Identification of miRNA Master Regulators in Breast Cancer

 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
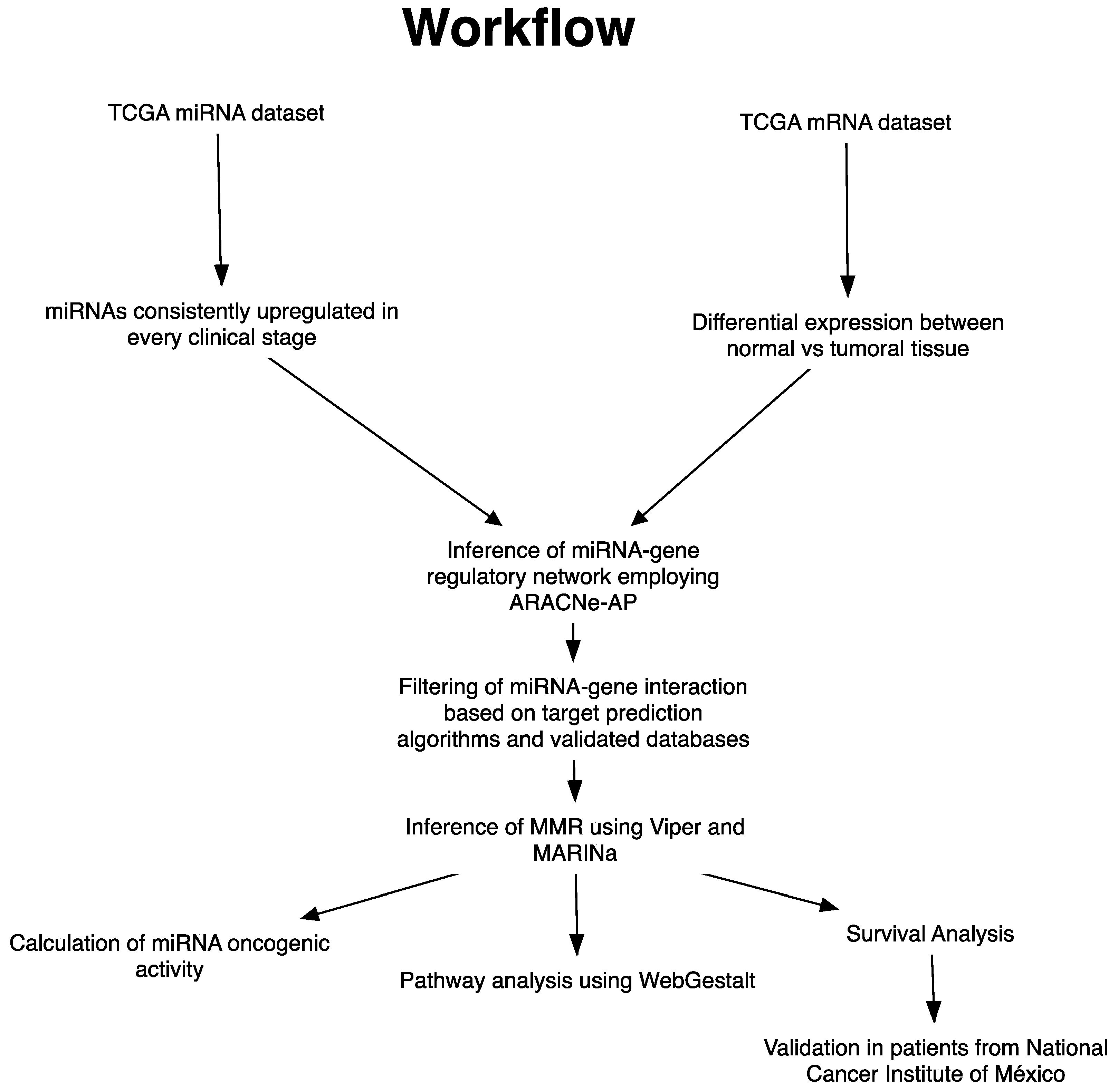
2. Materials and Methods
2.1. Analysis of Upregulated miRNAs in Clinical Stages
2.2. Identification of miRNA Master Regulators
2.3. Oncogenic Activity of miRNAs MMR
2.4. Pathway Analysis
2.5. Intrinsic Subtype Classification and Differential Expression
2.6. Survival Analysis
2.7. Patient Samples
2.8. RNA Isolation and Real-Time qPCR
2.9. Tissue Expression Meta-Analysis
3. Results
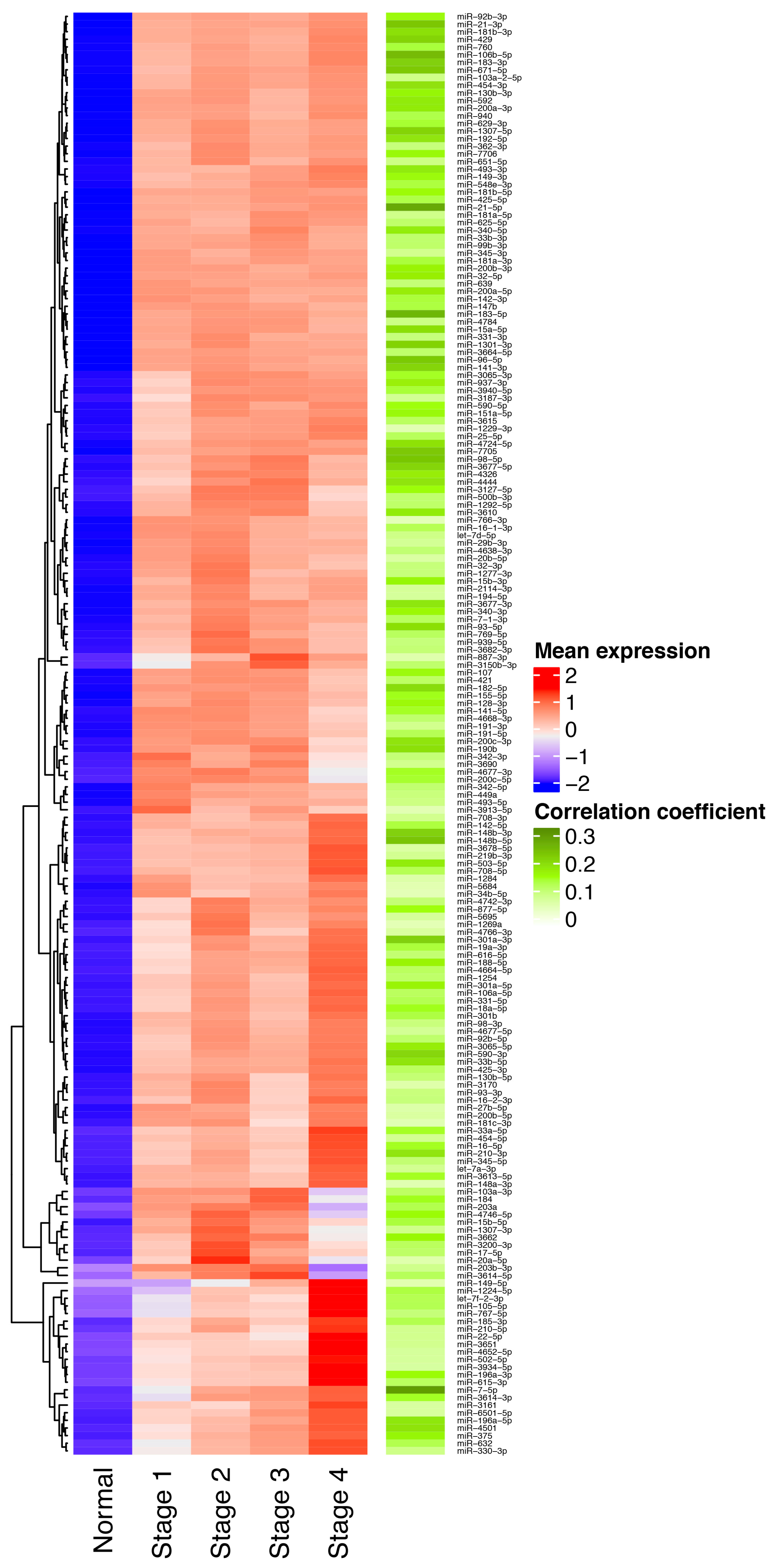
3.1. Identification of miRNAs Consistently Upregulated in Breast Tumor Tissues
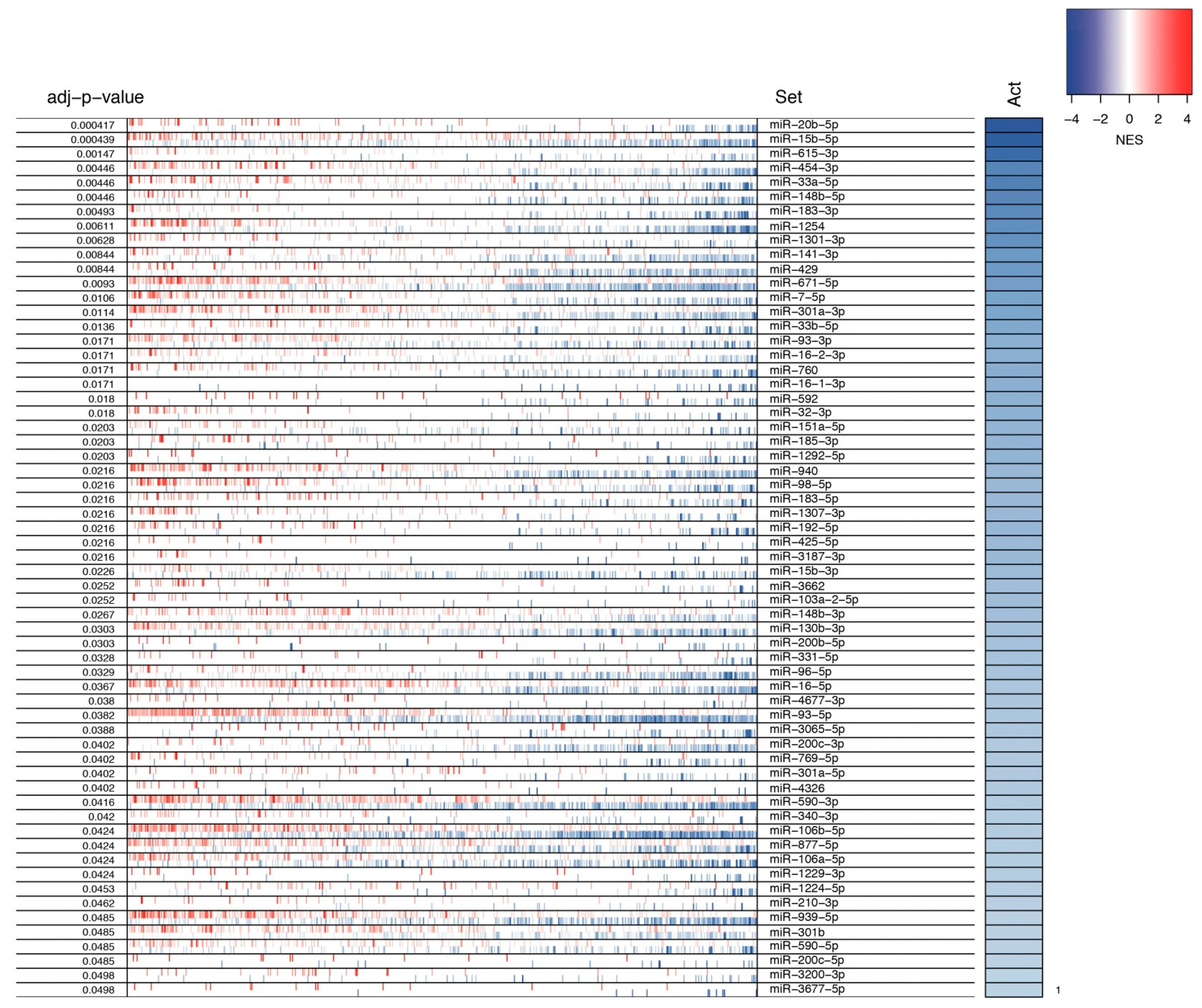
3.2. Identification of MMRs
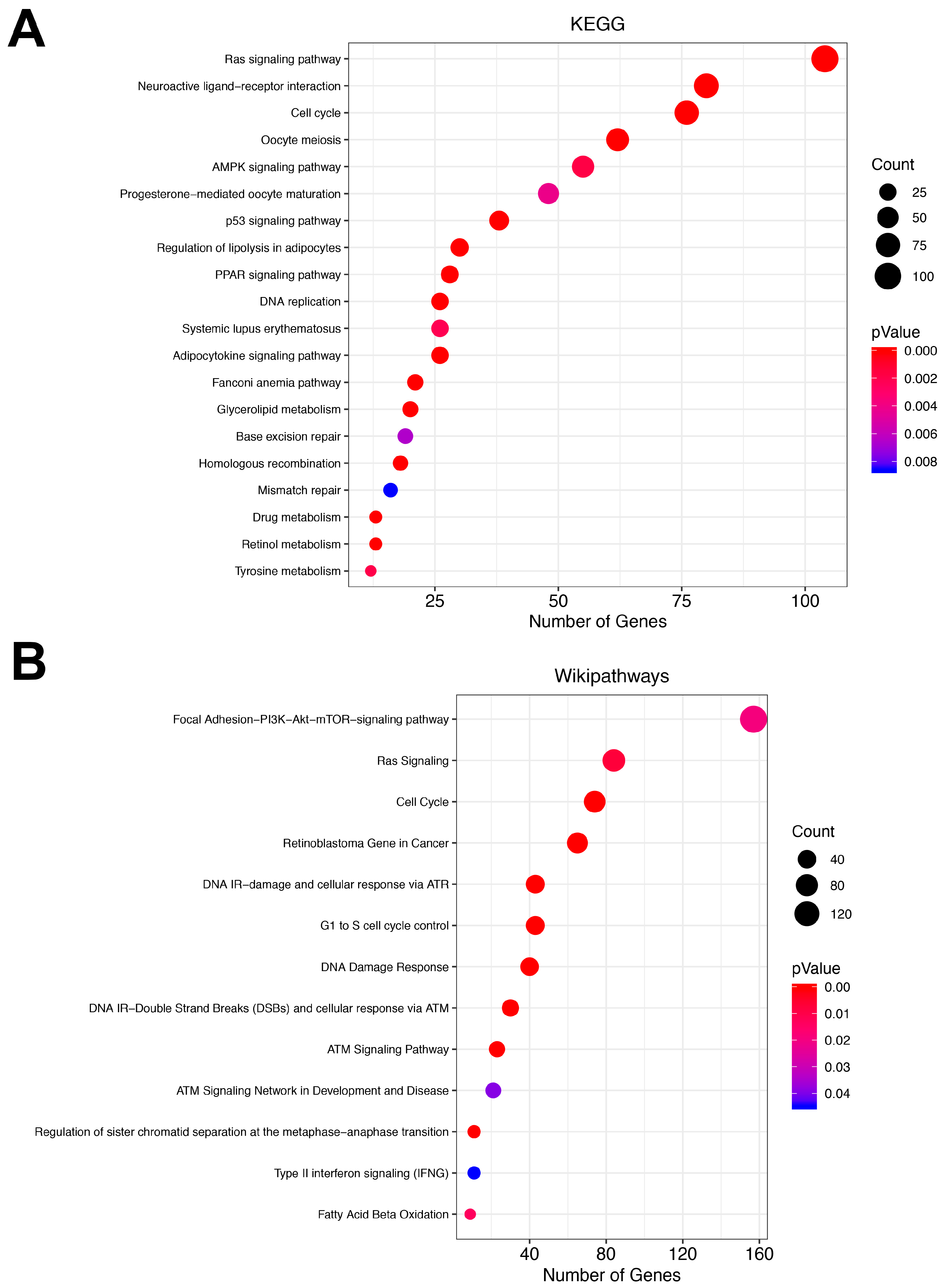
3.3. Pathway Analysis
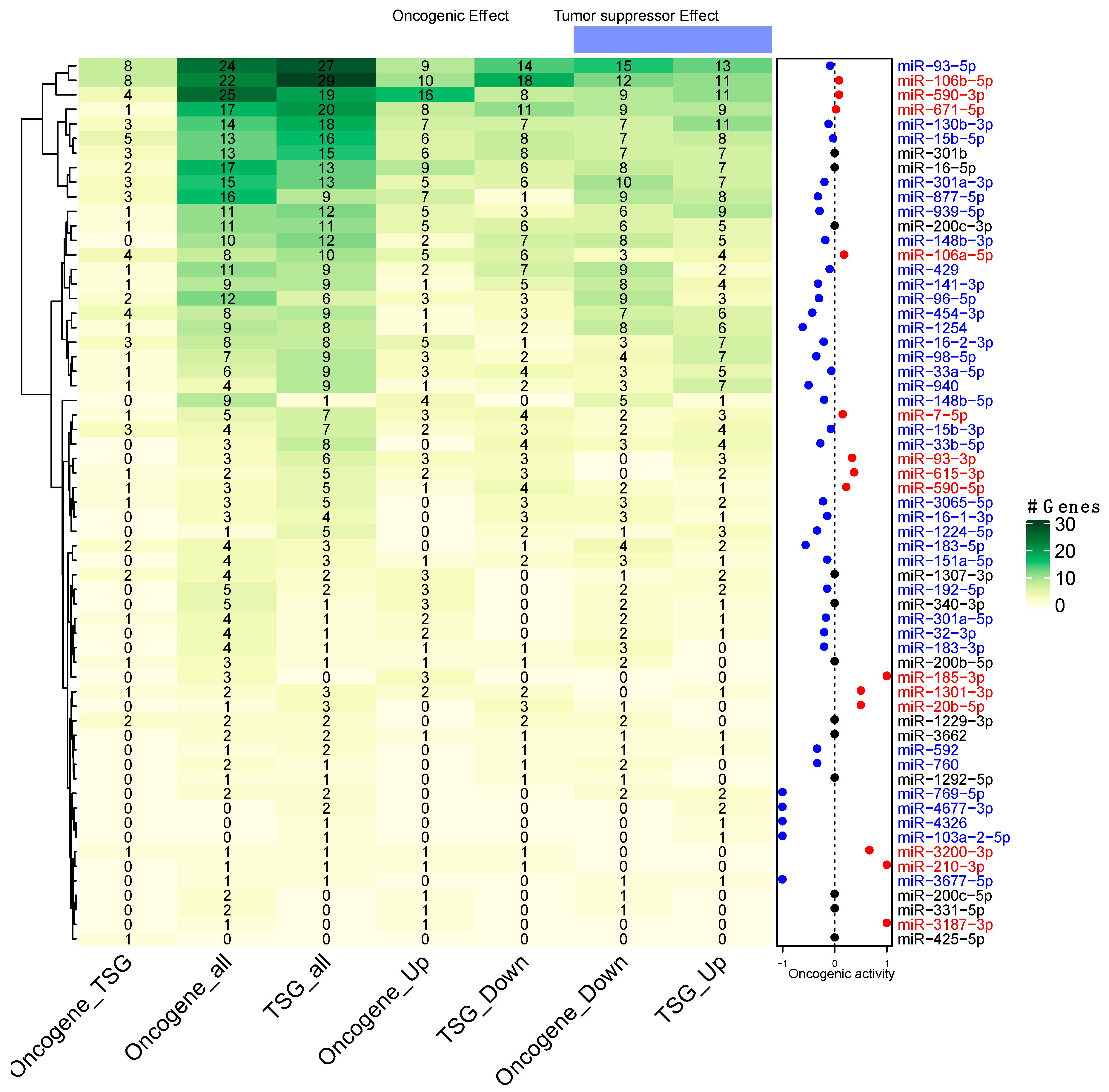
3.4. Prediction of miRNAs Oncogenic Activity
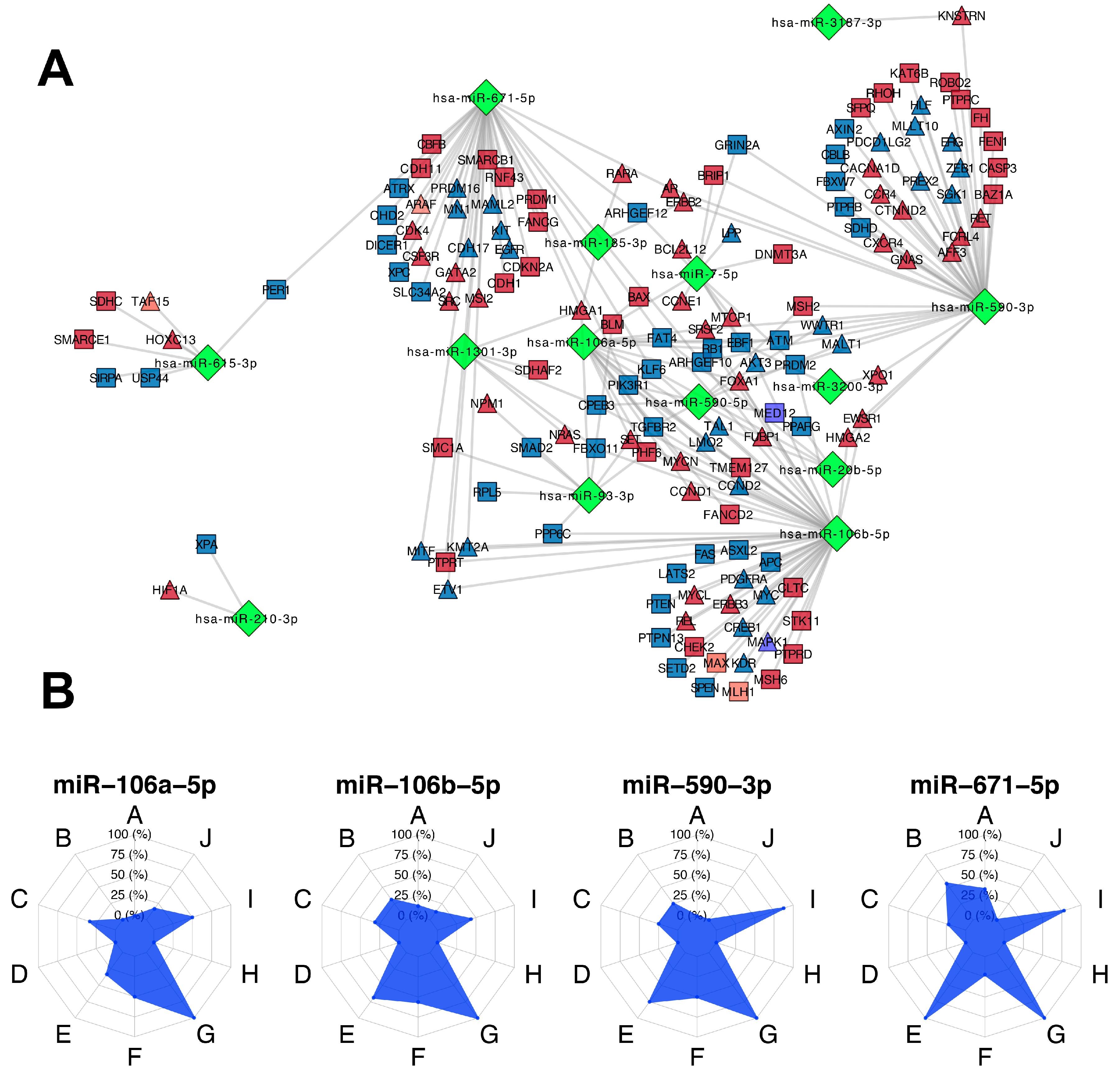
3.5. Participation of miRNAs Acting as Oncogenic Drivers
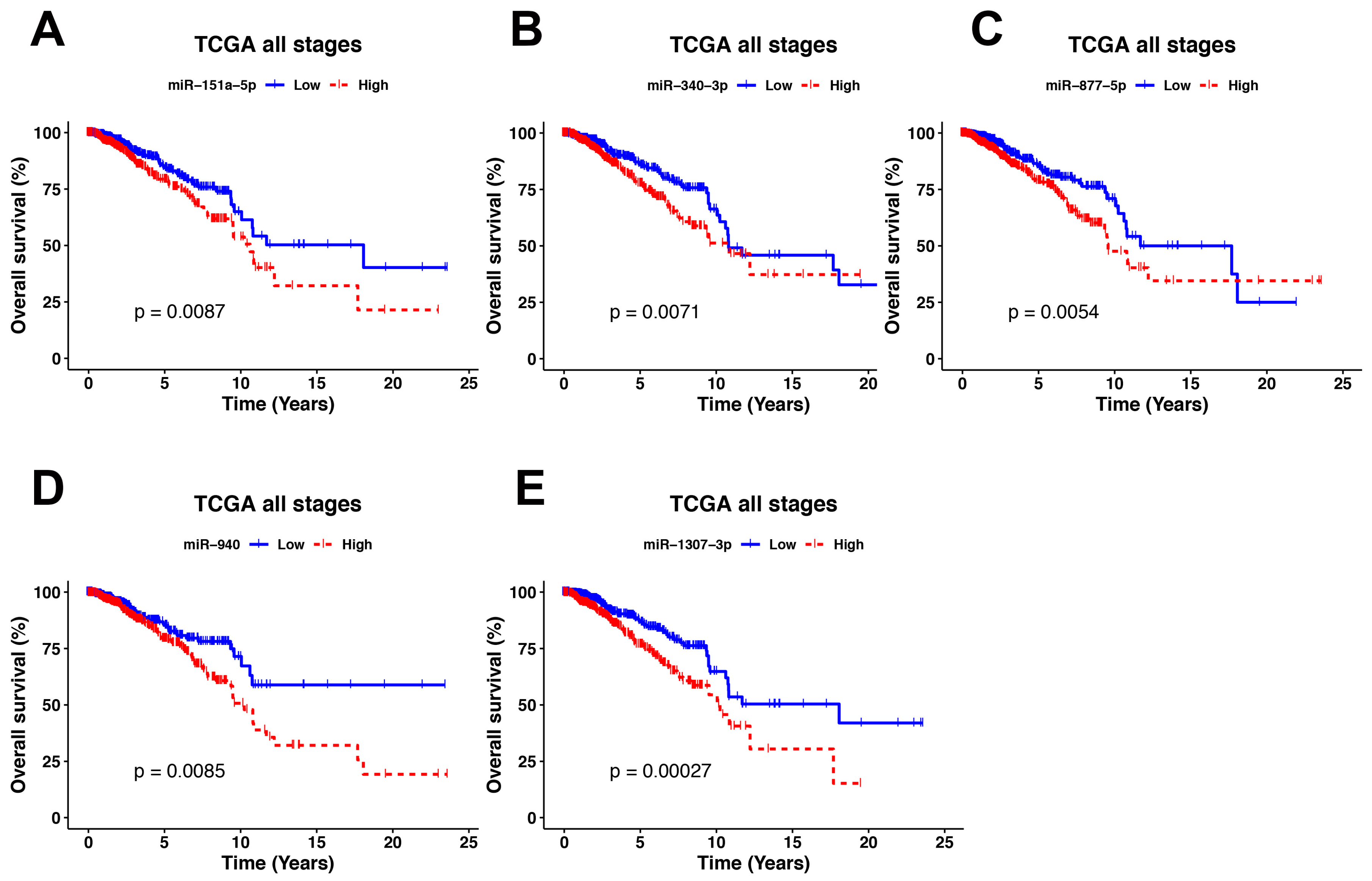
3.6. Identification of miRNA Master Regulators with Clinical Relevance
3.7. Impact of MMRs in the BC Intrinsic Subtypes
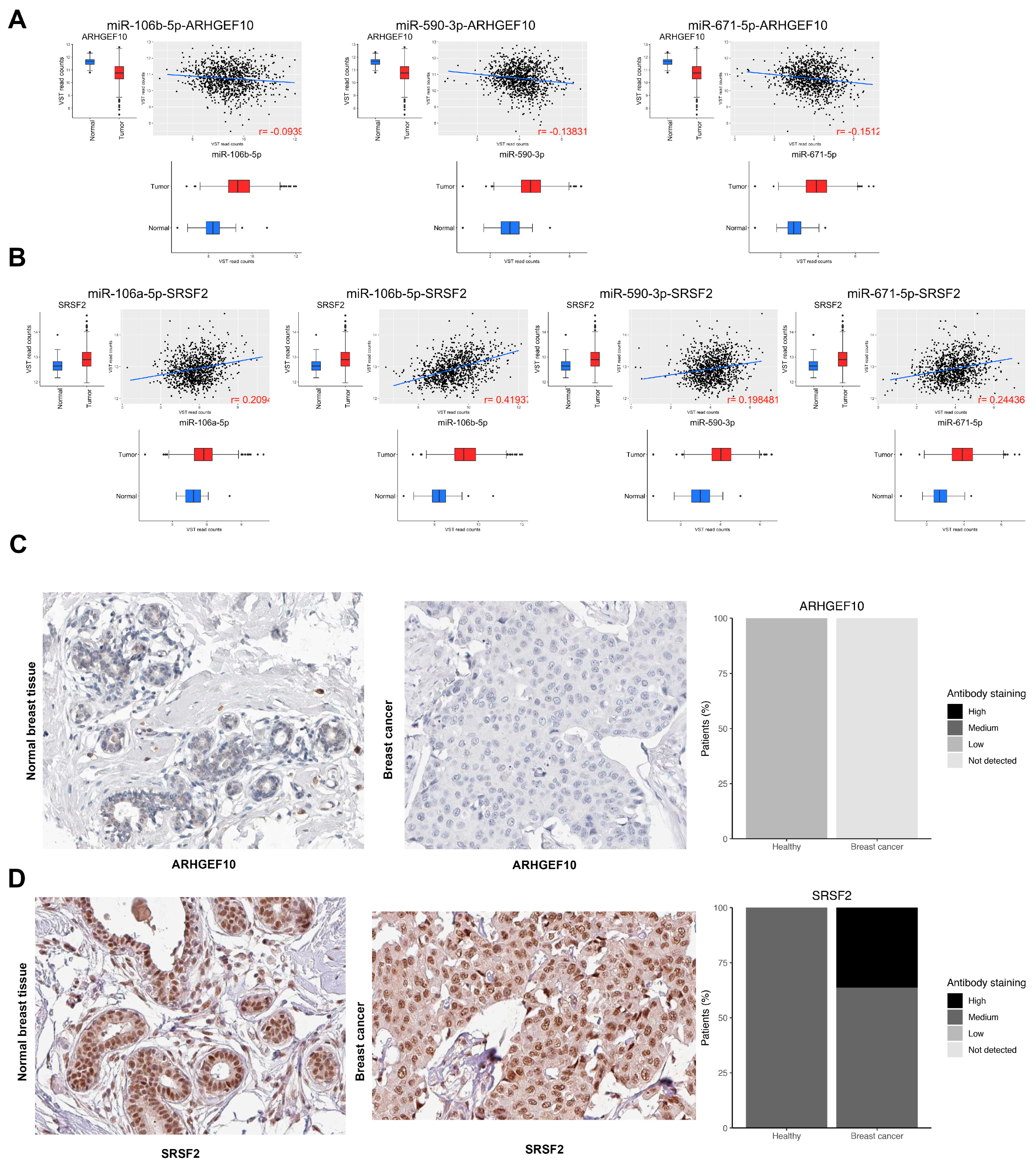
3.8. Validation in Locally Advanced Tumoral Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Conti, C.J. Mechanisms of Tumor Progression. Compr. Toxicol. 2010, 335–347. [Google Scholar] [CrossRef]
- Williams, M.; Cheng, Y.Y.; Blenkiron, C.; Reid, G. Exploring Mechanisms of MicroRNA Downregulation in Cancer. MicroRNA 2017, 6, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Casalini, P.; Tagliabue, E.; Ménard, S.; Croce, C.M. MicroRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur. J. Cancer 2008, 44, 2753–2759. [Google Scholar] [CrossRef]
