Show Me Your Friends and I Tell You Who You Are: The Many Facets of Prion Protein in Stroke



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Evidence That PrPC Has a Protective Role after Ischemic Damage
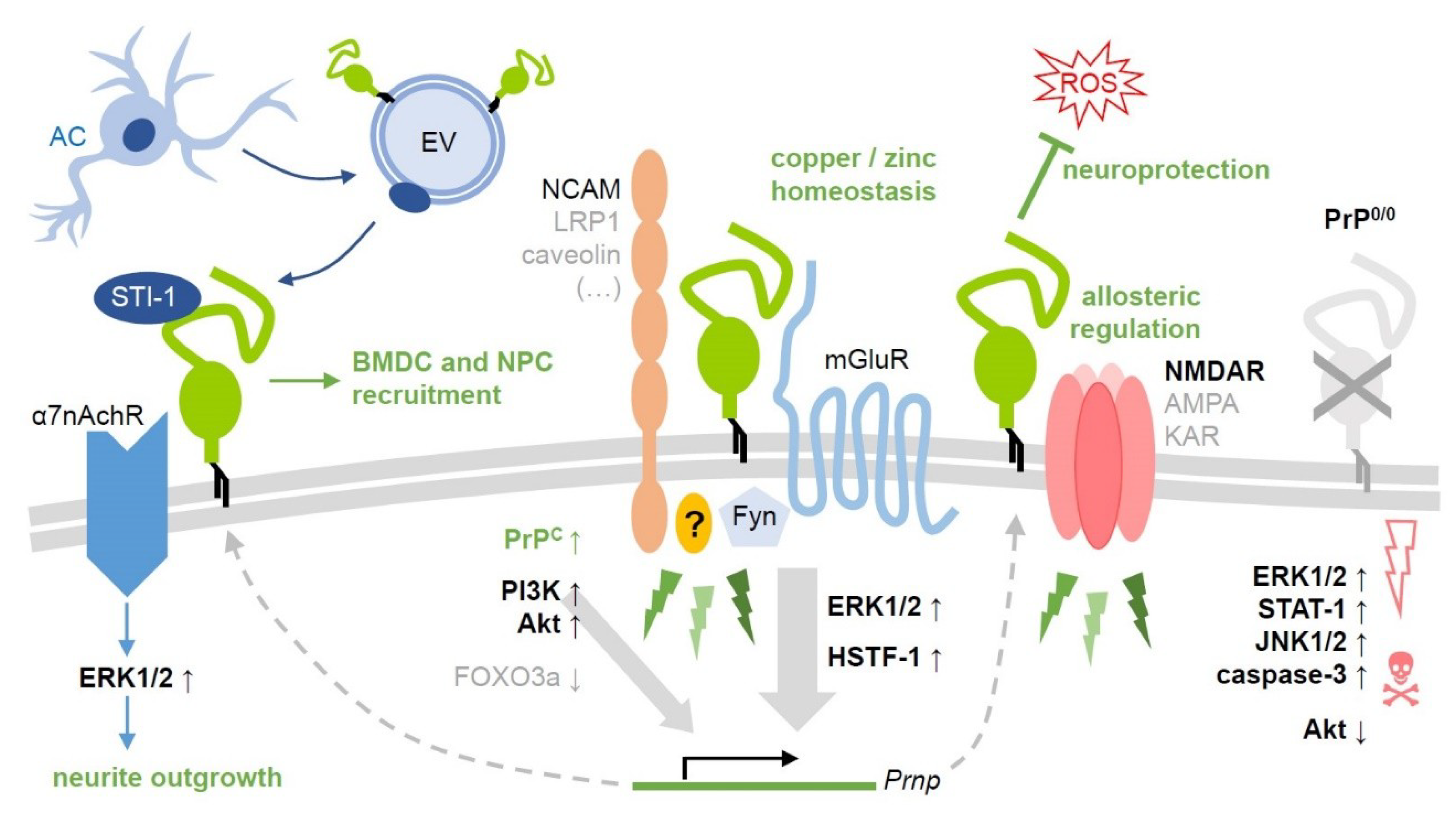
3. An Overview of PrP-Associated Signaling Pathways and Their (Potential) Role in Ischemia
3.1. The Mitogen-Activated Protein Kinase (MAPK) Pathway
3.2. The Phosphatidylinositol 3-Kinase/Protein Kinase B/Akt (PI3K/Akt) Pathway and PrPC
3.3. The Stress-Inducible Protein-1 (STI-1)
3.4. PrP Signaling and Glutamate (Receptors)
3.5. The Src Family Kinases (SFKs): Critical PrPC Interactors in Neurodegeneration—A Possible Role in Cerebral Ischemia?
4. PrP and Hyperpolarization-Activated Nucleotide-Gated Channel (HCN)
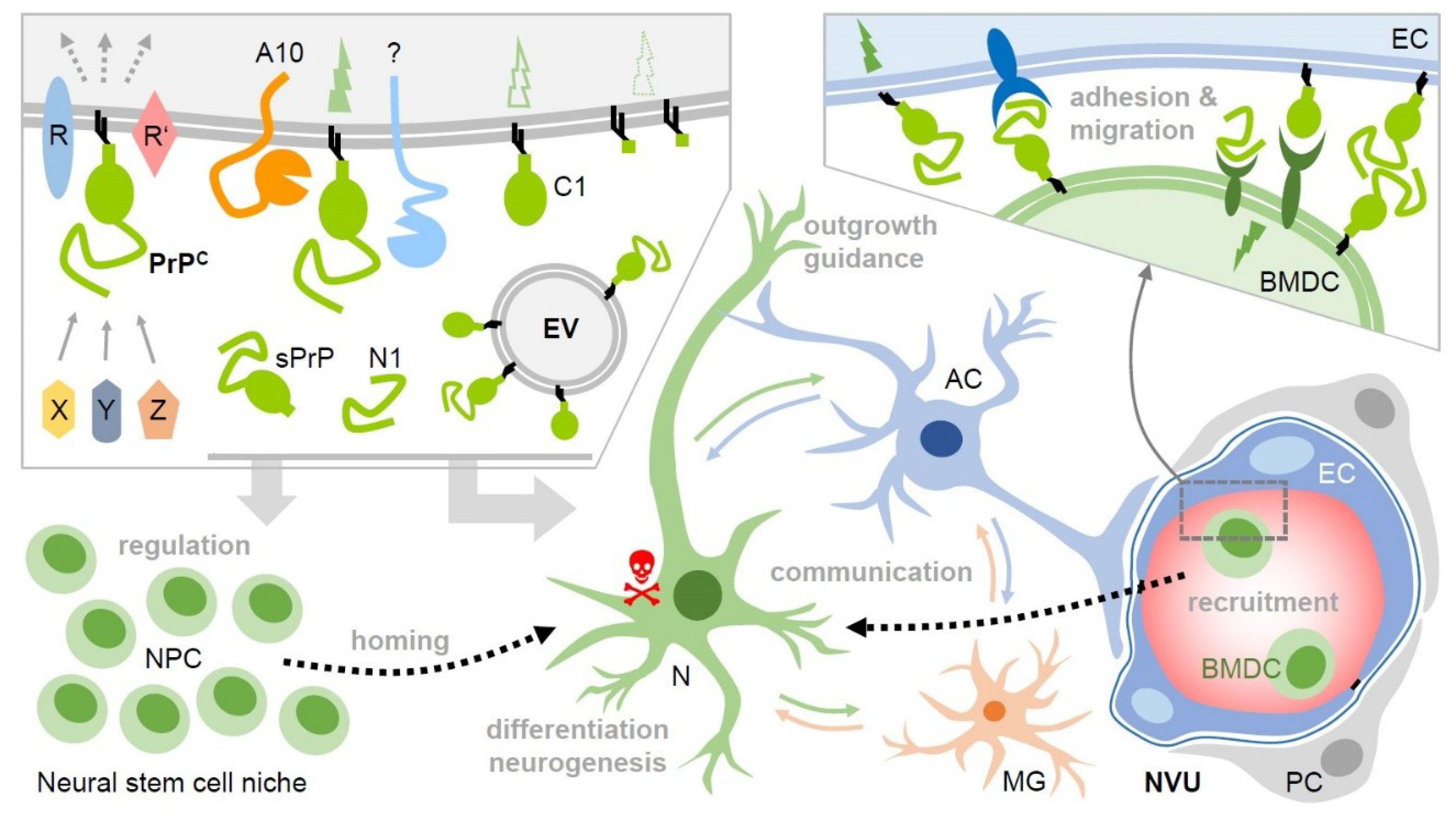
5. Homing Mechanisms in Cerebral Ischemia: PrP-Related Neurogenesis and Angiogenesis
6. Proteolytic Fragmentation Enables Functional Diversity–PrPC Derivatives and Stroke
7. Another Way of Releasing Information: EVs, PrP and Its C1 Fragment, and Stroke
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796. [Google Scholar] [CrossRef]
- Puig, B.; Brenna, S.; Magnus, T. Molecular Communication of a Dying Neuron in Stroke. Int. J. Mol. Sci. 2018, 19, 2834. [Google Scholar] [CrossRef]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med. 2008, 14, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Bastan, B. Identifying and utilizing the ischemic penumbra. Neurology 2012, 79, S79–S85. [Google Scholar] [CrossRef] [PubMed]
- Emberson, J.; Lees, K.R.; Lyden, P.; Blackwell, L.; Albers, G.; Bluhmki, E.; Brott, T.; Cohen, G.; Davis, S.; Donnan, G.; et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: A meta-analysis of individual patient data from randomised trials. Lancet 2014, 384, 1929–1935. [Google Scholar] [CrossRef]
- Mokin, M.; Ansari, S.A.; McTaggart, R.A.; Bulsara, K.R.; Goyal, M.; Chen, M.; Fraser, J.F. Indications for thrombectomy in acute ischemic stroke from emergent large vessel occlusion (ELVO): Report of the SNIS Standards and Guidelines Committee. J. Neurointerv. Surg. 2019, 11, 215–220. [Google Scholar] [CrossRef]
- Puig, J.; Shankar, J.; Liebeskind, D.; Terceño, M.; Nael, K.; Demchuk, A.M.; Menon, B.; Dowlatshahi, D.; Leiva-Salinas, C.; Wintermark, M.; et al. From “Time is Brain” to “Imaging is Brain”: A Paradigm Shift in the Management of Acute Ischemic Stroke. J. Neuroimaging 2020. [Google Scholar] [CrossRef] [PubMed]
- Phipps, M.S.; Cronin, C.A. Management of acute ischemic stroke. BMJ 2020, 368, l6983. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.B.; Yoon, B.W. Mechanisms of Functional Recovery after Stroke. Front. Neurol. Neurosci. 2013, 32, 1–8. [Google Scholar] [PubMed]
- Carmichael, S.T. Translating the frontiers of brain repair to treatments: Starting not to break the rules. Neurobiol. Dis. 2010, 37, 237–242. [Google Scholar] [CrossRef]
- Carmichael, S.T. Emergent properties of neural repair: Elemental biology to therapeutic concepts. Ann. Neurol. 2016, 79, 895–906. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149–156. [Google Scholar] [CrossRef]
- Puig, B.; Altmeppen, H.; Glatzel, M. The GPI-anchoring of PrP: Implications in sorting and pathogenesis. Prion 2014, 8, 11–18. [Google Scholar] [CrossRef]
- Biasini, E.; Turnbaugh, J.A.; Unterberger, U.; Harris, D.A. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012, 35, 92–103. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Weissmann, C. The state of the prion. Nat. Rev. Microbiol. 2004, 2, 861–871. [Google Scholar] [CrossRef]
- Schätzl, H.M.; Costa, M.D.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion Protein Gene Variation Among Primates. J. Mol. Biol. 1995, 245, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Cotto, E.; André, M.; Forgue, J.; Fleury, H.J.; Babin, P.J. Molecular characterization, phylogenetic relationships, and developmental expression patterns of prion genes in zebrafish (Danio rerio). FEBS J. 2005, 272, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, V.C.; Leighton, P.L.A.; Wang, H.; Pillay, L.M.; Ritzel, R.G.; Bhinder, G.; Roy, B.; Tierney, K.B.; Ali, D.W.; Waskiewicz, A.J.; et al. Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 2013, 55, 11–25. [Google Scholar] [CrossRef]
- Rivera-Milla, E.; Oidtmann, B.; Panagiotidis, C.H.; Baier, M.; Sklaviadis, T.; Hoffmann, R.; Zhou, Y.; Solis, G.P.; Stuermer, C.A.; Málaga-Trillo, E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J. 2006, 20, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.L.A.; Nadolski, N.J.; Morrill, A.; Hamilton, T.J.; Allison, W.T. An ancient conserved role for prion protein in learning and memory. Biol. Open 2018, 7, bio025734. [Google Scholar] [CrossRef]
- Wulf, M.-A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Linsenmeier, L.; Altmeppen, H.C.; Wetzel, S.; Mohammadi, B.; Saftig, P.; Glatzel, M. Diverse functions of the prion protein – Does proteolytic processing hold the key? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2128–2137. [Google Scholar] [CrossRef]
- Bakkebo, M.K.; Mouillet-Richard, S.; Espenes, A.; Goldmann, W.; Tatzelt, J.; Tranulis, M.A. The Cellular Prion Protein: A Player in Immunological Quiescence. Front. Immunol. 2015, 6, 450. [Google Scholar] [CrossRef]
- Hoshino, S.; Inoue, K.; Yokoyama, T.; Kobayashi, S.; Asakura, T.; Teramoto, A.; Itohara, S. Prions prevent brain damage after experimental brain injury: A preliminary report. Acta neurochirurgica. Supplement 2003, 86, 297–299. [Google Scholar]
