Highlights on Genomics Applications for Lysosomal Storage Diseases



Abstract
1. Introduction
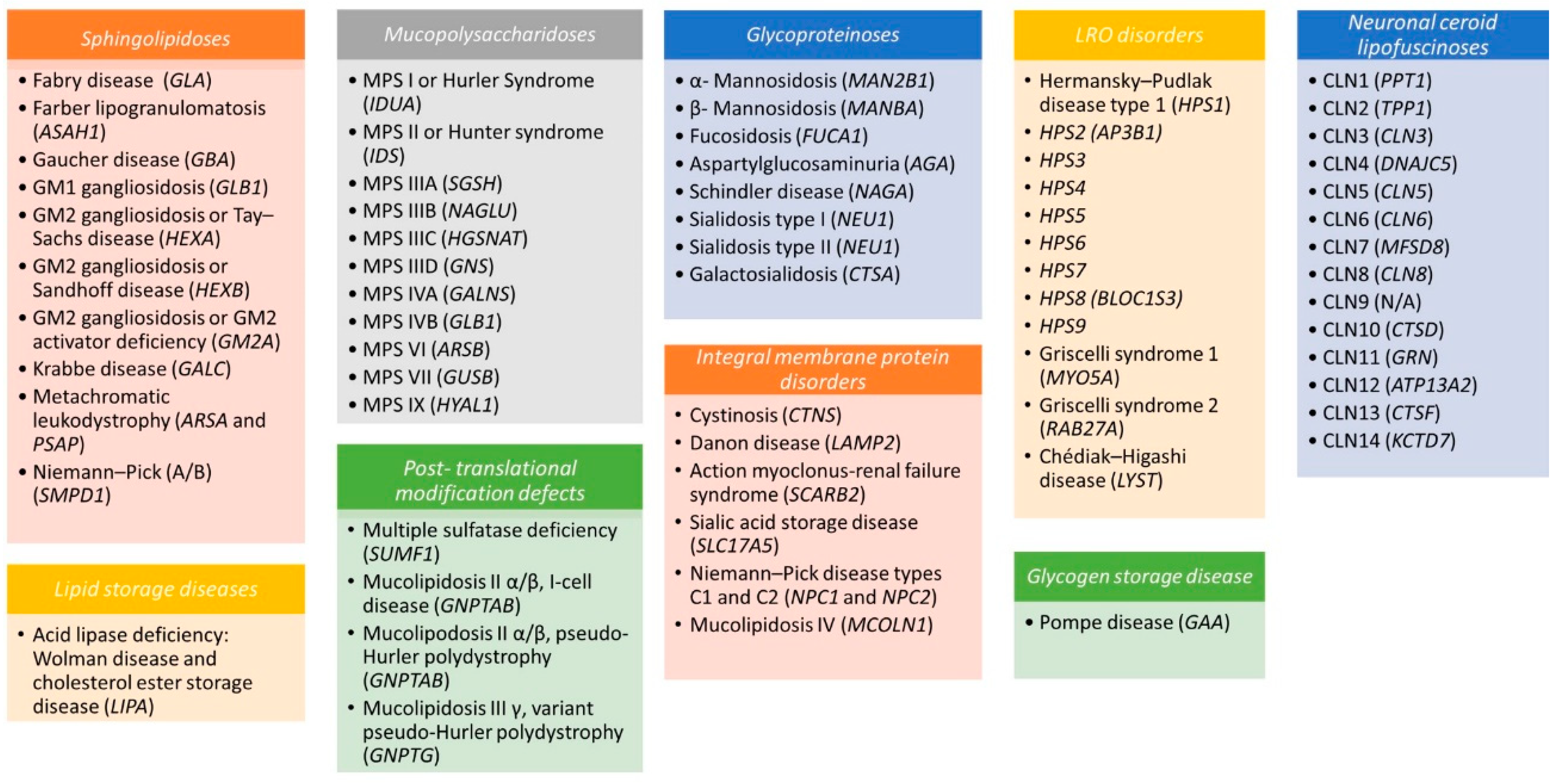
2. The Biology of Lysosomes
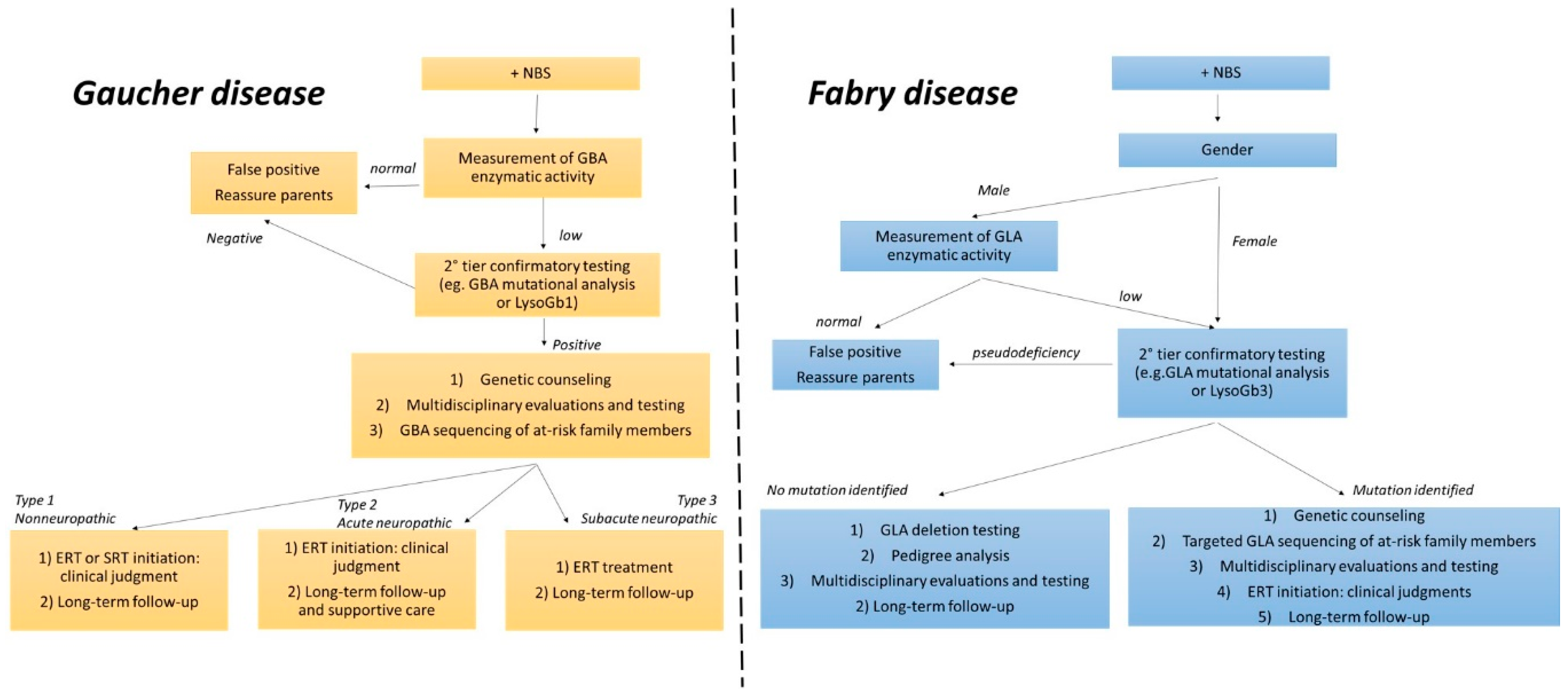
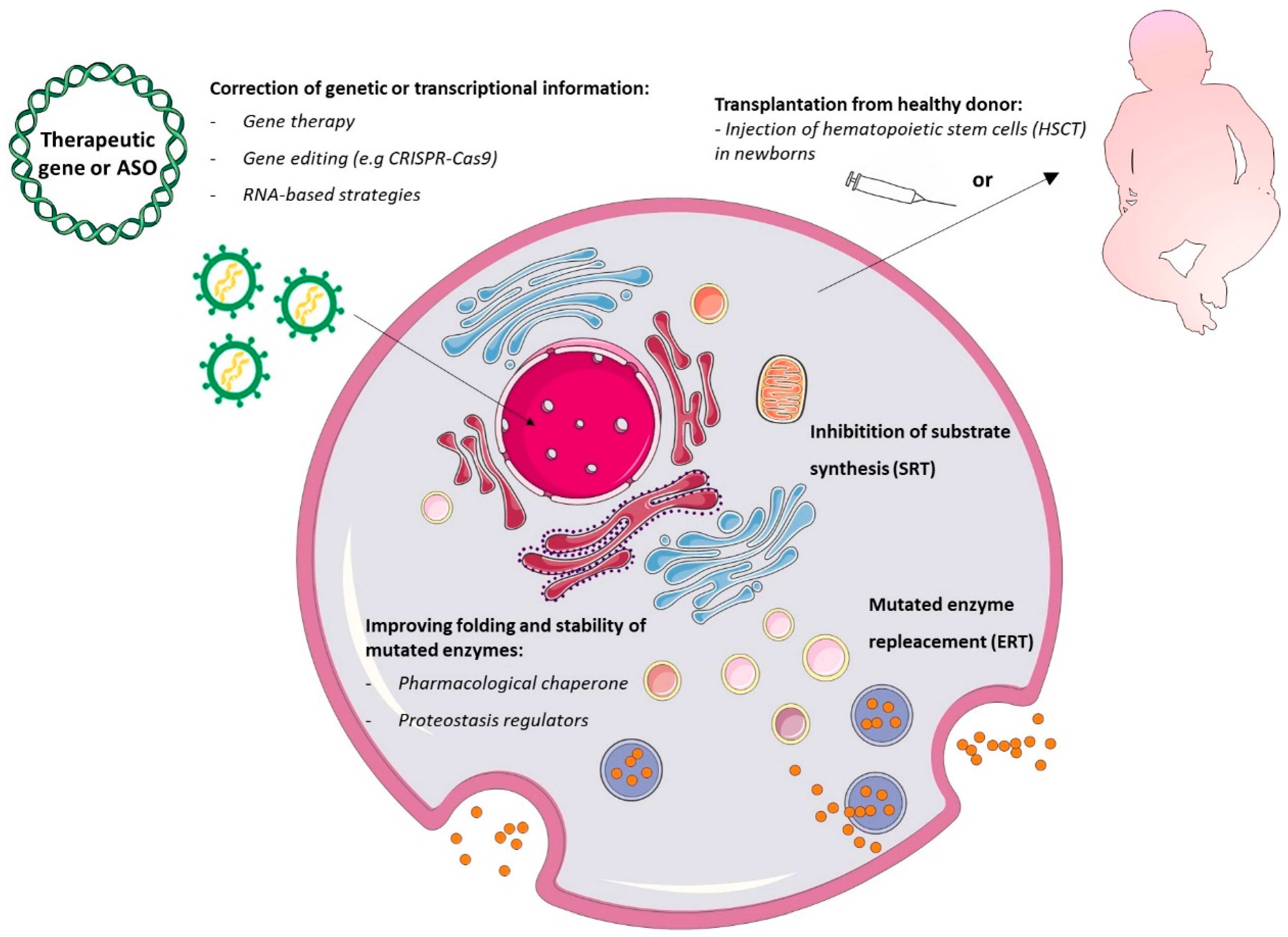
3. Diagnosis and Therapeutic Strategies
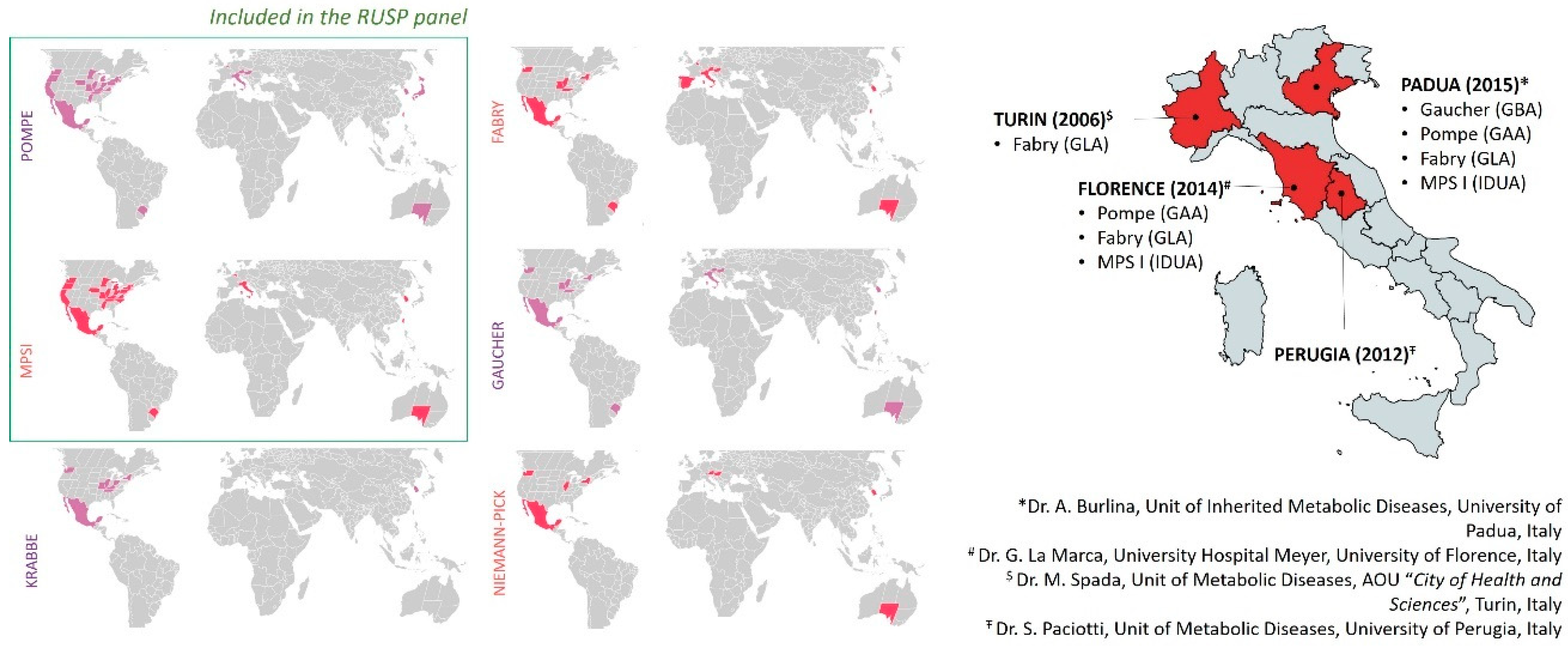
4. LSD Worldwide Newborn Screenings and Methodological Approaches
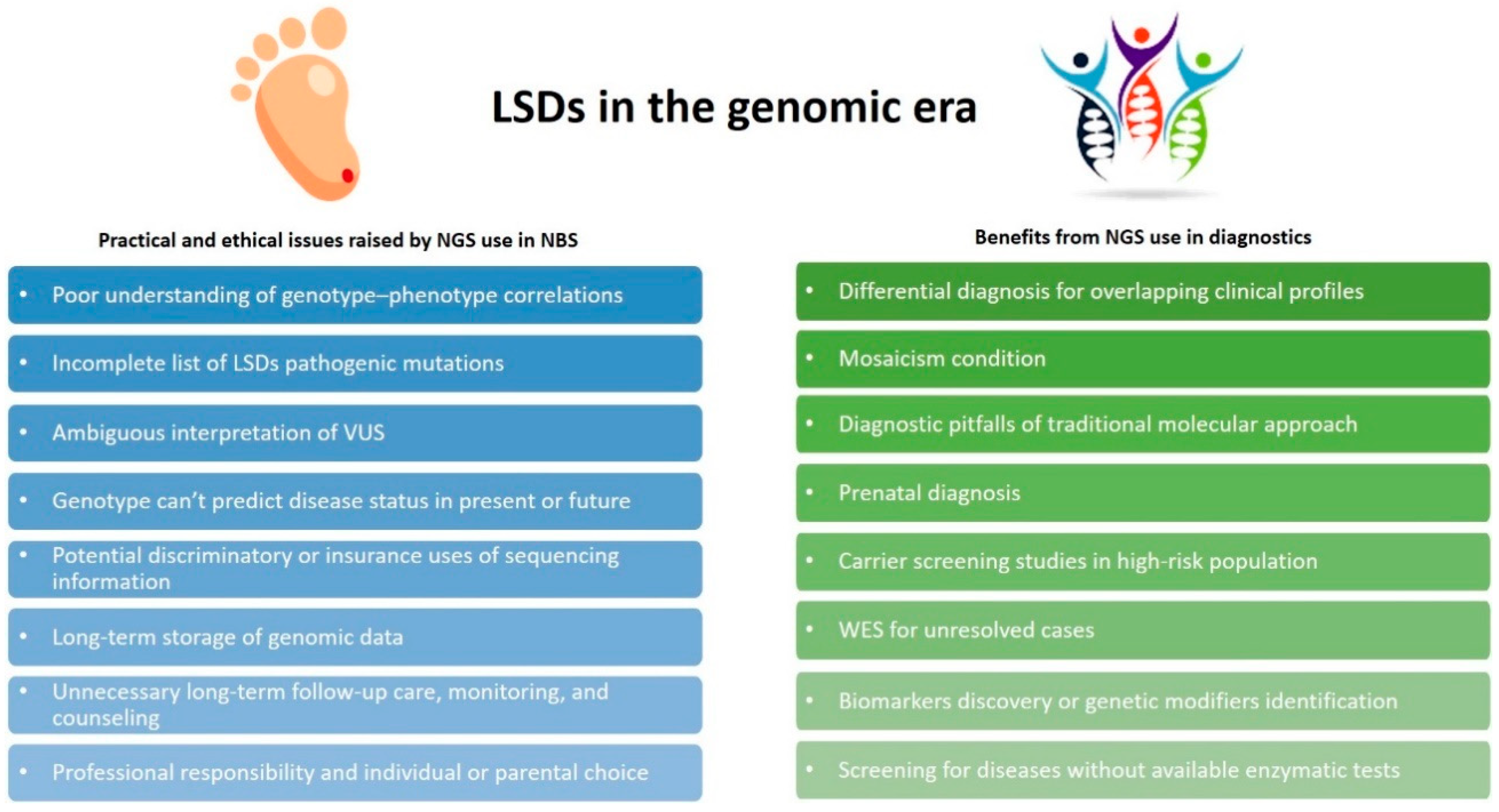
5. Opportunities and Challenges for Genomics in LSDs
6. Second-Tier Confirmatory Biomarkers: Which One?
7. The Importance of a Timely Diagnosis
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.F.G.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.M.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Wolf, P.; Levy, O.A.; Kang, U.J.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.H.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Lysosomal storage disorders: 4 big questions. Nature 2016, 537, S165. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front Pharm. 2017, 8, 448. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Kingma, S.D.K.; Bodamer, O.A.; Wijburg, F.A. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 145–157. [Google Scholar] [CrossRef]
- Mokhtariye, A.; Hagh-Nazari, L.; Varasteh, A.R.; Keyfi, F. Diagnostic methods for Lysosomal Storage Disease. Rep. Biochem. Mol. Biol. 2019, 7, 119–128. [Google Scholar] [PubMed]
- Verma, J.; Bijarnia-Mahay, S.; Verma, I.C. Prenatal Diagnosis of Lysosomal Storage Disorders Using Chorionic Villi. In Lysosomes; Humana Press: New York, NY, USA, 2017; Volume 1594, pp. 265–291. [Google Scholar]
- Li, D.; Lin, Y.; Huang, Y.; Zhang, W.; Jiang, M.; Li, X.; Zhao, X.; Sheng, H.; Yin, X.; Su, X.; et al. Early prenatal diagnosis of lysosomal storage disorders by enzymatic and molecular analysis. Prenat. Diagn. 2018, 38, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Pignata, C.; Vajro, P.; Salerno, M. New strategies for the treatment of lysosomal storage diseases (review). Int. J. Mol. Med. 2013, 31, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr. Ann. 2018, 47, e191–e197. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [Google Scholar] [CrossRef]
- Poswar, F.O.; Vairo, F.; Burin, M.; Michelin-Tirelli, K.; Brusius-Facchin, A.C.; Kubaski, F.; Souza, C.F.M.; Baldo, G.; Giugliani, R. Lysosomal diseases: Overview on current diagnosis and treatment. Genet. Mol. Biol. 2019, 42, 165–177. [Google Scholar] [CrossRef]