- Volinia, S.; Galasso, M.; Sana, M.E.; Wise, T.F.; Palatini, J.; Huebner, K.; Croce, C.M. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, 3024–3029. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Croce, C.M. Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 7413–7417. [Google Scholar] [CrossRef]
- Cascione, L.; Gasparini, P.; Lovat, F.; Carasi, S.; Pulvirenti, A.; Ferro, A.; Alder, H.; He, G.; Vecchione, A.; Croce, C.M.; et al. Integrated MicroRNA and mRNA Signatures Associated with Survival in Triple Negative Breast Cancer. PLoS ONE 2013, 8, e55910. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M. MicroRNAs: Oncogenes, tumor suppressors or master regulators of cancer heterogeneity? Biochim. Biophys. Acta 2010, 1805, 72–86. [Google Scholar] [CrossRef]
- Dragomir, M.; Mafra, A.C.P.; Dias, S.M.G.; Vasilescu, C.; Calin, G.A. Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 2018, 19, 1871. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Git, A.; Gräf, S.; Salmon-Divon, M.; Curtis, C.; Sottoriva, A.; Zhao, Y.; Hirst, M.; Armisen, J.; Miska, E.A.; et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature 2013, 497, 378–382. [Google Scholar] [CrossRef]
- Cantini, L.; Bertoli, G.; Cava, C.; Dubois, T.; Zinovyev, A.; Caselle, M.; Castiglioni, I.; Barillot, E.; Martignetti, L. Identification of microRNA clusters cooperatively acting on epithelial to mesenchymal transition in triple negative breast cancer. Nucleic Acids Res. 2019, 47, 2205–2215. [Google Scholar] [CrossRef]