- Megra, B.W.; Eugenin, E.A.; Berman, J.W. Inflammatory mediators reduce surface PrP(c) on human BMVEC resulting in decreased barrier integrity. Lab. Investig. 2018, 98, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.-P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schulz-Schaeffer, W.J.; Schmidt, B.; Kretzschmar, H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 1997, 146, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Nicholas, R.S.J.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef]
- Sakudo, A.; Lee, D.-C.; Saeki, K.; Nakamura, Y.; Inoue, K.; Matsumoto, Y.; Itohara, S.; Onodera, T. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2003, 308, 660–667. [Google Scholar] [CrossRef]
- Davies, P.; Brown, D.R. The chemistry of copper binding to PrP: Is there sufficient evidence to elucidate a role for copper in protein function? Biochem. J. 2008, 410, 237–244. [Google Scholar] [CrossRef]
- White, A.R.; Collins, S.J.; Maher, F.; Jobling, M.F.; Stewart, L.R.; Thyer, J.M.; Beyreuther, K.; Masters, C.L.; Cappai, R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 1999, 155, 1723–1730. [Google Scholar] [CrossRef]
- Hutter, G.; Heppner, F.L.; Aguzzi, A. No superoxide dismutase activity of cellular prion protein in vivo. Biol. Chem. 2003, 384, 1279–1285. [Google Scholar] [CrossRef]
- Guentchev, M.; Voigtländer, T.; Haberler, C.; Groschup, M.H.; Budka, H. Evidence for oxidative stress in experimental prion disease. Neurobiol. Dis. 2000, 7, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Klamt, F.; Dal-Pizzol, F.; Frota, M.; Walz, R.; Andrades, M.; Silva, E.; Brentani, R.; Izquierdo, I.; Moreira, J.C. Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic. Biol. Med. 2001, 30, 1137–1144. [Google Scholar] [CrossRef]
- Wong, B.-S.; Liu, T.; Li, R.; Pan, T.; Petersen, R.B.; Smith, M.A.; Gambetti, P.; Perry, G.; Manson, J.C.; Brown, D.R.; et al. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J. Neurochem. 2001, 76, 565–572. [Google Scholar] [CrossRef] [PubMed]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Mitsios, N.; Saka, M.; Krupinski, J.; Pennucci, R.; Sanfeliu, C.; Miguel Turu, M.; Gaffney, J.; Kumar, P.; Kumar, S.; Sullivan, M.; et al. Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J. Neurosci. Res. 2007, 85, 602–611. [Google Scholar] [CrossRef]
- Uzdensky, A.; Demyanenko, S.; Fedorenko, G.; Lapteva, T.; Fedorenko, A. Protein Profile and Morphological Alterations in Penumbra after Focal Photothrombotic Infarction in the Rat Cerebral Cortex. Mol. Neurobiol. 2017, 54, 4172–4188. [Google Scholar] [CrossRef]
- Weise, J.; Crome, O.; Sandau, R.; Schulz-Schaeffer, W.; Bähr, M.; Zerr, I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 2004, 372, 146–150. [Google Scholar] [CrossRef]
- Shyu, W.C.; Lin, S.Z.; Chiang, M.F.; Ding, D.C.; Li, K.W.; Chen, S.F.; Yang, H.I.; Li, H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 2005, 25, 8967–8977. [Google Scholar] [CrossRef]
- Spudich, A.; Frigg, R.; Kilic, E.; Kilic, U.; Oesch, B.; Raeber, A.; Bassetti, C.L.; Hermann, D.M. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 2005, 20, 442–449. [Google Scholar] [CrossRef]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nölting, S.; Hamann, G.F.; Kretzschmar, H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Bähr, M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Daude, N.; Gapeshina, H.; Dong, B.; Winship, I.; Westaway, D. Neuroprotective properties of the PrP-like Shadoo glycoprotein assessed in the middle cerebral artery occlusion model of ischemia. Prion 2015, 9, 376–393. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weise, J.; Doeppner, T.R.; Muller, T.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Witte, O.W.; Bahr, M. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor. Neurol. Neurosci. 2008, 26, 57–64. [Google Scholar] [PubMed]
- Doeppner, T.R.; Kaltwasser, B.; Schlechter, J.; Jaschke, J.; Kilic, E.; Bähr, M.; Hermann, D.M.; Weise, J. Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis. 2015, 6, e2024. [Google Scholar] [CrossRef]
- Zanetti, F.; Carpi, A.; Menabò, R.; Giorgio, M.; Schulz, R.; Valen, G.; Baysa, A.; Massimino, M.L.; Sorgato, M.C.; Bertoli, A.; et al. The cellular prion protein counteracts cardiac oxidative stress. Cardiovasc. Res. 2014, 104, 93–102. [Google Scholar] [CrossRef]
- Zhang, B.; Cowden, D.; Zhang, F.; Yuan, J.; Siedlak, S.; Abouelsaad, M.; Zeng, L.; Zhou, X.; O’Toole, J.; Das, A.S.; et al. Prion Protein Protects against Renal Ischemia/Reperfusion Injury. PloS One 2015, 10, e0136923. [Google Scholar] [CrossRef]
- Liang, J.; Bai, F.; Luo, G.; Wang, J.; Liu, J.; Ge, F.; Pan, Y.; Yao, L.; Du, R.; Li, X.; et al. Hypoxia induced overexpression of PrPC in gastric cancer cell lines. Cancer Biol.Ther. 2007, 6, 769–774. [Google Scholar] [CrossRef]
- Schuck, S.; Honsho, M.; Ekroos, K.; Shevchenko, A.; Simons, K. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA 2003, 100, 5795–5800. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.-L.; Lehmann, S.; Launay, j.-m.; Kellermann, O. Signal Transduction Through Prion Protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef]
- Chen, S.; Mangé, A.; Dong, L.; Lehmann, S.; Schachner, M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 2003, 22, 227–233. [Google Scholar] [CrossRef]
- Lewis, V.; Hooper, N.M. The role of lipid rafts in prion protein biology. Front. Biosci. 2011, 16, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Hooper, N.M. The prion protein and lipid rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Altmeppen, H.C.; Linsenmeier, L.; Chakroun, K.; Wegwitz, F.; Piontek, U.K.; Tatzelt, J.; Bate, C.; Magnus, T.; Glatzel, M. GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 2019, 15, e1007520. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.R.; Linden, R.; Prado, M.A.M.; Walz, R.; Sakamoto, A.C.; Izquierdo, I.; Brentani, R.R. Cellular prion protein: On the road for functions. FEBS Lett. 2002, 512, 25–28. [Google Scholar] [CrossRef]