- Beck, M. Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 2018, 60, 13–18. [Google Scholar] [CrossRef]
- Lund, T.C. Hematopoietic stem cell transplant for lysosomal storage diseases. Pediatr. Endocrinol. Rev. 2013, 11, 91–98. [Google Scholar]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Kishnani, P.; Tarnopolsky, M.; Roberts, M.; Sivakumar, K.; Dasouki, M.; Dimachkie, M.M.; Finanger, E.; Goker-Alpan, O.; Guter, K.A.; Mozaffar, T.; et al. Duvoglustat HCl Increases Systemic and Tissue Exposure of Active Acid α-Glucosidase in Pompe Patients Co-administered with Alglucosidase α. Mol. Ther. 2017, 25, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Nagree, M.S.; Scalia, S.; McKillop, W.M.; Medin, J.A. An update on gene therapy for lysosomal storage disorders. Expert Opin. Biol. Ther. 2019, 19, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.S.; Davidson, B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006, 13, 839–849. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, T.G.; da Silveira Matte, U.; Giugliani, R.; Baldo, G. Genome Editing: Potential Treatment for Lysosomal Storage Diseases. Curr. Stem Cell Rep. 2015, 1, 9–15. [Google Scholar] [CrossRef]
- Christensen, C.; Choy, F. A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells. Diseases 2017, 5, 6. [Google Scholar] [CrossRef]
- Poletto, E.; Baldo, G.; Gomez-Ospina, N. Genome Editing for Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 500. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Tăbăran, A.-F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay–Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef]
- Dardis, A.; Buratti, E. Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders. Genes 2018, 9, 73. [Google Scholar] [CrossRef]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef]
- Ferri, L.; Covello, G.; Caciotti, A.; Guerrini, R.; Denti, M.A.; Morrone, A. Double-target Antisense U1snRNAs Correct Mis-splicing Due to c.639+861C>T and c.639+919G>A GLA Deep Intronic Mutations. Mol. Ther. Nucleic Acids 2016, 5, e380. [Google Scholar] [CrossRef]
- Rodríguez-Pascau, L.; Coll, M.J.; Vilageliu, L.; Grinberg, D. Antisense oligonucleotide treatment for a pseudoexon-generating mutation in theNPC1gene causing Niemann-Pick type C disease. Hum. Mutat. 2009, 30, E993–E1001. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; Ham, K.A.; Tchan, M.; Johnsen, R.; Schnell, F.J.; Fletcher, S.; Wilton, S.D. Splice modulating antisense oligonucleotides restore some acid-alpha-glucosidase activity in cells derived from patients with late-onset Pompe disease. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, E.; Bergsma, A.J.; Pijnenburg, J.M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol. Ther. Nucleic Acids 2017, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening for Disease; World Health Organization: Geneva, Switzerland, 1968; p. 164. [Google Scholar]
- Andermann, A. Revisting wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bull. World Health Organ. 2008, 86, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M. Newborn Screening for Lysosomal Storage Diseases: Methodologies, Screen Positive Rates, Normalization of Datasets, Second-Tier Tests, and Post-Analysis Tools. Int. J. Neonatal Screen. 2018, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S. Newborn Screening for Lysosomal Storage Disorders. J. Pediatric Health Care 2018, 32, 285–294. [Google Scholar] [CrossRef]
- Schielen, P.; Kemper, E.; Gelb, M. Newborn Screening for Lysosomal Storage Diseases: A Concise Review of the Literature on Screening Methods, Therapeutic Possibilities and Regional Programs. Int. J. Neonatal Screen. 2017, 3, 6. [Google Scholar] [CrossRef]
- Taruscio, D.; Carbone, P.; Polizzi, A. Expanded Newborn Screening: A Chess Board Motif in Public Health. J. Pediatr. Biochem. 2016, 6, 66–70. [Google Scholar]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef]
- Burlina, A.; Gueraldi, D.; Polo, G.; Rubert, L.; Cazzorla, C.; Giuliani, A.; Burlina, A. Newborn screening for MPS I: The clinical benefit. Mol. Genet. Metab. 2020, 129, S35–S36. [Google Scholar] [CrossRef]
- Burlina, A.; Polo, G.; Gueraldi, D.; Rubert, L.; Cazzorla, C.; Giuliani, A.; Burlina, A. High incidence of Gaucher disease in northeast Italy: Results from lysosomal newborn screening. Mol. Genet. Metab. 2020, 129, S36. [Google Scholar] [CrossRef]
- Paciotti, S.; Persichetti, E.; Pagliardini, S.; Deganuto, M.; Rosano, C.; Balducci, C.; Codini, M.; Filocamo, M.; Menghini, A.R.; Pagliardini, V.; et al. First pilot newborn screening for four lysosomal storage diseases in an Italian region: Identification and analysis of a putative causative mutation in the GBA gene. Clin. Chim. Acta. 2012, 413, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Scolamiero, E.; Casetta, B.; Malvagia, S.; Tanigawa, T.; Forni, G.; Funghini, S.; Mura, M.; Raspini, F.; Poggiali, S.; la Marca, G. Development of a fast LC-MS/MS protocol for combined measurement of six LSDs on dried blood spot in a newborn