- Lachmann, A.; Giorgi, F.M.; Lopez, G.; Califano, A. ARACNe-AP: Gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 2016, 32, 2233–2235. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rajbhandari, P.; Alvarez, M.J.; Bandaru, P.; Lim, W.K.; Sato, M.; Wang, K.; Sumazin, P.; Kustagi, M.; Bisikirska, B.C.; et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010, 6, 377. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Enviroment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Gendoo, D.M.A.; Ratanasirigulchai, N.; Schröder, M.S.; Paré, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2016, 32, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Bovolenta, L.A.; Nachtigall, P.G.; Herkenhoff, M.E.; Lemke, N.; Pinhal, D. Combining Results from Distinct MicroRNA Target Prediction Tools Enhances the Performance of Analyses. Front. Genet. 2017, 8, 59. [Google Scholar] [CrossRef]
- Cooke, S.L.; Pole, J.C.M.; Chin, S.F.; Ellis, I.O.; Caldas, C.; Edwards, P.A.W. High-resolution array CGH clarifies events occurring on 8p in carcinogenesis. BMC Cancer 2008, 8, 288. [Google Scholar] [CrossRef]
- Anczuków, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef]
- Eroles, P.; Bosch, A.; Alejandro Pérez-Fidalgo, J.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Tarabichi, M.; Antoniou, A.; Saiselet, M.; Pita, J.M.; Andry, G.; Dumont, J.E.; Detours, V.; Maenhaut, C. Systems biology of cancer: Entropy, disorder, and selection-driven evolution to independence, invasion and “swarm intelligence”. Cancer Metastasis Rev. 2013, 32, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Alvarez, M.J.; Talos, F.; Dhruv, H.; Rieckhof, G.E.; Iyer, A.; Diefes, K.L.; Aldape, K.; Berens, M.; Shen, M.M.; et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 2014, 159, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Califano, A.; Alvarez, M.J. