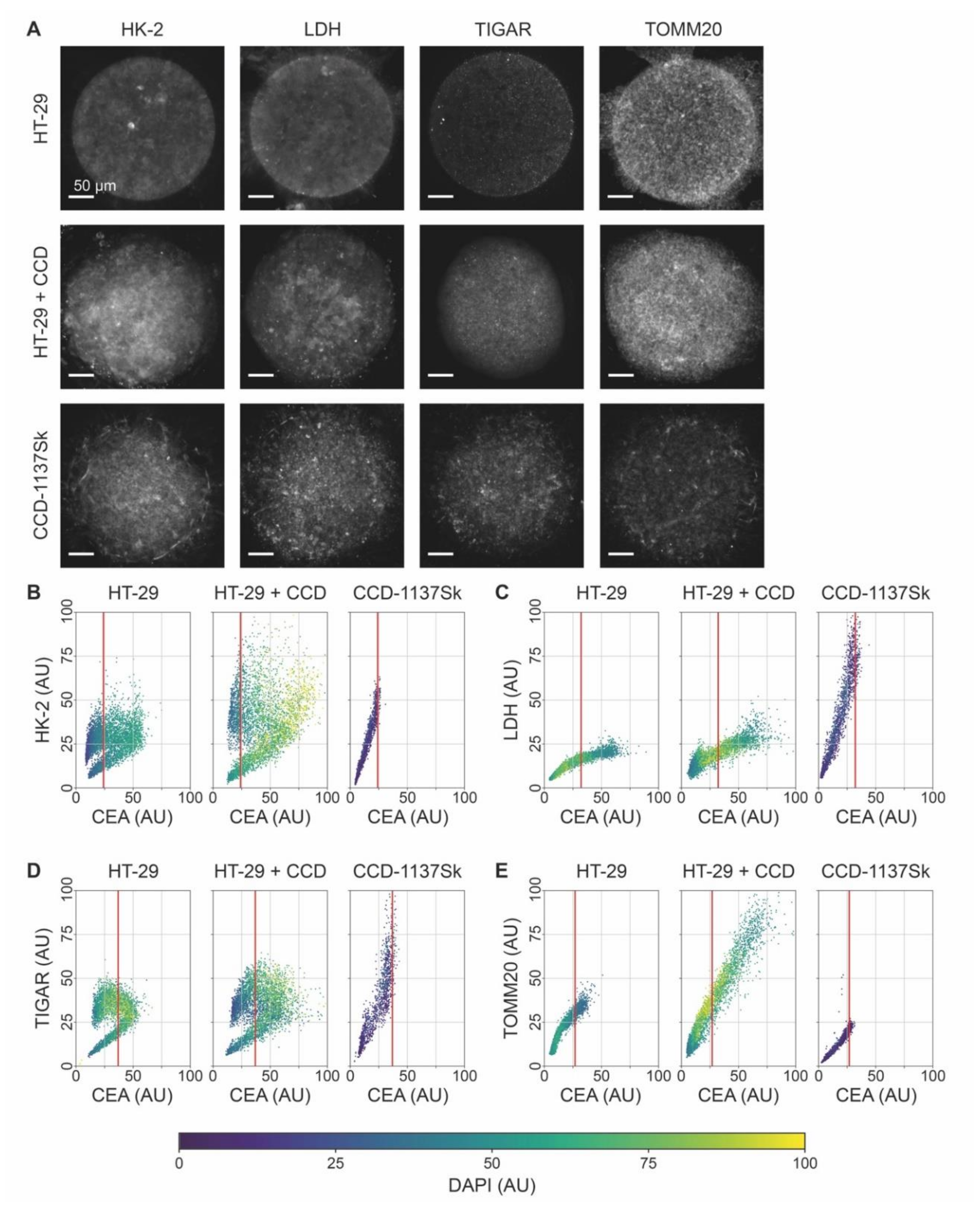
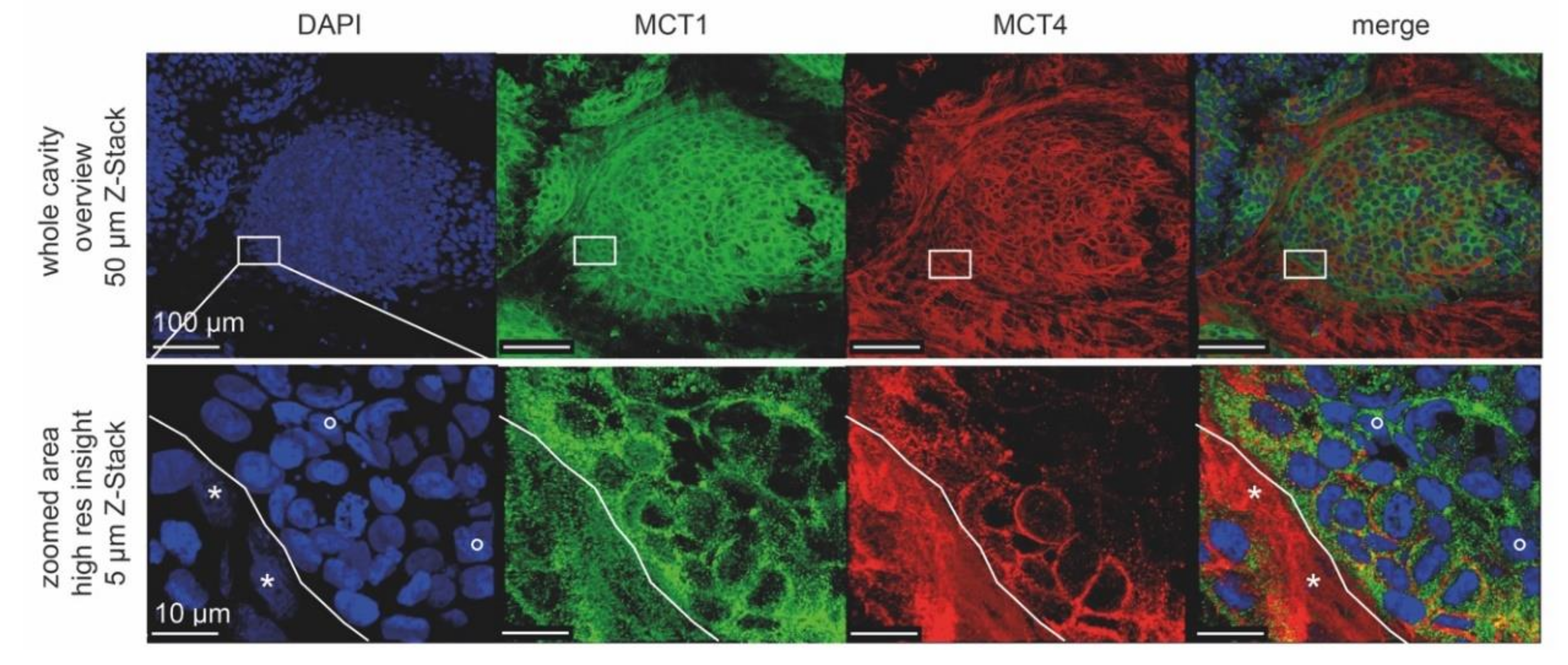
3.5. Fluorescence Signals of Metabolic Marker Proteins Show Differential Regulation in Dynarray Co-Cultures
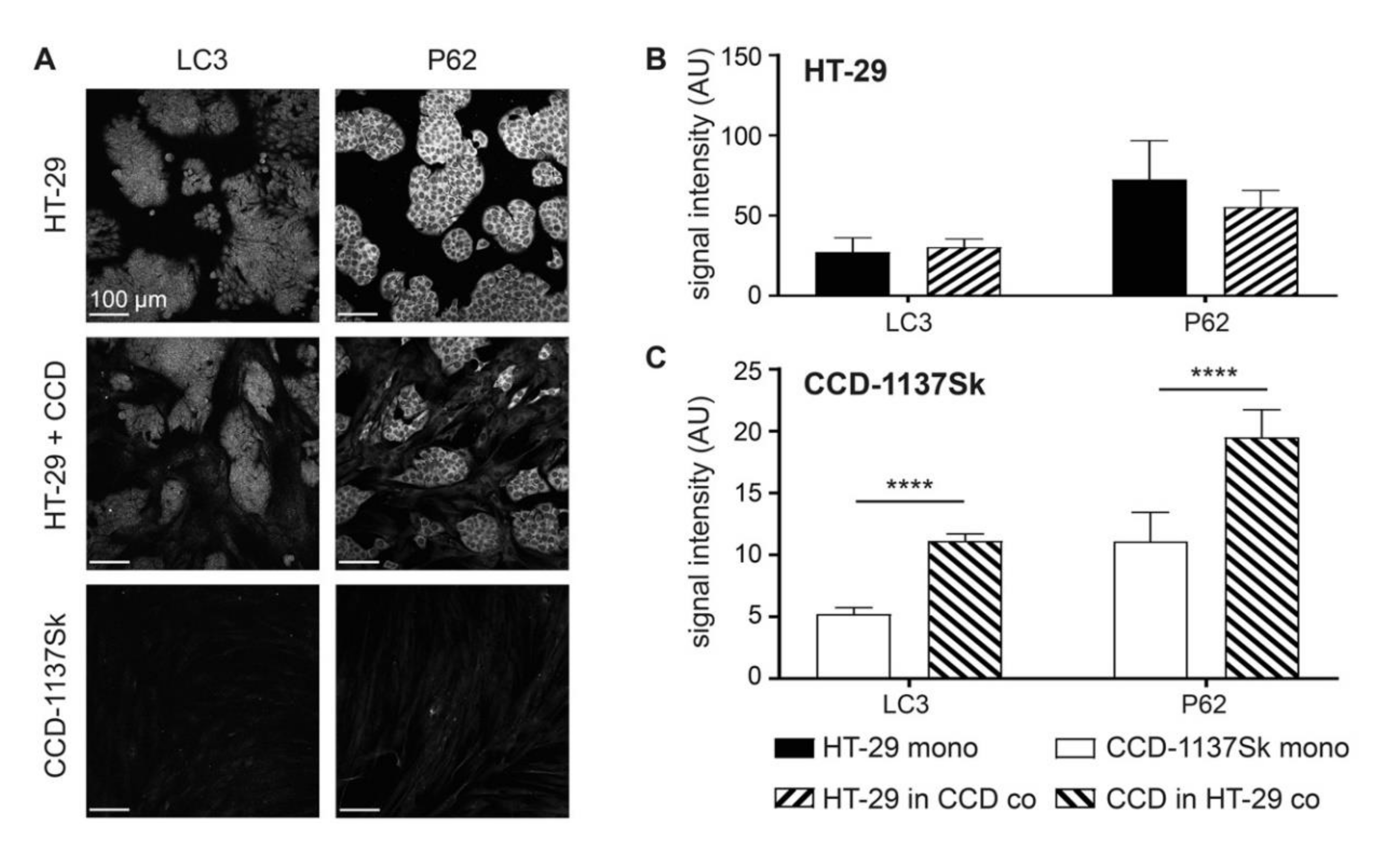
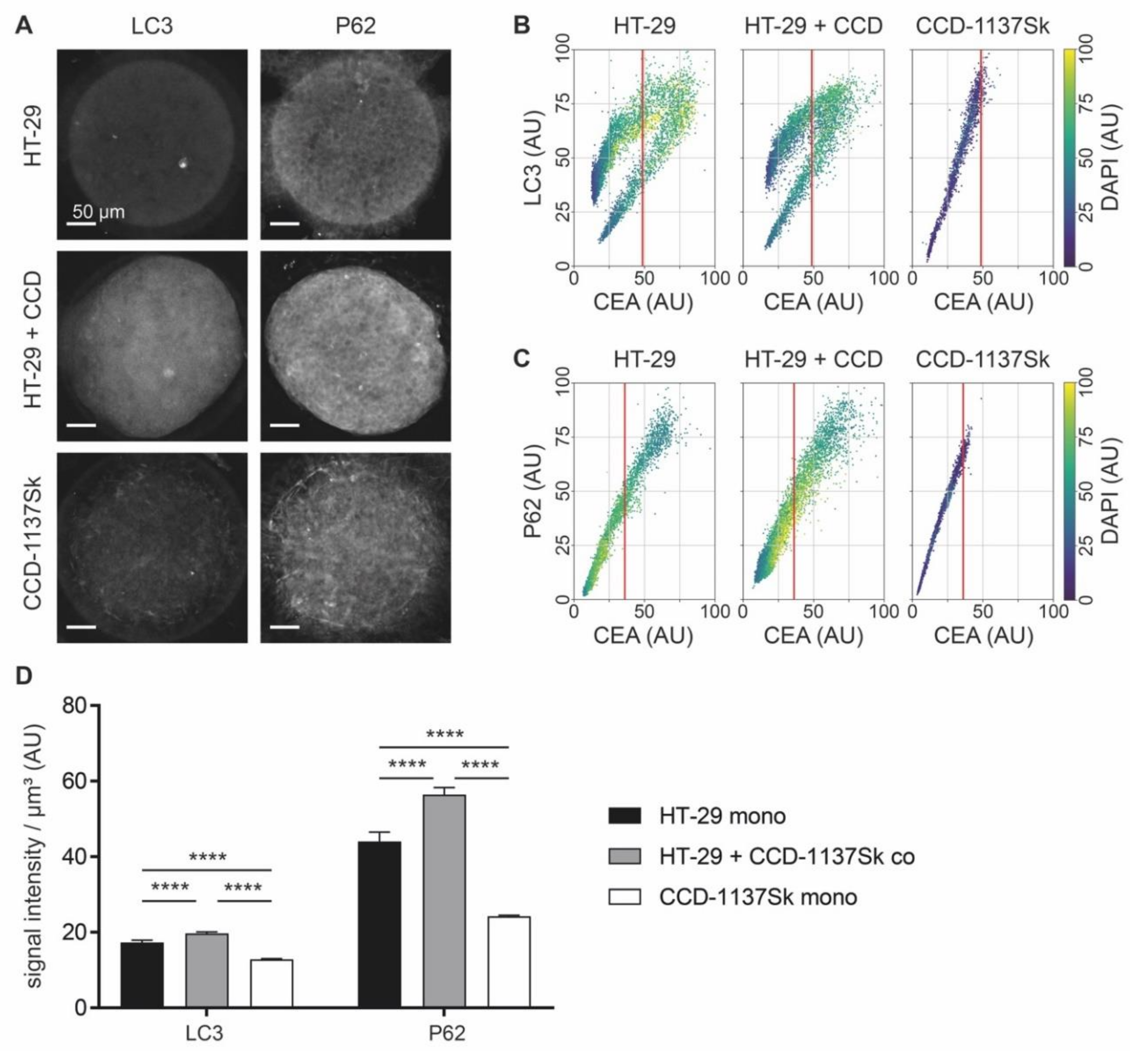
3.6. Autophagy is Enhanced in Dynarray Co-Cultures of HT-29 and CCD-1137Sk Cells
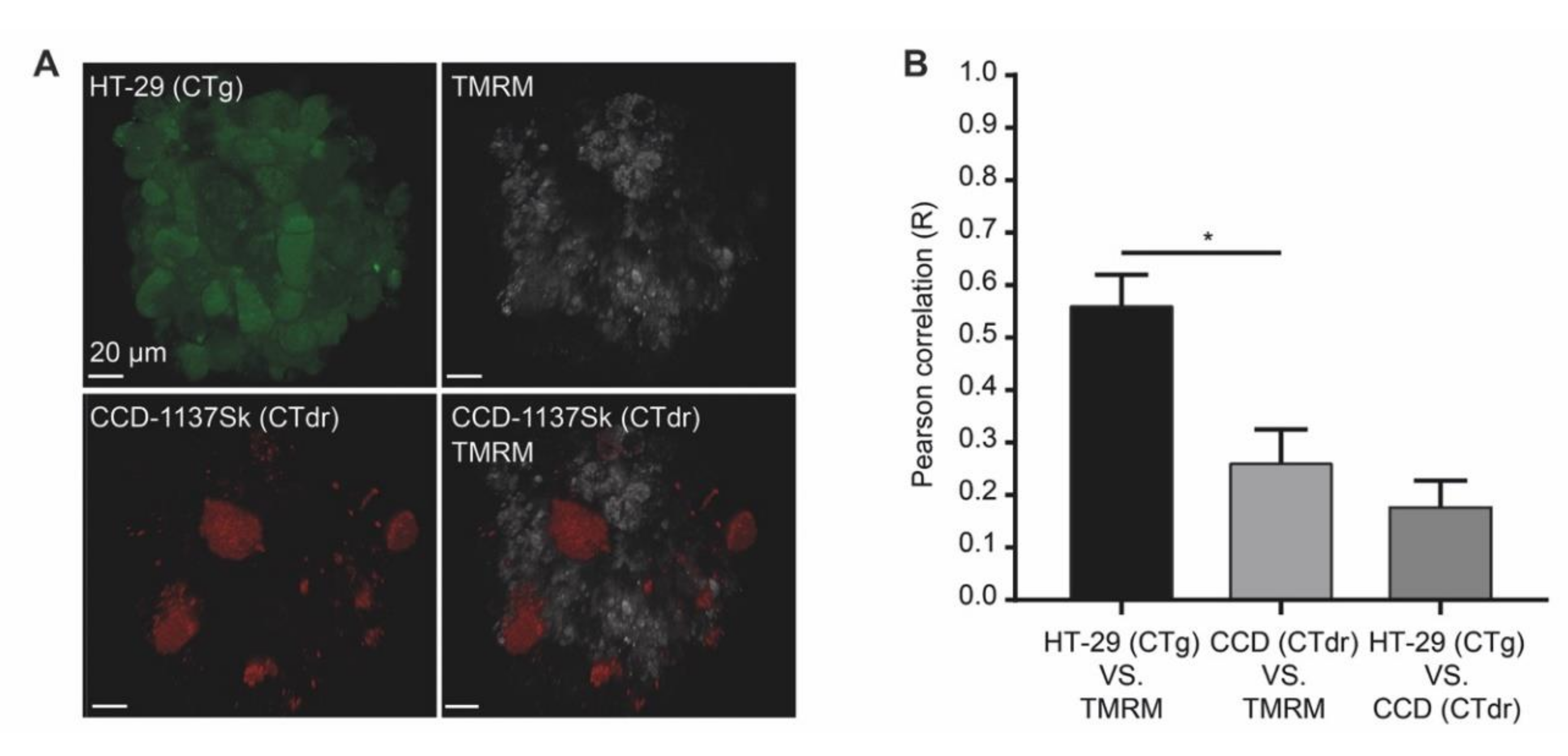
3.7. In Microarray Chip Co-Cultures, CCD-1137Sk Fibroblasts Show Low Mitochondrial Membrane Potential
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Løberg, M.; Kalager, M.; Holme, Ø.; Hoff, G.; Adami, H.-O.; Bretthauer, M. Long-term colorectal-cancer mortality after adenoma removal. N. Engl. J. Med. 2014, 371, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Sensi, F.; D’Angelo, E.; D’Aronco, S.; Molinaro, R.; Agostini, M. Preclinical three-dimensional colorectal cancer model: The next generation of in vitro drug efficacy evaluation. J. Cell. Physiol. 2018, 234, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, L.; Wang, P.P.; Roebothan, B.; Zhao, J.; Dicks, E.; Cotterchio, M.; Buehler, S.; Campbell, P.T.; McLaughlin, J.R.; et al. Association of total energy intake and macronutrient consumption with colorectal cancer risk: Results from a large population-based case-control study in Newfoundland and Labrador and Ontario, Canada. Nutr. J. 2012, 11, 18. [Google Scholar] [CrossRef]