- Linden, R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef]
- Hirsch, T.; Hernandez-Rapp, J.; Martin-Lannerée, S.; Launay, j.-m.; Mouillet-Richard, S. PrPC signalling in neurons: From basics to clinical challenges. Biochimie 2014, 104, 2–11. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Nozaki, K.; Nishimura, M.; Hashimoto, N. Mitogen-activated protein kinases and cerebral ischemia. Mol. Neurobiol. 2001, 23, 1–19. [Google Scholar] [CrossRef]
- Irving, E.A.; Bamford, M. Role of Mitogen- and Stress-Activated Kinases in Ischemic Injury. J. Cereb. Blood Flow Metab. 2002, 22, 631–647. [Google Scholar] [CrossRef]
- Ferrer, I.; Friguls, B.; Dalfó, E.; Planas, A.M. Early modifications in the expression of mitogen-activated protein kinase (MAPK/ERK), stress-activated kinases SAPK/JNK and p38, and their phosphorylated substrates following focal cerebral ischemia. Acta Neuropathol. 2003, 105, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Mutel, V.; Pietri, M.; Ermonval, M.; Mouillet-Richard, S.; Kellermann, O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13326–13331. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997, 20, 132–139. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Mou, R.-T.; Feng, D.-X.; Wang, Z.; Chen, G. The role of nitric oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar]
- Wang, V.; Chuang, T.-C.; Hsu, Y.-D.; Chou, W.-Y.; Kao, M.-C. Nitric oxide induces prion protein via MEK and p38 MAPK signaling. Biochem. Biophys. Res. Commun. 2005, 333, 95–100. [Google Scholar] [CrossRef]
- Shyu, W.C.; Chen, C.P.; Saeki, K.; Kubosaki, A.; Matusmoto, Y.; Onodera, T.; Ding, D.C.; Chiang, M.F.; Lee, Y.J.; Lin, S.Z.; et al. Hypoglycemia enhances the expression of prion protein and heat-shock protein 70 in a mouse neuroblastoma cell line. J. Neurosci. Res. 2005, 80, 887–894. [Google Scholar] [CrossRef]
- Shyu, W.C.; Harn, H.J.; Saeki, K.; Kubosaki, A.; Matsumoto, Y.; Onodera, T.; Chen, C.J.; Hsu, Y.D.; Chiang, Y.H. Molecular modulation of expression of prion protein by heat shock. Mol. Neurobiol. 2002, 26, 1–12. [Google Scholar] [CrossRef]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.-V.; Sunyach, C.; Druon, C.; Scarzello, S.; Checler, F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009, 284, 35973–35986. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Altmeppen, H.C.; Ulbrich, S.; Linsenmeier, L.; Krasemann, S.; Chakroun, K.; Acevedo-Morantes, C.Y.; Wille, H.; Tatzelt, J.; Glatzel, M. Secretory pathway retention of mutant prion protein induces p38-MAPK activation and lethal disease in mice. Sci. Rep. 2016, 6, 24970. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wu, B.; Le, N.T.T.; Imberdis, T.; Mercer, R.C.C.; Harris, D.A. Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog. 2018, 14, e1007283. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway as a Discovery Target in Stroke. J. Mol. Neurosci. 2016, 59, 90–98. [Google Scholar] [CrossRef] [PubMed]
- 86 Brazil, D.P.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664. [Google Scholar] [CrossRef]
- 87 Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harbor Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-kinase/Akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006, 34, 249–269. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Steinberg, G.; Zhao, H. The Akt pathway is involved in rapid ischemic tolerance in focal ischemia in Rats. Transl. Stroke Res. 2010, 1, 202–209. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Takahashi, T.; Hsieh, J.; Liao, J.; Steinberg, G.K.; Zhao, H. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 2008, 105, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H. Activation of phosphatidylinositol in 3-kinase by cellular prion protein and its role cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, T.; Li, B.; Rong, Y.; Verkhratsky, A.; Peng, L. Crosstalk Between MAPK/ERK and PI3K/AKT Signal Pathways During Brain Ischemia/Reperfusion. ASN Neuro 2015, 7, 1759091415602463. [Google Scholar] [CrossRef]
- Chen, R.; Ovbiagele, B.; Feng, W. Diabetes and Stroke: Epidemiology, Pathophysiology, Pharmaceuticals and Outcomes. Am. J. Med. Sci. 2016, 351, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.; Dhar, A.; Khalaj, S.; Desai, K.; Taghibiglou, C. Down regulation of brain cellular prion protein in an animal model of insulin resistance: Possible implication in increased prevalence of stroke in pre-diabetics/diabetics. Biochem. Biophys. Res. Commun. 2014, 448, 151–156. [Google Scholar] [CrossRef]
- Bake, S.; Okoreeh, A.; Khosravian, H.; Sohrabji, F. Insulin-like Growth Factor (IGF)-1 treatment stabilizes the microvascular cytoskeleton under ischemic conditions. Exp. Neurol. 2019, 311, 162–172. [Google Scholar] [CrossRef]
- Liu, T.; Yi, W.; Feng, B.; Zhou, Z.; Xiao, G. IGF-1-Induced Enhancement of PRNP Expression Depends on the Negative Regulation of Transcription Factor FOXO3a. PLoS ONE 2013, 8, e71896. [Google Scholar] [CrossRef]
- Hernández, M.P.; Sullivan, W.P.; Toft, D.O. The Assembly and Intermolecular Properties of the hsp70-Hop-hsp90 Molecular Chaperone Complex. J. Biol. Chem. 2002, 277, 38294–38304. [Google Scholar] [CrossRef] [PubMed]