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 2017, 17, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, Y.; Hu, L.; Zheng, H.; Ji, P.; Pecot, C.V.; Zhao, Y.; Reynolds, S.; Cheng, H.; Rupaimoole, R.; et al. Integrated Analyses Identify a Master MicroRNA Regulatory Network for the Mesenchymal Subtype in Serous Ovarian Cancer. Cancer Cell 2013, 23, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Cantini, L.; Isella, C.; Petti, C.; Picco, G.; Chiola, S.; Ficarra, E.; Caselle, M.; Medico, E. MicroRNA–mRNA interactions underlying colorectal cancer molecular subtypes. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Tovar, H.; García-Herrera, R.; Espinal-Enríquez, J. Transcriptional master regulator analysis in breast cancer genetic networks. Comput. Biol. Chem. 2015, 59, 67–77. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef]
- Oliveto, S.; Mancino, M.; Manfrini, N.; Biffo, S. Role of microRNAs in translation regulation and cancer. World J. Biol. Chem. 2017, 8, 45–56. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.E.; Song, Y.-S.; Cho, E.Y.; Lee, A. miR-106b-5p and miR-17-5p could predict recurrence and progression in breast ductal carcinoma in situ based on the transforming growth factor-beta pathway. Breast Cancer Res. Treat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; He, J.; Huang, C.; Chen, L.; Tao, D.; Wu, X.; Wang, M.; Luo, G.; Xiao, X.; Zeng, F.; et al. miR-106b-5p targets tumor suppressor gene SETD2 to inactive its function in clear cell renal cell carcinoma. Oncotarget 2015, 6, 4066–4079. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Gu, S.; Zhang, X.; Zhang, S.; Zhang, D.; Lin, J.; Hasengbayi, S.; Han, W. miR-106b-5p promotes aggressive progression of hepatocellular carcinoma via targeting RUNX3. Cancer Med. 2019. [Google Scholar] [CrossRef]
