The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Search Strategy
3. The Innate Immune System during the Aging Process
3.1. Adaptive Immunity
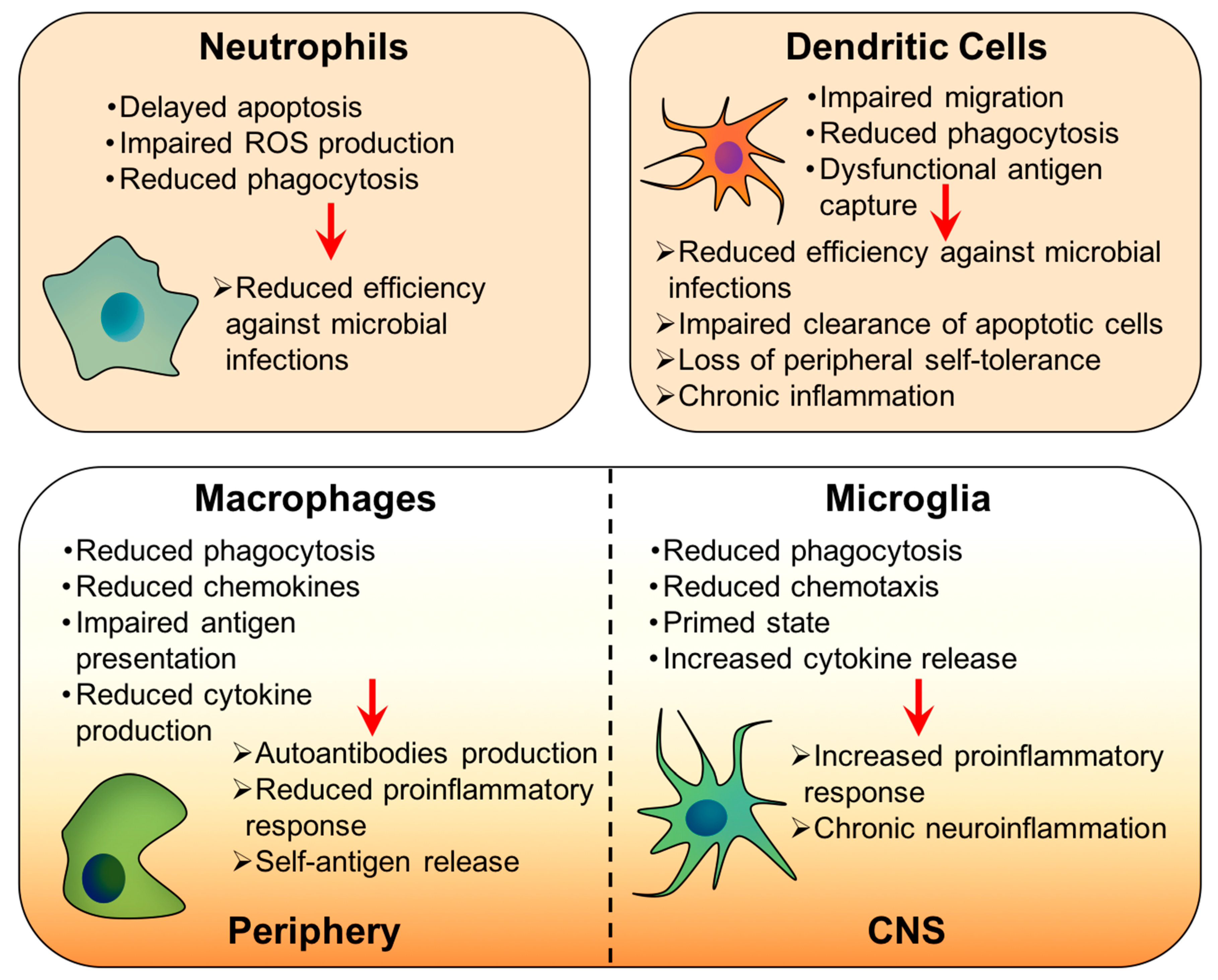
3.2. Innate Immunity
4. Inflammaging or Age-Related Inflammation
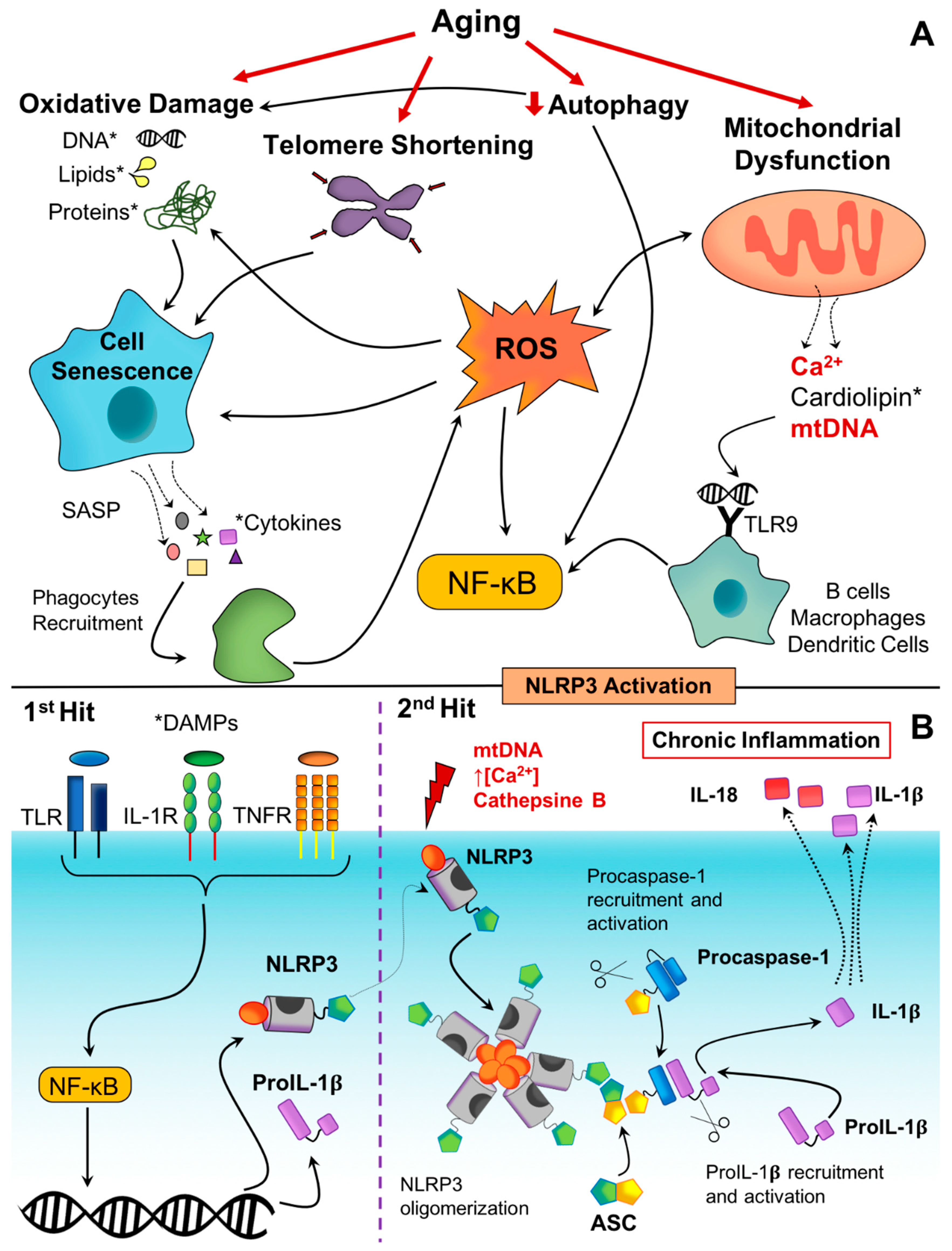
4.1. Molecular and Cellular Mechanisms in Inflammaging
4.1.1. Inflammasomes as Molecular Sensors of Danger Signals
4.1.2. Cellular Senescence and Immunosenescence
4.1.3. Activation of the DNA Damage Response
4.1.4. Mitochondrial Dysfunction
4.1.5. Defective Autophagy and Mitophagy
5. Age-Associated Diseases: Implication of the NLRP3 Inflammasome
5.1. Cancer
5.2. Metabolic Disorders
Type-2 Diabetes and Obesity
5.3. Neurodegenerative Disorders
5.3.1. Alzheimer’s Disease
5.3.2. Parkinson’s Disease
5.3.3. Other Age-Related Diseases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Ferrando-Martínez, S.; Ruiz-Mateos, E.; Hernandez, A.; Gutiérrez, E.; Rodríguez-Méndez, M.D.M.; Ordóñez-Fernández, A.; Leal, M. Age-related deregulation of naive T cell homeostasis in elderly humans. AGE 2010, 33, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.-P. Aging of the Immune System: How Much Can the Adaptive Immune System Adapt? Immunity 2006, 24, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C. Innate Immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, R.F. C-Reactive Protein, Inflammation, and Innate Immunity. Immunol. Res. 2001, 24, 163–176. [Google Scholar] [CrossRef]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Tortorella, C.; Piazzolla, G.; Spaccavento, F.; Jirillo, E.; Antonaci, S. Age-related effects of oxidative metabolism and cyclic AMP signaling on neutrophil apoptosis. Mech. Ageing Dev. 1999, 110, 195–205. [Google Scholar] [CrossRef]