- Odunuga, O.O.; Longshaw, V.M.; Blatch, G.L. Hop: More than an Hsp70/Hsp90 adaptor protein. BioEssays 2004, 26, 1058–1068. [Google Scholar] [CrossRef]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.M.; Muras, A.G.; Mancini, G.L.; Castro, R.M.P.S.; Ribeiro, K.C.B.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of Cellular Prion and Stress-Inducible Protein 1 Promotes Neuritogenesis and Neuroprotection by Distinct Signaling Pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Caetano, F.A.; Lopes, M.H.; Hajj, G.N.; Machado, C.F.; Pinto Arantes, C.; Magalhães, A.C.; Vieira Mde, P.; Américo, T.A.; Massensini, A.R.; Priola, S.A.; et al. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 2008, 28, 6691–6702. [Google Scholar] [CrossRef] [PubMed]
- Sakudo, A.; Lee, D.-C.; Li, S.; Nakamura, T.; Matsumoto, Y.; Saeki, K.; Itohara, S.; Ikuta, K.; Onodera, T. PrP cooperates with STI1 to regulate SOD activity in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2005, 328, 14–19. [Google Scholar] [CrossRef]
- Roffé, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Queiroz, N.G.T.; Young, K.; Rylett, R.J.; Markus, R.P.; Prado, M.A.M.; Martins, V.R. Role of α7 Nicotinic Acetylcholine Receptor in Calcium Signaling Induced by Prion Protein Interaction with Stress-inducible Protein 1. J. Biol. Chem. 2010, 285, 36542–36550. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, F.H.; Ostapchenko, V.G.; Xu, J.Z.; Di Guglielmo, G.M.; Fan, J.; Nicholls, P.J.; Caron, M.G.; Prado, V.F.; Prado, M.A.M. Mechanisms of neuroprotection against ischemic insult by stress-inducible phosphoprotein-1/prion protein complex. J. Neurochem. 2018, 145, 68–79. [Google Scholar] [CrossRef]
- Hajj, G.N.M.; Arantes, C.P.; Dias, M.V.S.; Roffé, M.; Costa-Silva, B.; Lopes, M.H.; Porto-Carreiro, I.; Rabachini, T.; Lima, F.R.; Beraldo, F.H.; et al. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci. 2013, 70, 3211–3227. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Soares, I.N.; Goncalves, D.F.; Fan, J.; Thomas, A.A.; Santos, T.G.; Mohammad, A.H.; Roffe, M.; Calder, M.D.; Nikolova, S.; et al. Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 2013, 27, 3594–3607. [Google Scholar] [CrossRef]
- Lee, S.-D.; Lai, T.W.; Lin, S.-Z.; Lin, C.-H.; Hsu, Y.-H.; Li, C.-Y.; Wang, H.-J.; Lee, W.; Su, C.-Y.; Yu, Y.-L.; et al. Role of stress-inducible protein-1 in recruitment of bone marrow derived cells into the ischemic brains. EMBO Mol. Med. 2013, 5, 1227–1246. [Google Scholar] [CrossRef]
- Bergeron, M.; Yu, A.Y.; Solway, K.E.; Semenza, G.L.; Sharp, F.R. Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur. J. Neurosci. 1999, 11, 4159–4170. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.F.; Hirata, P.H.; Magalhaes, A.C.; Lamour, G.; Li, H.; Maciejewski, A.; Belrose, J.C.; et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef] [PubMed]
- Soundarapandian, M.M.; Tu, W.H.; Peng, P.L.; Zervos, A.S.; Lu, Y. AMPA receptor subunit GluR2 gates injurious signals in ischemic stroke. Mol. Neurobiol. 2005, 32, 145–155. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Hardingham, G.E. Pro-survival signalling from the NMDA receptor. Biochem. Soc. Trans. 2006, 34, 936–938. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, W.; Axerio-Cilies, P.; Wang, Y.T. NMDARs in Cell Survival and Death: Implications in Stroke Pathogenesis and Treatment. Trends Mol. Med. 2020, 26, 533–551. [Google Scholar] [CrossRef]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Black, S.A.G.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 45. [Google Scholar] [CrossRef]
- Khosravani, H.; Zhang, Y.; Zamponi, G.W. Cellular prion protein null mice display normal AMPA receptor mediated long term depression. Prion 2008, 2, 48–50. [Google Scholar] [CrossRef]
- Vyklický, L., Jr.; Benveniste, M.; Mayer, M.L. Modulation of N-methyl-D-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990, 428, 313–331. [Google Scholar] [CrossRef]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.T.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef]
- Stys, P.K.; You, H.; Zamponi, G.W. Copper-dependent regulation of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012, 590, 1357–1368. [Google Scholar] [CrossRef]
- Huang, S.; Chen, L.; Bladen, C.; Stys, P.K.; Zamponi, G.W. Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol. Brain 2018, 11, 62. [Google Scholar] [CrossRef]
- Terpolilli, N.A.; Moskowitz, M.A.; Plesnila, N. Nitric oxide: Considerations for the treatment of ischemic stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1332–1346. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal 2015, 22, 772–784. [Google Scholar] [CrossRef]
- Akins, P.T.; Atkinson, R.P. Glutamate AMPA Receptor Antagonist Treatment for Ischaemic Stroke. Curr. Med. Res. Opin. 2002, 18, s9–s13. [Google Scholar] [CrossRef]
- Weiser, T. AMPA receptor antagonists for the treatment of stroke. Curr. Drug Targets. CNS Neurol. Dis. 2005, 4, 153–159. [Google Scholar] [CrossRef]
- De Mario, A.; Peggion, C.; Massimino, M.L.; Viviani, F.; Castellani, A.; Giacomello, M.; Lim, D.; Bertoli, A.; Sorgato, M.C. The prion protein regulates glutamate-mediated Ca(2+) entry and mitochondrial Ca(2+) accumulation in neurons. J. Cell Sci. 2017, 130, 2736–2746. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Kerrigan, T.L.; Griffiths, H.H.; Rushworth, J.V.; Whitehouse, I.J.; Hooper, N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012, 3, 1134. [Google Scholar] [CrossRef]
- Galasso, S.L.; Dyck, R.H. The role of zinc in cerebral ischemia. Mol. Med. 2007, 13, 380–387. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, F.; Yin, J.; Pan, R.; Shi, W.; Qi, Z.; Fang, Y.; Huang, Y.; Li, S.; Luo, Y.; et al. Synergistic Interaction Between Zinc and Reactive Oxygen Species Amplifies Ischemic Brain Injury in Rats. Stroke 2018, 49, 2200–2210. [Google Scholar] [CrossRef]