- Benz, M.R.; Czernin, J.; Dry, S.M.; Tap, W.D.; Allen-Auerbach, M.S.; Elashoff, D.; Phelps, M.E.; Weber, W.A.; Eilber, F.C. Quantitative F18-fluorodeoxyglucose positron emission tomography accurately characterizes peripheral nerve sheath tumors as malignant or benign. Cancer 2010, 116, 451–458. [Google Scholar] [CrossRef]
- Xiao, Y.; Kim, D.; Dura, B.; Zhang, K.; Yan, R.; Li, H.; Han, E.; Ip, J.; Zou, P.; Liu, J.; et al. Ex vivo Dynamics of Human Glioblastoma Cells in a Microvasculature-on-a-Chip System Correlates with Tumor Heterogeneity and Subtypes. Adv. Sci. 2019, 6, 1801531. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Otto, A.M. Warburg effect(s)-a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016, 4, 5. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Chiavarina, B.; Whitaker-Menezes, D.; Migneco, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Tanowitz, H.B.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle 2010, 9, 3534–3551. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front. Oncol. 2014, 4, 820. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Zorzano, A.; Zughaier, S.M. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Smith, C.P.; Wilbur, R.R.; Cain, C.P.; Kallu, S.R.; Valasapalli, S.; Sahoo, A.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. An overview of MCT1 and MCT4 in GBM: Small molecule transporters with large implications. Am. J. Cancer Res. 2018, 8, 1967–1976. [Google Scholar] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Cannino, G.; Ciscato, F.; Masgras, I.; Sánchez-Martín, C.; Rasola, A. Metabolic plasticity of tumor cell mitochondria. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef]
- Georges, L.M.C.; de Wever, O.; Galván, J.A.; Dawson, H.; Lugli, A.; Demetter, P.; Zlobec, I. Cell Line Derived Xenograft Mouse Models Are a Suitable in vivo Model for Studying Tumor Budding in Colorectal Cancer. Front. Med. 2019, 6, 139. [Google Scholar] [CrossRef]
- Benton, G.; Kleinman, H.K.; George, J.; Arnaoutova, I. Multiple uses of basement membrane-like matrix (BME/Matrigel) in vitro and in vivo with cancer cells. Int. J. Cancer 2011, 128, 1751–1757. [Google Scholar] [CrossRef]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs. 