screening program. J. Pharm. Biomed. Anal. 2019, 165, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.; Malvagia, S.; Funghini, S.; Scolamiero, E.; Mura, M.; Della Bona, M.; Villanelli, F.; Damiano, R.; la Marca, G. LC-MS/MS method for simultaneous quantification of heparan sulfate and dermatan sulfate in urine by butanolysis derivatization. Clin. Chim. Acta 2019, 488, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; Lukacs, Z.; Ranieri, E.; Schielen, P. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2019, 5, 1. [Google Scholar] [CrossRef]
- Gelb, M.H.; Scott, C.R.; Turecek, F. Newborn screening for lysosomal storage diseases. Clin. Chem. 2015, 61, 335–346. [Google Scholar] [CrossRef]
- Yu, C.; Sun, Q.; Zhou, H. Enzymatic Screening and Diagnosis of Lysosomal Storage Diseases. N. Am. J. Med. Sci. 2013, 6, 186–193. [Google Scholar] [CrossRef]
- Millington, D.; Bali, D. Current State of the Art of Newborn Screening for Lysosomal Storage Disorders. Int. J. Neonatal Screen. 2018, 4, 24. [Google Scholar] [CrossRef]
- Berg, J.S.; Agrawal, P.B.; Bailey, D.B., Jr.; Beggs, A.H.; Brenner, S.E.; Brower, A.M.; Cakici, J.A.; Ceyhan-Birsoy, O.; Chan, K.; Chen, F.; et al. Newborn Sequencing in Genomic Medicine and Public Health. Pediatrics 2017, 139, e20162252. [Google Scholar] [CrossRef]
- Friedman, E. Next generation sequencing for newborn screening: Are we there yet? Genet. Res. 2015, 97, e17. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Yubero, D.; Brandi, N.; Ormazabal, A.; Garcia-Cazorla, À.; Pérez-Dueñas, B.; Campistol, J.; Ribes, A.; Palau, F.; Artuch, R.; et al. Targeted Next Generation Sequencing in Patients with Inborn Errors of Metabolism. PLoS ONE 2016, 11, e0156359. [Google Scholar]
- Málaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Saraiva-Pereira, M.L.; de Souza, C.F.M.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019, 42, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Seneca, S.; Stouffs, K.; Lissens, W.; Jansen, A.; Laeremans, H.; Verloo, P.; Schoonjans, A.S.; Meuwissen, M.; Barca, D.; et al. Clinical implementation of gene panel testing for lysosomal storage diseases. Mol. Genet. Genom. Med. 2018, e00527. [Google Scholar] [CrossRef]
- Komlosi, K.; Sólyom, A.; Beck, M. The Role of Next-Generation Sequencing in the Diagnosis of Lysosomal Storage Disorders. J. Inborn Errors Metab. Screen. 2016, 4, 232640981666937. [Google Scholar] [CrossRef]