- You, F.; Luan, H.; Sun, D.; Cui, T.; Ding, P.; Tang, H.; Sun, D. MiRNA-106a Promotes Breast Cancer Cell Proliferation, Clonogenicity, Migration, and Invasion Through Inhibiting Apoptosis and Chemosensitivity. DNA Cell Biol. 2019, 38, 198–207. [Google Scholar] [CrossRef]
- Salem, M.; Shan, Y.; Bernaudo, S.; Peng, C. miR-590-3p Targets Cyclin G2 and FOXO3 to Promote Ovarian Cancer Cell Proliferation, Invasion, and Spheroid Formation. Int. J. Mol. Sci. 2019, 20, 1810. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-Q.; Shi, K.; Zhou, Q.-B.; Zeng, X.-Y.; Liu, J.; Yang, S.-X.; Wang, Q.-S.; Li, Z.; Wang, G.-X.; Song, J.-M.; et al. MiR-590-3p promotes proliferation and metastasis of colorectal cancer via Hippo pathway. Oncotarget 2017, 8, 58061–58071. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Chen, M.; Zhao, X.; Li, J.; Deng, S.; Hu, J.; Yang, H.; Jiang, J. FAT4 functions as a tumor suppressor in triple-negative breast cancer. Tumor Biol. 2016, 37, 16337–16343. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, F.; Koksal, M.; Ozmen, V.; Aydin, I.; Buyru, N. Mutations and Krüppel-like factor 6 (KLF6) expression levels in breast cancer. Tumor Biol. 2014, 35, 5219–5225. [Google Scholar] [CrossRef]
- Gao, Y.; Li, H.; Ma, X.; Fan, Y.; Ni, D.; Zhang, Y.; Huang, Q.; Liu, K.; Li, X.; Wang, L.; et al. KLF6 suppresses metastasis of clear cell renal cell carcinoma via transcriptional repression of E2F1. Cancer Res. 2017, 77, 330–342. [Google Scholar] [CrossRef]