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Chen, W.; Wong, C.; Vosburgh, E.; Levine, A.J.; Foran, D.J.; Xu, E.Y. High-throughput image analysis of tumor spheroids: A user-friendly software application to measure the size of spheroids automatically and accurately. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Keller, F.; Rudolf, R.; Hafner, M. Towards optimized breast cancer 3D spheroid mono- and co-culture models for pharmacological research and screening. J. Cell. Biotechnol. 2019, 5, 89–101. [Google Scholar] [CrossRef]
- Nürnberg, E.; Vitacolonna, M.; Klicks, J.; von Molitor, E.; Cesetti, T.; Keller, F.; Bruch, R.; Ertongur-Fauth, T.; Riedel, K.; Scholz, P.; et al. Routine Optical Clearing of 3D-Cell Cultures: Simplicity Forward. Front. Mol. Biosci. 2020, 7, 441. [Google Scholar] [CrossRef]
- Smith, P.; Reid, D.B.; Environment, C.; Palo, L.; Alto, P.; Smith, P.L. Otsu_1979_otsu_method. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef]
- Tiernan, J.P.; Perry, S.L.; Verghese, E.T.; West, N.P.; Yeluri, S.; Jayne, D.G.; Hughes, T.A. Carcinoembryonic antigen is the preferred biomarker for in vivo colorectal cancer targeting. Br. J. Cancer 2013, 108, 662–667. [Google Scholar] [CrossRef]