- Mori, M.; Haskell, G.; Kazi, Z.; Zhu, X.; DeArmey, S.M.; Goldstein, J.L.; Bali, D.; Rehder, C.; Cirulli, E.T.; Kishnani, P.S. Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol. Genet. Metab. 2017, 122, 189–197. [Google Scholar] [CrossRef]
- Di Fruscio, G.; Schulz, A.; De Cegli, R.; Savarese, M.; Mutarelli, M.; Parenti, G.; Banfi, S.; Braulke, T.; Nigro, V.; Ballabio, A. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 2015, 11, 928–938. [Google Scholar] [CrossRef]
- Arjunan, A.; Litwack, K.; Collins, N.; Charrow, J. Carrier screening in the era of expanding genetic technology. Genet. Med. 2016, 18, 1214–1217. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Greger, V.; Strovel, E.T.; Blitzer, M.G.; Umbarger, M.A.; Kennedy, C.; Bishop, B.; Saunders, P.; Porreca, G.J.; Schienda, J.; et al. Next-generation DNA sequencing of HEXA: A step in the right direction for carrier screening. Mol. Genet. Genom. Med. 2013, 1, 260–268. [Google Scholar] [CrossRef]
- Yoshida, S.; Kido, J.; Matsumoto, S.; Momosaki, K.; Mitsubuchi, H.; Shimazu, T.; Sugawara, K.; Endo, F.; Nakamura, K. Prenatal diagnosis of Gaucher disease using next-generation sequencing. Pediatr. Int. 2016, 58, 946–949. [Google Scholar] [CrossRef]
- Tsai, A.C.-H.; Hung, Y.-W.; Harding, C.; Koeller, D.M.; Wang, J.; Wong, L.-J.C. Next generation deep sequencing corrects diagnostic pitfalls of traditional molecular approach in a patient with prenatal onset of Pompe disease. Am. J. Med Genet. Part A 2017, 173, 2500–2504. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, A.; D’Avanzo, F.; Bertoldi, L.; Zampieri, G.; Feltrin, E.; De Pascale, F.; Rampazzo, A.; Forzan, M.; Valle, G.; Tomanin, R. Setup and Validation of a Targeted Next-Generation Sequencing Approach for the Diagnosis of Lysosomal Storage Disorders. J. Mol. Diagn. 2020, 22, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, Y.; Gedvilaite, E.; Loh, J.W.; Lin, T.; Liu, X.; Liu, C.-G.; Kumar, D.; Donnelly, R.; Raymond, K.; et al. Using whole-exome sequencing to investigate the genetic bases of lysosomal storage diseases of unknown etiology. Hum. Mutat. 2017, 38, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, J.; Linpeng, S.; Li, Z.; Tan, H.; Wu, L. Identification of Five Novel Mutations Causing Rare Lysosomal Storage Diseases. Med. Sci. Monit. 2019, 25, 7634–7644. [Google Scholar] [CrossRef]