- Sun, Y.; Feng, X.; Ding, Y.; Li, M.; Yao, J.; Wang, L.; Gao, Z. Phased Treatment Strategies for Cerebral Ischemia Based on Glutamate Receptors. Front. Cell. Neurosci. 2019, 13, 168. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate Receptors in Health and Disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Puig, B.; Ferrer, I. Caspase-3-associated apoptotic cell death in excitotoxic necrosis of the entorhinal cortex following intraperitoneal injection of kainic acid in the rat. Neurosci. Lett. 2002, 321, 182–186. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B.; Domínguez, I.; Viñals, F. Active, phosphorylation-dependent MAP kinases, MAPK/ERK, SAPK/JNK and p38, and specific transcription factor substrates are differentially expressed following systemic administration of kainic acid to the adult rat. Acta Neuropathol. 2002, 103, 391–407. [Google Scholar]
- Carulla, P.; Bribián, A.; Rangel, A.; Gavín, R.; Ferrer, I.; Caelles, C.; Del Río, J.A.; Llorens, F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 2011, 22, 3041–3054. [Google Scholar] [CrossRef]
- Striebel, J.F.; Race, B.; Chesebro, B. Prion protein and susceptibility to kainate-induced seizures. Prion 2013, 7, 280–285. [Google Scholar] [CrossRef]
- Carulla, P.; Llorens, F.; Matamoros-Angles, A.; Aguilar-Calvo, P.; Espinosa, J.C.; Gavín, R.; Ferrer, I.; Legname, G.; Torres, J.M.; del Río, J.A. Involvement of PrPC in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 2015, 5, 11971. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Machado, C.F.; Roffe, M.; Hajj, G.N.; Lee, K.S.; Magalhães, A.C.; Caetano, F.A.; Mancini, G.L.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar] [CrossRef]
- Matsubara, T.; Satoh, K.; Homma, T.; Nakagaki, T.; Yamaguchi, N.; Atarashi, R.; Sudo, Y.; Uezono, Y.; Ishibashi, D.; Nishida, N. Prion protein interacts with the metabotropic glutamate receptor 1 and regulates the organization of Ca2+ signaling. Biochem. Biophys. Res. Commun. 2020, 525, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef]
- Hu, N.-W.; Nicoll, A.J.; Zhang, D.; Mably, A.J.; O’Malley, T.; Purro, S.A.; Terry, C.; Collinge, J.; Walsh, D.M.; Rowan, M.J. mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 2014, 5, 3374. [Google Scholar] [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Um, J.W.; Strittmatter, S.M. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013, 7, 37–41. [Google Scholar] [CrossRef]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Muller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Tuttle, M.D.; Lee, S.; Klein, L.E.; Takahashi, H.; Cox, T.O.; Gunther, E.C.; Zilm, K.W.; Strittmatter, S.M. Liquid and Hydrogel Phases of PrPC Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-beta Oligomers. Mol. Cell 2018, 72, 426–443. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nature Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Ostapchenko, V.G.; Caetano, F.A.; Guimaraes, A.L.S.; Ferretti, G.D.S.; Daude, N.; Bertram, L.; Nogueira, K.O.P.C.; Silva, J.L.; Westaway, D.; et al. Regulation of Amyloid β Oligomer Binding to Neurons and Neurotoxicity by the Prion Protein-mGluR5 Complex. J. Biol. Chem. 2016, 291, 21945–21955. [Google Scholar] [CrossRef]
- Rushworth, J.V.; Griffiths, H.H.; Watt, N.T.; Hooper, N.M. Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 2013, 288, 8935–8951. [Google Scholar] [CrossRef]
- Takagi, N.; Cheung, H.H.; Bissoon, N.; Teves, L.; Wallace, M.C.; Gurd, J.W. The effect of transient global ischemia on the interaction of Src and Fyn with the N-methyl-D-aspartate receptor and postsynaptic densities: Possible involvement of Src homology 2 domains. J. Cereb. Blood Flow Metab. 1999, 19, 880–888. [Google Scholar] [CrossRef]
- Cheung, H.H.; Takagi, N.; Teves, L.; Logan, R.; Wallace, M.C.; Gurd, J.W. Altered association of protein tyrosine kinases with postsynaptic densities after transient cerebral ischemia in the rat brain. J. Cereb. Blood Flow Metab. 2000, 20, 505–512. [Google Scholar] [CrossRef]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef]
- Knox, R.; Jiang, X. Fyn in Neurodevelopment and Ischemic Brain Injury. Dev. Neurosci. 2015, 37, 311–320. [Google Scholar] [CrossRef]
- Hou, X.Y.; Liu, Y.; Zhang, G.Y. PP2, a potent inhibitor of Src family kinases, protects against hippocampal CA1 pyramidal cell death after transient global brain ischemia. Neurosci. Lett. 2007, 420, 235–239. [Google Scholar] [CrossRef]
- Knox, R.; Brennan-Minnella, A.M.; Lu, F.; Yang, D.; Nakazawa, T.; Yamamoto, T.; Swanson, R.A.; Ferriero, D.M.; Jiang, X. NR2B phosphorylation at tyrosine 1472 contributes to brain injury in a rodent model of neonatal hypoxia-ischemia. Stroke 2014, 45, 3040–3047. [Google Scholar] [CrossRef]
- Du, C.P.; Gao, J.; Tai, J.M.; Liu, Y.; Qi, J.; Wang, W.; Hou, X.Y. Increased tyrosine phosphorylation of PSD-95 by Src family kinases after brain ischaemia. Biochem. J. 2009, 417, 277–285. [Google Scholar] [CrossRef]
- Knox, R.; Zhao, C.; Miguel-Perez, D.; Wang, S.; Yuan, J.; Ferriero, D.; Jiang, X. Enhanced NMDA receptor tyrosine phosphorylation and increased brain injury following neonatal hypoxia-ischemia in mice with neuronal Fyn overexpression. Neurobiol. Dis. 2013, 51, 113–119. [Google Scholar] [CrossRef]
- Fan, J.; Stemkowski, P.L.; Gandini, M.A.; Black, S.A.; Zhang, Z.; Souza, I.A.; Chen, L.; Zamponi, G.W. Reduced Hyperpolarization-Activated Current Contributes to Enhanced Intrinsic Excitability in Cultured Hippocampal Neurons from PrP(-/-) Mice. Front. Cell. Neurosci. 2016, 10, 74. [Google Scholar] [CrossRef]