- Pastor-Pareja, J.C. Atypical basement membranes and basement membrane diversity—What is normal anyway? J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- da Cunha, B.R.; Domingos, C.; Stefanini, A.C.B.; Henrique, T.; Polachini, G.M.; Castelo-Branco, P.; Tajara, E.H. Cellular Interactions in the Tumor Microenvironment: The Role of Secretome. J. Cancer 2019, 10, 4574–4587. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, G.S.; Phillips, K.A. Precision medicine: From science to value. Health Aff. 2018, 37, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Kumar, C.; Bohl, S.; Klingmueller, U.; Mann, M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell. Proteom. 2009, 8, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Clevers, H. Studying cellular heterogeneity and drug sensitivity in colorectal cancer using organoid technology. Curr. Opin. Genet. Dev. 2018, 52, 117–122. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Rustamov, V.; Keller, F.; Klicks, J.; Hafner, M.; Rudolf, R. Bone Sialoprotein Shows Enhanced Expression in Early, High-Proliferation Stages of Three-Dimensional Spheroid Cell Cultures of Breast Cancer Cell Line MDA-MB-231. Front. Oncol. 2019, 9, 36. [Google Scholar] [CrossRef]
- Cruz-Acuña, R.; García, A.J. Engineered materials to model human intestinal development and cancer using organoids. Exp. Cell Res. 2019, 337, 109–114. [Google Scholar] [CrossRef]
- Shen, Y.; Zhu, D.; Lu, W.; Liu, B.; Li, Y.; Cao, S. The characteristics of intrinsic fluorescence of type I collagen influenced by collagenase I. Appl. Sci. 2018, 8, 1947. [Google Scholar] [CrossRef]
- Solanki, A.; King, D.; Thibault, G.; Wang, L.; Gibbs, S.L. Quantification of fluorophore distribution and therapeutic response in matched in vivo and ex vivo pancreatic cancer model systems. PLoS ONE 2020, 15, 1–18. [Google Scholar] [CrossRef]
- Sharick, J.T.; Jeffery, J.J.; Karim, M.R.; Walsh, C.M.; Esbona, K.; Cook, R.S.; Skala, M.C. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia 2019, 21, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Blank, A.; Roberts, D.E.; Dawson, H.; Zlobec, I.; Lugli, A. Tumor Heterogeneity in Primary Colorectal Cancer and Corresponding Metastases. Does the Apple Fall Far From the Tree? Front. Med. 2018, 5, 234. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Dueñas-González, A. Understanding tumor anabolism and patient catabolism in cancer-associated cachexia. Am. J. Cancer Res. 2017, 7, 1107–1135. [Google Scholar] [PubMed]