- Davidson, B.A.; Hassan, S.; Garcia, E.J.; Tayebi, N.; Sidransky, E. Exploring genetic modifiers of Gaucher disease: The next horizon. Hum. Mutat. 2018, 39, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, J.A.; Guecaimburu Ehuletche, M.d.R.; Perna, A.; Dubrovsky, A.; Franca, M.C.; Vargas, S.; Hegde, M.; Claeys, K.G.; Straub, V.; Daba, N.; et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J. Rare Dis. 2020, 15, 11. [Google Scholar] [CrossRef]
- Lévesque, S.; Auray-Blais, C.; Gravel, E.; Boutin, M.; Dempsey-Nunez, L.; Jacques, P.-E.; Chenier, S.; Larue, S.; Rioux, M.-F.; Al-Hertani, W.; et al. Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing. Orphanet J. Rare Dis. 2016, 11, 8. [Google Scholar] [CrossRef]
- Mahdieh, N.; Mikaeeli, S.; Tavasoli, A.R.; Rezaei, Z.; Maleki, M.; Rabbani, B. Genotype, phenotype and in silico pathogenicity analysis of HEXB mutations: Panel based sequencing for differential diagnosis of gangliosidosis. Clin. Neurol. Neurosurg. 2018, 167, 43–53. [Google Scholar] [CrossRef]
- in’t Groen, S.L.M.; de Faria, D.O.S.; Iuliano, A.; van den Hout, J.M.P.; Douben, H.; Dijkhuizen, T.; Cassiman, D.; Witters, P.; Barba Romero, M.-Á.; de Klein, A.; et al. Novel GAA Variants and Mosaicism in Pompe Disease Identified by Extended Analyses of Patients with an Incomplete DNA Diagnosis. Mol. Ther. Methods Clin. Dev. 2020, 17, 337–348. [Google Scholar] [CrossRef]
- Pianese, L.; Fortunato, A.; Silvestri, S.; Solano, F.G.; Burlina, A.; Burlina, A.P.; Ragno, M. Maternal germline mosaicism in Fabry disease. Neurol. Sci. 2019, 40, 1279–1281. [Google Scholar] [CrossRef]
- Johnston, J.; Lantos, J.D.; Goldenberg, A.; Chen, F.; Parens, E.; Koenig, B.A.; members of the, N.E.; Policy Advisory, B. Sequencing Newborns: A Call for Nuanced Use of Genomic Technologies. Hastings Cent. Rep. 2018, 48, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- van Campen, J.C.; Sollars, E.S.A.; Thomas, R.C.; Bartlett, C.M.; Milano, A.; Parker, M.D.; Dawe, J.; Winship, P.R.; Peck, G.; Grafham, D.; et al. Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. Int. J. Neonatal Screen. 2019, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; Tan, T.Y.; Chong, B.; Brett, G.R.; Yap, P.; Walsh, M.; Yeung, A.; Peters, H.; Mordaunt, D.; Cowie, S.; et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med. 2016, 18, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lim, J.; Shin, J.E.; Eun, H.S.; Park, M.S.; Park, K.I.; Namgung, R.; Lee, J.S. Implementation of a Targeted Next-Generation Sequencing Panel for Constitutional Newborn Screening in High-Risk Neonates. Yonsei Med. J. 2019, 60, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F. Newborn testing and screening by whole-genome sequencing. Genet. Med. 2016, 18, 214–216. [Google Scholar] [CrossRef][Green Version]