- Adams, K.L.; Gallo, V. The diversity and disparity of the glial scar. Nat. Neurosci. 2018, 21, 9–15. [Google Scholar] [CrossRef]
- Honsa, P.; Pivonkova, H.; Harantova, L.; Butenko, O.; Kriska, J.; Dzamba, D.; Rusnakova, V.; Valihrach, L.; Kubista, M.; Anderova, M. Increased expression of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in reactive astrocytes following ischemia. Glia 2014, 62, 2004–2021. [Google Scholar] [CrossRef]
- Ehling, P.; Göb, E.; Bittner, S.; Budde, T.; Ludwig, A.; Kleinschnitz, C.; Meuth, S.G. Ischemia-induced cell depolarization: Does the hyperpolarization-activated cation channel HCN2 affect the outcome after stroke in mice? Exp. Transl. Stroke Med. 2013, 5, 16. [Google Scholar] [CrossRef][Green Version]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef]
- Zhang, R.L.; Zhang, Z.G.; Chopp, M. Ischemic stroke and neurogenesis in the subventricular zone. Neuropharmacology 2008, 55, 345–352. [Google Scholar] [CrossRef]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef]
- Stępień, T.; Tarka, S.; Chutorański, D.; Felczak, P.; Acewicz, A.; Wierzba-Bobrowicz, T. Neurogenesis in adult human brain after hemorrhage and ischemic stroke. Folia Neuropathol. 2018, 56, 293–300. [Google Scholar] [CrossRef]
- Kanazawa, M.; Takahashi, T.; Ishikawa, M.; Onodera, O.; Shimohata, T.; Del Zoppo, G.J. Angiogenesis in the ischemic core: A potential treatment target? J. Cereb. Blood Flow Metab. 2019, 39, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Hill, W.D.; Martin-Studdard, A.; Carothers, J.; Brailer, J.; Carroll, J. Blood into brain after stroke. Trends Mol. Med. 2002, 8, 452–453. [Google Scholar] [CrossRef]
- Shyu, W.C.; Lee, Y.J.; Liu, D.D.; Lin, S.Z.; Li, H. Homing genes, cell therapy and stroke. Front. Biosci. 2006, 11, 899–907. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Borlongan, C.V.; Glover, L.E.; Tajiri, N.; Kaneko, Y.; Freeman, T.B. The great migration of bone marrow-derived stem cells toward the ischemic brain: Therapeutic implications for stroke and other neurological disorders. Prog. Neurobiol. 2011, 95, 213–228. [Google Scholar] [CrossRef]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef]
- Parrie, L.E.; Crowell, J.A.E.; Moreno, J.A.; Suinn, S.S.; Telling, G.C.; Bessen, R.A. The cellular prion protein promotes neuronal regeneration after acute nasotoxic injury. Prion 2020, 14, 31–41. [Google Scholar] [CrossRef]
- Cirit, M.; Grant, K.G.; Haugh, J.M. Systemic perturbation of the ERK signaling pathway by the proteasome inhibitor, MG132. PLoS ONE 2012, 7, e50975. [Google Scholar] [CrossRef]
- Tanimoto, Y.; Kizaki, H. Proteasome inhibitors block Ras/ERK signaling pathway resulting in the downregulation of Fas ligand expression during activation-induced cell death in T cells. J. Biochem. 2002, 131, 319–326. [Google Scholar] [CrossRef]
- Sharp, F.R.; Bernaudin, M. HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 2004, 5, 437–448. [Google Scholar] [CrossRef]
- Ohtaki, H.; Nakamachi, T.; Dohi, K.; Shioda, S. Role of PACAP in Ischemic Neural Death. J. Mol. Neurosci. 2008, 36, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chiu, L.; Lee, H.-T.; Chiang, C.-W.; Liu, S.-P.; Hsu, Y.-H.; Lin, S.-Z.; Hsu, C.Y.; Hsieh, C.-H.; Shyu, W.-C. PACAP38/PAC1 signaling induces bone marrow-derived cells homing to ischemic brain. Stem Cells 2015, 33, 1153–1172. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.G.; Silva, I.R.; Costa-Silva, B.; Lepique, A.P.; Martins, V.R.; Lopes, M.H. Enhanced Neural Progenitor/Stem Cells Self-Renewal via the Interaction of Stress-Inducible Protein 1 with the Prion Protein. Stem Cells 2011, 29, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Molnár, Z.; Taguchi, A.; Nakano-Doi, A.; Lu, S.; Kasahara, Y.; Nakagomi, N.; Matsuyama, T. Leptomeningeal-derived doublecortin-expressing cells in poststroke brain. Stem Cells Dev. 2012, 21, 2350–2354. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Lee, H.-T.; Chen, C.-L.; Chang, C.-H.; Hsu, C.Y.; Shyu, W.-C. Role of FOXC1 in regulating APSCs self-renewal via STI-1/PrPC signaling. Theranostics 2019, 9, 6443–6465. [Google Scholar] [CrossRef]
- Bifari, F.; Berton, V.; Pino, A.; Kusalo, M.; Malpeli, G.; Di Chio, M.; Bersan, E.; Amato, E.; Scarpa, A.; Krampera, M.; et al. Meninges harbor cells expressing neural precursor markers during development and adulthood. Front. Cell. Neurosci. 2015, 9, 383. [Google Scholar] [CrossRef]
- Nakagomi, T.; Matsuyama, T. Leptomeninges: A novel stem cell niche with neurogenic potential. Stem Cell Investig. 2017, 4, 22. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Rogers, M.; Stahl, N.; Telling, G.; Prusiner, S.B. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 1993, 3, 319–329. [Google Scholar] [CrossRef]
- Chen, S.G.; Teplow, D.B.; Parchi, P.; Teller, J.K.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995, 270, 19173–19180. [Google Scholar] [CrossRef]
- Harris, D.A.; Huber, M.T.; Van Dijken, P.; Shyng, S.L.; Chait, B.T.; Wang, R. Processing of a cellular prion protein: Identification of N- and C-terminal cleavage sites. Biochemistry 1993, 32, 1009–1016. [Google Scholar] [CrossRef]
- Tagliavini, F.; Prelli, F.; Porro, M.; Salmona, M.; Bugiani, O.; Frangione, B. A soluble form of prion protein in human cerebrospinal fluid: Implications for prion-related encephalopathies. Biochem. Biophys. Res. Commun. 1992, 184, 1398–1404. [Google Scholar] [CrossRef]
- Haigh, C.L.; Marom, S.Y.; Collins, S.J. Copper, endoproteolytic processing of the prion protein and cell signalling. Front. Biosci. 2010, 15, 1086–1104. [Google Scholar] [CrossRef]
- McDonald, A.J.; Millhauser, G.L. PrP overdrive: Does inhibition of α-cleavage contribute to PrP(C) toxicity and prion disease? Prion 2014, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar] [PubMed]