- Bovenzi, C.D.; Hamilton, J.; Tassone, P.; Johnson, J.; Cognetti, D.M.; Luginbuhl, A.; Keane, W.M.; Zhan, T.; Tuluc, M.; Bar-Ad, V.; et al. Prognostic Indications of Elevated MCT4 and CD147 across Cancer Types: A Meta-Analysis. Biomed Res. Int. 2015, 2015, 242437. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, F.; Bruch, R.; Schneider, R.; Meier-Hubberten, J.; Hafner, M.; Rudolf, R. A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro. Cells 2020, 9, 1900. https://doi.org/10.3390/cells9081900
Keller F, Bruch R, Schneider R, Meier-Hubberten J, Hafner M, Rudolf R. A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro. Cells. 2020; 9(8):1900. https://doi.org/10.3390/cells9081900
Chicago/Turabian StyleKeller, Florian, Roman Bruch, Richard Schneider, Julia Meier-Hubberten, Mathias Hafner, and Rüdiger Rudolf. 2020. "A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro" Cells 9, no. 8: 1900. https://doi.org/10.3390/cells9081900
APA StyleKeller, F., Bruch, R., Schneider, R., Meier-Hubberten, J., Hafner, M., & Rudolf, R. (2020). A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro. Cells, 9(8), 1900. https://doi.org/10.3390/cells9081900