- Fleige, T.; Burggraf, S.; Czibere, L.; Häring, J.; Glück, B.; Keitel, L.M.; Landt, O.; Harms, E.; Hohenfellner, K.; Durner, J.; et al. Next generation sequencing as second-tier test in high-throughput newborn screening for nephropathic cystinosis. Eur. J. Hum. Genet. 2019, 28, 193–201. [Google Scholar] [CrossRef]
- Bobillo Lobato, J.; Jiménez Hidalgo, M.; Jiménez Jiménez, L. Biomarkers in Lysosomal Storage Diseases. Diseases 2016, 4, 40. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin. Chem. Lab. Med. 2019, 57, 1863–1874. [Google Scholar] [CrossRef]
- Johnson, B.; Mascher, H.; Mascher, D.; Legnini, E.; Hung, C.Y.; Dajnoki, A.; Chien, Y.-H.; Maródi, L.; Hwu, W.-L.; Bodamer, O.A. Analysis of Lyso-Globotriaosylsphingosine in Dried Blood Spots. Ann. Lab. Med. 2013, 33, 274. [Google Scholar] [CrossRef]
- Spada, M.; Kasper, D.; Pagliardini, V.; Biamino, E.; Giachero, S.; Porta, F. Metabolic progression to clinical phenotype in classic Fabry disease. Ital. J. Pediatr. 2017, 43, 1. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Bodamer, O.A.; Chiang, S.-C.; Mascher, H.; Hung, C.; Hwu, W.-L. Lyso-globotriaosylsphingosine (lyso-Gb3) levels in neonates and adults with the Fabry disease later-onset GLA IVS4+919G>A mutation. J. Inherit. Metab. Dis. 2012, 36, 881–885. [Google Scholar] [CrossRef]
- Kuchar, L.; Sikora, J.; Gulinello, M.E.; Poupetova, H.; Lugowska, A.; Malinova, V.; Jahnova, H.; Asfaw, B.; Ledvinova, J. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem. 2017, 525, 73–77. [Google Scholar] [CrossRef]
- Tarallo, A.; Carissimo, A.; Gatto, F.; Nusco, E.; Toscano, A.; Musumeci, O.; Coletta, M.; Karali, M.; Acampora, E.; Damiano, C.; et al. microRNAs as biomarkers in Pompe disease. Genet. Med. 2018, 21, 591–600. [Google Scholar] [CrossRef]
- Carrasco-Rozas, A.; Fernández-Simón, E.; Lleixà, M.C.; Belmonte, I.; Pedrosa-Hernandez, I.; Montiel-Morillo, E.; Nuñez-Peralta, C.; Llauger Rossello, J.; Segovia, S.; De Luna, N.; et al. Identification of serum microRNAs as potential biomarkers in Pompe disease. Ann. Clin. Transl. Neurol. 2019, 6, 1214–1224. [Google Scholar] [CrossRef]