- Liang, J.; Kong, Q. α-Cleavage of cellular prion protein. Prion 2012, 6, 453–460. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.E.; Mangé, A.; Nishida, N.; Créminon, C.; Casanova, D.; Lehmann, S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J. Biol. Chem. 2001, 276, 2286–2291. [Google Scholar] [CrossRef] [PubMed]
- Mangé, A.; Béranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef]
- Lau, A.; McDonald, A.; Daude, N.; Mays, C.E.; Walter, E.D.; Aglietti, R.; Mercer, R.C.; Wohlgemuth, S.; van der Merwe, J.; Yang, J.; et al. Octarepeat region flexibility impacts prion function, endoproteolysis and disease manifestation. EMBO Mol. Med. 2015, 7, 339–356. [Google Scholar] [CrossRef]
- Lewis, V.; Johanssen, V.A.; Crouch, P.J.; Klug, G.M.; Hooper, N.M.; Collins, S.J. Prion protein “gamma-cleavage”: Characterizing a novel endoproteolytic processing event. Cell. Mol. Life Sci. 2016, 73, 667–683. [Google Scholar] [CrossRef]
- Mohammadi, B.; Linsenmeier, L.; Shafiq, M.; Puig, B.; Galliciotti, G.; Giudici, C.; Willem, M.; Eden, T.; Koch-Nolte, F.; Lin, Y.H.; et al. Transgenic Overexpression of the Disordered Prion Protein N1 Fragment in Mice Does Not Protect Against Neurodegenerative Diseases Due to Impaired ER Translocation. Mol. Neurobiol. 2020, 57, 2812–2829. [Google Scholar] [CrossRef]
- Taylor, D.R.; Parkin, E.T.; Cocklin, S.L.; Ault, J.R.; Ashcroft, A.E.; Turner, A.J.; Hooper, N.M. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J. Biol. Chem. 2009, 284, 22590–22600. [Google Scholar] [CrossRef] [PubMed]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Kluth, M.A.; Bernreuther, C.; Thurm, D.; Jorissen, E.; Petrowitz, B.; Bartsch, U.; De Strooper, B.; et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol. Neurodegener. 2011, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Linsenmeier, L.; Mohammadi, B.; Wetzel, S.; Puig, B.; Jackson, W.S.; Hartmann, A.; Uchiyama, K.; Sakaguchi, S.; Endres, K.; Tatzelt, J.; et al. Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol. Neurodegener. 2018, 13, 18. [Google Scholar] [CrossRef]
- Castle, A.R.; Daude, N.; Gilch, S.; Westaway, D. Application of high-throughput, capillary-based Western analysis to modulated cleavage of the cellular prion protein. J. Biol. Chem. 2019, 294, 2642–2650. [Google Scholar] [CrossRef]
- Lewis, V. Analysis of Cellular Prion Protein Endoproteolytic Processing. Methods Mol. Biol. 2017, 1658, 119–132. [Google Scholar]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Piccoli, L.; Delle Monache, S.; Angelucci, A.; Misasi, R.; Sorice, M.; Mattei, V. Cellular and Molecular Mechanisms Mediated by recPrP(C) Involved in the Neuronal Differentiation Process of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 345. [Google Scholar] [CrossRef]
- Amin, L.; Nguyen, X.T.; Rolle, I.G.; D’Este, E.; Giachin, G.; Tran, T.H.; Šerbec, V.; Cojoc, D.; Legname, G. Characterization of prion protein function by focal neurite stimulation. J. Cell. Sci. 2016, 129, 3878–3891. [Google Scholar] [CrossRef]
- Megra, B.W.; Eugenin, E.A.; Berman, J.W. The Role of Shed PrPc in the Neuropathogenesis of HIV Infection. J. Immunol. 2017, 199, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Drew, S.C.; Boland, M.P.; Masters, C.L.; Barnham, K.J.; Lawson, V.A.; Collins, S.J. Dominant roles of the polybasic proline motif and copper in the PrP23-89-mediated stress protection response. J. Cell. Sci. 2009, 122, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Tumpach, C.; Drew, S.C.; Collins, S.J. The Prion Protein N1 and N2 Cleavage Fragments Bind to Phosphatidylserine and Phosphatidic Acid; Relevance to Stress-Protection Responses. PLoS ONE 2015, 10, e0134680. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; McGlade, A.R.; Collins, S.J. MEK1 transduces the prion protein N2 fragment antioxidant effects. Cell. Mol. Life Sci. 2015, 72, 1613–1629. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Tumpach, C.; Groveman, B.R.; Drew, S.C.; Haigh, C.L. Prion protein cleavage fragments regulate adult neural stem cell quiescence through redox modulation of mitochondrial fission and SOD2 expression. Cell. Mol. Life Sci. 2018, 75, 3231–3249. [Google Scholar] [CrossRef] [PubMed]
- Sunyach, C.; Cisse, M.A.; da Costa, C.A.; Vincent, B.; Checler, F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J. Biol. Chem. 2007, 282, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Groveman, B.R.; Williams, K.; Moore, R.; Race, B.; Haigh, C.L. Prion protein N1 cleavage peptides stimulate microglial interaction with surrounding cells. Sci. Rep. 2020, 10, 6654. [Google Scholar] [CrossRef]
- Haigh, C.L.; Lewis, V.A.; Vella, L.J.; Masters, C.L.; Hill, A.F.; Lawson, V.A.; Collins, S.J. PrPC-related signal transduction is influenced by copper, membrane integrity and the alpha cleavage site. Cell Res. 2009, 19, 1062–1078. [Google Scholar] [CrossRef]
- Schätzl, H.M.; Laszlo, L.; Holtzman, D.M.; Tatzelt, J.; DeArmond, S.J.; Weiner, R.I.; Mobley, W.C.; Prusiner, S.B. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 1997, 71, 8821–8831. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef]
- Pimpinelli, F.; Lehmann, S.; Maridonneau-Parini, I. The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. Eur. J. Neurosci. 2005, 21, 2063–2072. [Google Scholar] [CrossRef]
- Vella, L.; Sharples, R.; Lawson, V.; Masters, C.; Cappai, R.; Hill, A. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 2007, 211, 582–590. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, X.; Bai, Y.; Zhang, Z.; Zhao, D. Cellular prion protein released on exosomes from macrophages binds to Hsp70. Acta Biochim. Biophys. Sin. 2010, 42, 345–350. [Google Scholar] [CrossRef]