- Gasparini, P.; Cascione, L.; Fassan, M.; Lovat, F.; Guler, G.; Balci, S.; Irkkan, C.; Morrison, C.; Croce, C.M.; Shapiro, C.L.; et al. microRNA expression profiling identifies a four microRNA signature as a novel diagnostic and prognostic biomarker in triple negative breast cancers. Oncotarget 2014, 5, 1174–1184. [Google Scholar] [CrossRef]
- Chen, W.-T.; Yang, Y.-J.; Zhang, Z.-D.; An, Q.; Li, N.; Liu, W.; Yang, B. MiR-1307 promotes ovarian cancer cell chemoresistance by targeting the ING5 expression. J. Ovar. Res. 2017, 10, 1. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, J. Mir-1307 regulates cisplatin resistance by targeting Mdm4 in breast cancer expressing wild type P53. Thorac. Cancer 2018, 9, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, Y.; Guan, H.; Meng, H. Clinical potential of miR-940 as a diagnostic and prognostic biomarker in breast cancer patients. Cancer Biomark. 2018, 22, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Chen, M.; Yang, H.; Xing, T.; Li, J.; Li, G.; Zhang, L.; Deng, S.; Hu, J.; Zhao, X.; et al. MiR-940 Inhibited Cell Growth and Migration in Triple-Negative Breast Cancer. Med. Sci. Monit. 2016, 22, 3666–3672. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
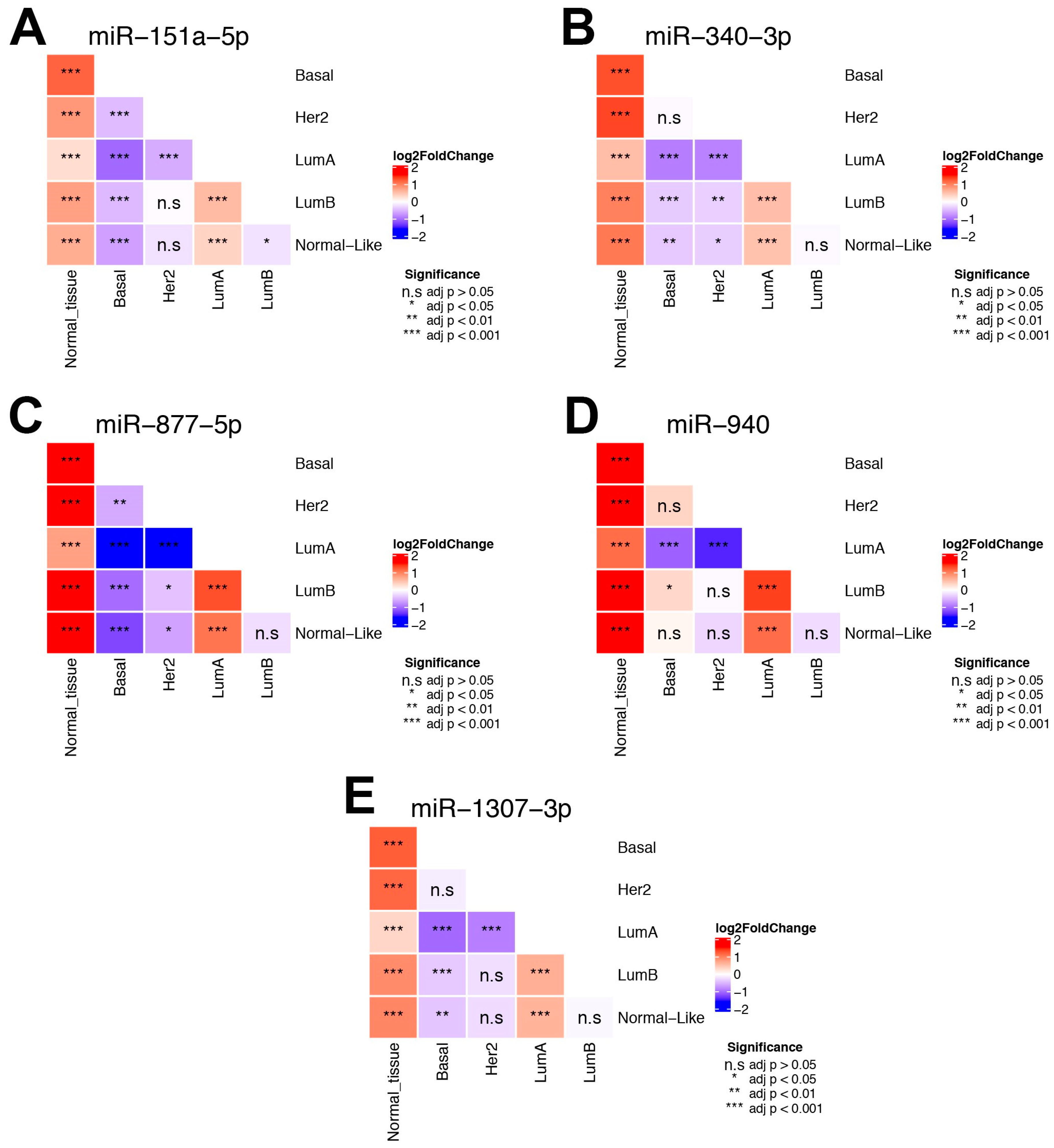{kind=link}
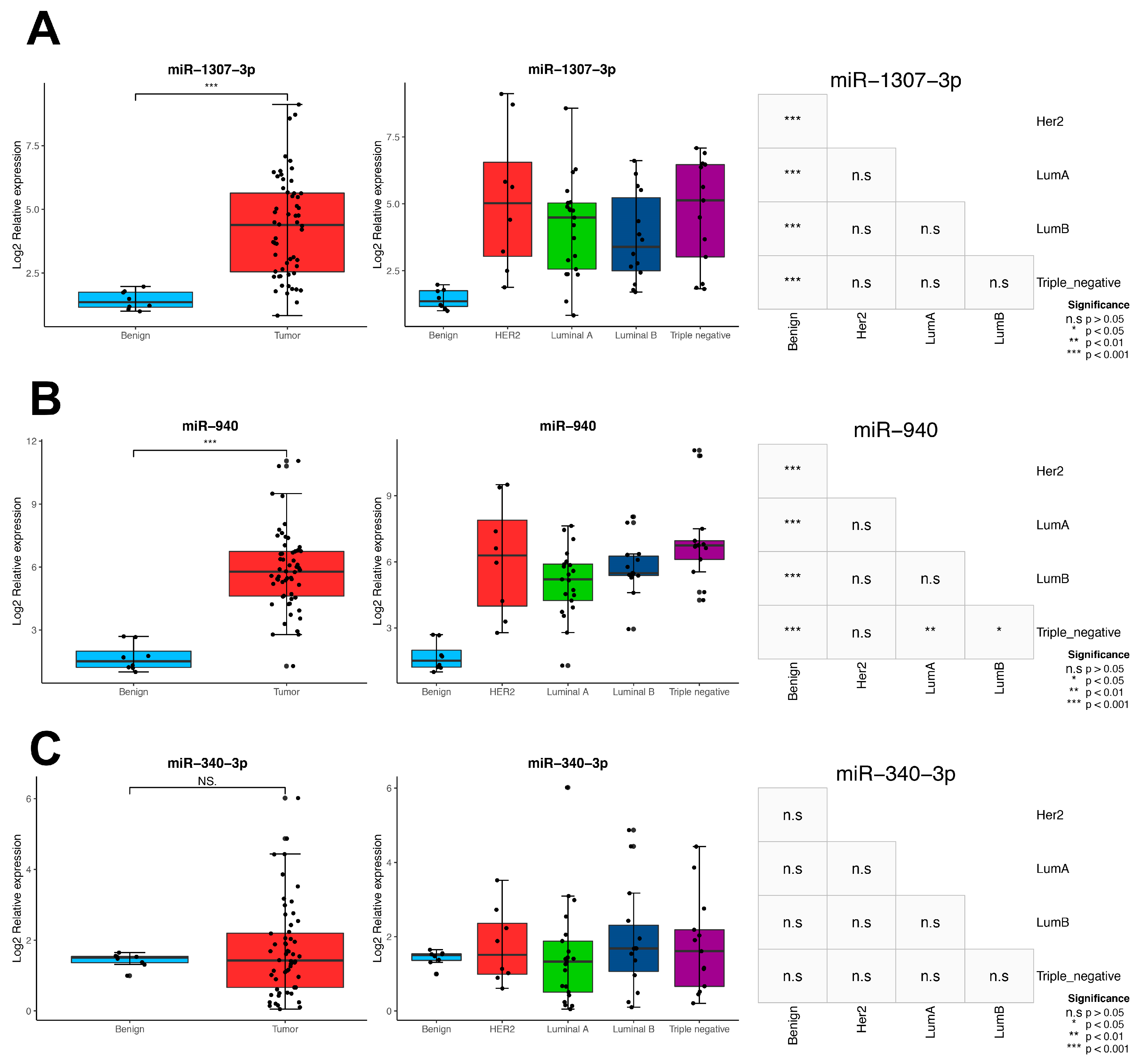{kind=link}
| miRNAs. | Genes | |||
|---|---|---|---|---|
| Name | Degree | Name | Degree | Gene log2FoldChange |
| hsa-miR-106b-5p | 1179 | RUNX1T1 | 24 | −1.715126046 |
| hsa-miR-590-3p | 996 | BNC2 | 20 | −0.440592219 |
| hsa-miR-93-5p | 970 | TNS1 | 20 | −3.104826764 |
| hsa-miR-671-5p | 921 | DLC1 | 18 | −1.789077079 |
| hsa-miR-939-5p | 711 | FOXP2 | 18 | −1.963889919 |
| hsa-miR-877-5p | 670 | IGF1 | 17 | −2.494558934 |
| hsa-miR-130b-3p | 585 | ZEB2 | 17 | −1.552276689 |