- Cammarata, G.; Scalia, S.; Colomba, P.; Zizzo, C.; Pisani, A.; Riccio, E.; Montalbano, M.; Alessandro, R.; Giordano, A.; Duro, G. A pilot study of circulating microRNAs as potential biomarkers of Fabry disease. Oncotarget 2018, 9, 27333–27345. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Head | Chest & Abdomen | Skeletal | Neurologic | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease name | Gene | Stored material | Onset of symptoms | Coarse facial features | Hearing loss | Teeth abnormalities | Retinal degeneration | Corneal clouding | Enlarged tongue | Heart valvular disease | Respiratory tract infections | Hernias | Hepatosplenomegaly | Short stature | Joint stiffness and contractures | Dysostosis multiplex | Developmental delay | Cognitive deficits | Neurodegeneration | Sleep disturbances | Hydrocephalus | Seizures |
| MPS IH | IDUA | DS, HS | 1 year | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||
| MPS IH/S | 3–8 years | + | + | + | + | + | + | + | + | |||||||||||||
| MPS IS | >5 years | + | + | + | + | |||||||||||||||||
| MPS II | IDS | DS, HS | 2–4 years | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| MPS IIIA | SGSH | HS | 2–6 years | + | + | + | + | + | + | + | + | + | + | |||||||||
| MPS IIIB | NAGLU | + | + | + | + | + | + | + | + | + | + | |||||||||||
| MPS IIIC | HGSNAT | + | + | + | + | + | + | + | + | + | + | + | ||||||||||
| MPS IIID | GNS | + | + | + | + | + | + | + | + | + | + | |||||||||||
| MPS IVA | GALNS | KS | 1–3 years | + | + | + | + | + | + | + | + | + | ||||||||||
| MPS IVB | GLB1 | + | + | + | + | + | + | + | + | + | ||||||||||||
| MPS VI | ARSB | DS | Infancy | + | + | + | + | + | + | + | + | + | ||||||||||
| MPS VII | GUSB | DS, HS | 1–4 years | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||
| MPS IX | HYAL1 | HA | <1 year | + | + | |||||||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Cognata, V.; Guarnaccia, M.; Polizzi, A.; Ruggieri, M.; Cavallaro, S. Highlights on Genomics Applications for Lysosomal Storage Diseases. Cells 2020, 9, 1902. https://doi.org/10.3390/cells9081902
La Cognata V, Guarnaccia M, Polizzi A, Ruggieri M, Cavallaro S. Highlights on Genomics Applications for Lysosomal Storage Diseases. Cells. 2020; 9(8):1902. https://doi.org/10.3390/cells9081902
Chicago/Turabian StyleLa Cognata, Valentina, Maria Guarnaccia, Agata Polizzi, Martino Ruggieri, and Sebastiano Cavallaro. 2020. "Highlights on Genomics Applications for Lysosomal Storage Diseases" Cells 9, no. 8: 1902. https://doi.org/10.3390/cells9081902
APA StyleLa Cognata, V., Guarnaccia, M., Polizzi, A., Ruggieri, M., & Cavallaro, S. (2020). Highlights on Genomics Applications for Lysosomal Storage Diseases. Cells, 9(8), 1902. https://doi.org/10.3390/cells9081902