- Ritchie, A.J.; Crawford, D.M.; Ferguson, D.J.P.; Burthem, J.; Roberts, D.J. Normal prion protein is expressed on exosomes isolated from human plasma. Br. J. Haematol. 2013, 163, 678–680. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef]
- Vella, L.J.; Greenwood, D.L.V.; Cappai, R.; Scheerlinck, J.-P.Y.; Hill, A.F. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 2008, 124, 385–393. [Google Scholar] [CrossRef]
- Falker, C.; Hartmann, A.; Guett, I.; Dohler, F.; Altmeppen, H.; Betzel, C.; Schubert, R.; Thurm, D.; Wegwitz, F.; Joshi, P.; et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem. 2016, 137, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Brenna, S.; Altmeppen, H.C.; Mohammadi, B.; Rissiek, B.; Schlink, F.; Ludewig, P.; Failla, A.V.; Schneider, C.; Glatzel, M.; Puig, B.; et al. Brain-Derived Extracellular Vesicles are Highly Enriched in the Prion Protein and Its C1 Fragment: Relevance for Cellular Uptake and Implications in Stroke. bioRxiv 2019. [Google Scholar] [CrossRef]
- Guitart, K.; Loers, G.; Buck, F.; Bork, U.; Schachner, M.; Kleene, R. Improvement of neuronal cell survival by astrocyte-derived exosomes under hypoxic and ischemic conditions depends on prion protein. Glia 2016, 64, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Ramasubramanian, L.; Kumar, P.; Wang, A. Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine. Biomolecules 2020, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Controll. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, T.R.; Herz, J.; Görgens, A.; Schlechter, J.; Ludwig, A.-K.; Radtke, S.; de Miroschedji, K.; Horn, P.A.; Giebel, B.; Hermann, D.M. Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl. Med. 2015, 4, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chopp, M. Exosome Therapy for Stroke. Stroke 2018, 49, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, T.R.; Bähr, M.; Hermann, D.M.; Giebel, B. Concise Review: Extracellular Vesicles Overcoming Limitations of Cell Therapies in Ischemic Stroke. Stem Cells Transl. Med. 2017, 6, 2044–2052. [Google Scholar] [CrossRef]
- Otero-Ortega, L.; Laso-García, F.; Gómez-de Frutos, M.; Fuentes, B.; Diekhorst, L.; Díez-Tejedor, E.; Gutiérrez-Fernández, M. Role of Exosomes as a Treatment and Potential Biomarker for Stroke. Transl. Stroke Res. 2019, 10, 241–249. [Google Scholar] [CrossRef]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Cunningham, C.J.; Lawrence, C.B.; Pinteaux, E.; Allan, S.M. Therapeutic potential of extracellular vesicles in preclinical stroke models: A systematic review and meta-analysis. BMJ Open Sci. 2020, 4, e100047. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.H.A.M.; Heijnen, H.F.G.; van Bergen en Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, L.; Kuang, Y.; Venkataramani, V.; Jin, F.; Hein, K.; Zafeiriou, M.P.; Lenz, C.; Moebius, W.; Kilic, E.; et al. Extracellular Vesicles Derived from Neural Progenitor Cells––A Preclinical Evaluation for Stroke Treatment in Mice. Transl. Stroke Res. 2020. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Katamine, S.; Nishida, N.; Moriuchi, R.; Shigematsu, K.; Sugimoto, T.; Nakatani, A.; Kataoka, Y.; Houtani, T.; Shirabe, S.; et al. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996, 380, 528–531. [Google Scholar] [CrossRef]
- Li, A.; Sakaguchi, S.; Atarashi, R.; Roy, B.C.; Nakaoke, R.; Arima, K.; Okimura, N.; Kopacek, J.; Shigematsu, K. Identification of a novel gene encoding a PrP-like protein expressed as chimeric transcripts fused to PrP exon 1/2 in ataxic mouse line with a disrupted PrP gene. Cell Mol. Neurobiol. 2000, 20, 553–567. [Google Scholar] [CrossRef]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef]
- Rossi, D.; Cozzio, A.; Flechsig, E.; Klein, M.A.; Rülicke, T.; Aguzzi, A.; Weissmann, C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001, 20, 694–702. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Ratté, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.R.; Collinge, J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002, 21, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Nuvolone, M.; Hermann, M.; Sorce, S.; Russo, G.; Tiberi, C.; Schwarz, P.; Minikel, E.; Sanoudou, D.; Pelczar, P.; Aguzzi, A. Strictly co-isogenic C57BL/6J-Prnp-/- mice: A rigorous resource for prion science. J. Exp. Med. 2016, 213, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Balkaya, M.G.; Trueman, R.C.; Boltze, J.; Corbett, D.; Jolkkonen, J. Behavioral outcome measures to improve experimental stroke research. Behav. Brain Res. 2018, 352, 161–171. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puig, B.; Yang, D.; Brenna, S.; Altmeppen, H.C.; Magnus, T. Show Me Your Friends and I Tell You Who You Are: The Many Facets of Prion Protein in Stroke. Cells 2020, 9, 1609. https://doi.org/10.3390/cells9071609
Puig B, Yang D, Brenna S, Altmeppen HC, Magnus T. Show Me Your Friends and I Tell You Who You Are: The Many Facets of Prion Protein in Stroke. Cells. 2020; 9(7):1609. https://doi.org/10.3390/cells9071609
Chicago/Turabian StylePuig, Berta, Denise Yang, Santra Brenna, Hermann Clemens Altmeppen, and Tim Magnus. 2020. "Show Me Your Friends and I Tell You Who You Are: The Many Facets of Prion Protein in Stroke" Cells 9, no. 7: 1609. https://doi.org/10.3390/cells9071609
APA StylePuig, B., Yang, D., Brenna, S., Altmeppen, H. C., & Magnus, T. (2020). Show Me Your Friends and I Tell You Who You Are: The Many Facets of Prion Protein in Stroke. Cells, 9(7), 1609. https://doi.org/10.3390/cells9071609