| hsa-miR-16-5p | 547 | ANK2 | 16 | −2.347077977 |
| hsa-miR-301a-3p | 546 | BACH2 | 16 | −1.35469313 |
| hsa-miR-15b-5p | 529 | TCF4 | 16 | −0.872983915 |
| hsa-miR-940 | 483 | ZCCHC24 | 16 | −1.552147667 |
| hsa-miR-301b | 469 | ZFHX4 | 16 | −1.12229256 |
| hsa-miR-106a-5p | 451 | NOVA1 | 15 | −0.712825148 |
| hsa-miR-1254 | 398 | TSHZ3 | 15 | −0.478344046 |
| hsa-miR-93-3p | 373 | BHLHE41 | 14 | −1.138290499 |
| hsa-miR-141-3p | 355 | CCND2 | 14 | −0.965637676 |
| hsa-miR-96-5p | 338 | CREBRF | 14 | −0.973637932 |
| hsa-miR-454-3p | 329 | PRICKLE2 | 14 | −1.12294618 |
| hsa-miR-200c-3p | 329 | QKI | 14 | −1.25158266 |
| hsa-miR-429 | 327 | RACGAP1 | 14 | 1.909634723 |
| OncoMMRs | ||
|---|---|---|
| Name | Degree | Oncogenic Activity |
| miR-106b-5p | 51 | 0.08 |
| miR-590-3p | 44 | 0.08 |
| miR-671-5p | 37 | 0.02 |
| miR-106a-5p | 18 | 0.18 |
| miR-7-5p | 12 | 0.15 |
| miR-93-3p | 9 | 0.33 |
| miR-590-5p | 8 | 0.22 |
| miR-615-3p | 7 | 0.37 |
| miR-1301-3p | 5 | 0.5 |
| miR-20b-5p | 4 | 0.5 |
| miR-185-3p | 3 | 1 |
| miR-3200-3p | 2 | 0.66 |
| miR-210-3p | 2 | 1 |
| miR-3187-3p | 1 | 1 |
| Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|
| Overall Survival | HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| miR-151a-5p | 0.64 (0.46–0.9) | 0.0093 | 0.76 (0.50–1.16) | 0.2172 | |
| miR-940 | High vs. low expression | 0.63 (0.44–0.89) | 0.0091 | 0.77 ( 0.53–1.14) | 0.2026 |
| miR-1307-3p | 0.53 (0.38–0.75) | 0.00033 | 0.63 (0.42–0.97) | 0.0357 | |
| miR-340-3p | 0.63 (0.45–0.89) | 0.0076 | 0.91 (0.61–1.34) | 0.6429 | |
| miR-877-5p | 0.62 (0.44–0.87) | 0.0059 | 0.98 (0.66–1.45) | 0.9272 | |
| Age at diagnosis | <58 vs. ≥58 | 1.9 (1.3–2.6) | 0.00038 | 2.12 (1.49–3.01) | 2.78 × 10−5 |
| ER | Negative vs. positive | 0.57 (0.4–0.81) | 0.0017 | 0.68 (0.39–1.19) | 0.1802 |
| PR | 0.65 (0.47–0.91) | 0.013 | 0.88 (0.51–1.50) | 0.6473 | |
| Clinical stage | 2.3 (1.8–2.8) | 2.6 × 10−12 | 2.48 (1.96–3.14) | 3.51 × 10−14 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Gutierrez, A.D.; Cantú de León, D.; Millan-Catalan, O.; Coronel-Hernandez, J.; Campos-Parra, A.D.; Porras-Reyes, F.; Exayana-Alderete, A.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Ramos-Payan, R.; et al. Identification of miRNA Master Regulators in Breast Cancer. Cells 2020, 9, 1610. https://doi.org/10.3390/cells9071610
Martinez-Gutierrez AD, Cantú de León D, Millan-Catalan O, Coronel-Hernandez J, Campos-Parra AD, Porras-Reyes F, Exayana-Alderete A, López-Camarillo C, Jacobo-Herrera NJ, Ramos-Payan R, et al. Identification of miRNA Master Regulators in Breast Cancer. Cells. 2020; 9(7):1610. https://doi.org/10.3390/cells9071610
Chicago/Turabian StyleMartinez-Gutierrez, Antonio Daniel, David Cantú de León, Oliver Millan-Catalan, Jossimar Coronel-Hernandez, Alma D. Campos-Parra, Fany Porras-Reyes, Angelica Exayana-Alderete, César López-Camarillo, Nadia J Jacobo-Herrera, Rosalio Ramos-Payan, and et al. 2020. "Identification of miRNA Master Regulators in Breast Cancer" Cells 9, no. 7: 1610. https://doi.org/10.3390/cells9071610
APA StyleMartinez-Gutierrez, A. D., Cantú de León, D., Millan-Catalan, O., Coronel-Hernandez, J., Campos-Parra, A. D., Porras-Reyes, F., Exayana-Alderete, A., López-Camarillo, C., Jacobo-Herrera, N. J., Ramos-Payan, R., & Pérez-Plasencia, C. (2020). Identification of miRNA Master Regulators in Breast Cancer. Cells, 9(7), 1610. https://doi.org/10.3390/cells9071610