- Fülöp, T.; Larbi, A.; Douziech, N.; Fortin, C.; Guérard, K.-P.; Lesur, O.; Khalil, A.; Dupuis, G. Signal transduction and functional changes in neutrophils with aging. Aging Cell 2004, 3, 217–226. [Google Scholar] [CrossRef]
- Ito, Y.; Kajkenova, O.; Feuers, R.J.; Udupa, K.B.; Desai, V.G.; Epstein, J.; Hart, R.W.; Lipschitz, D.A. Impaired glutathione peroxidase activity accounts for the age-related accumulation of hydrogen peroxide in activated human neutrophils. J. Gerontol. Ser. A Biol. Sci. Med Sci. 1998, 53, M169–M175. [Google Scholar] [CrossRef] [PubMed]
- Wenisch, C.; Patruta, S.; Daxböck, F.; Krause, R.; Hörl, W. Effect of age on human neutrophil function. J. Leukoc. Biol. 2000, 67, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hemmi, H. Dendritic cells: Translating innate to adaptive immunity. In Current Topics in Microbiology and Immunology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2006; Volume 311, pp. 17–58. [Google Scholar]
- Müller, L.; Fülöp, T.; Pawelec, G. Immunosenescence in vertebrates and invertebrates. Immun. Ageing 2013, 10, 12. [Google Scholar] [CrossRef]
- Corberand, J.; Ngyen, F.; Laharrague, P.; Fontanilles, A.M.; Gleyzes, B.; Gyrard, E.; Senegas, C. Polymorphonuclear Functions and Aging in Humans. J. Am. Geriatr. Soc. 1981, 29, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Agrawal, S.; Cao, J.-N.; Su, H.; Osann, K.; Gupta, S. Altered innate immune functioning of dendritic cells in elderly humans: A role of phosphoinositide 3-kinase-signaling pathway. J. Immunol. 2007, 178, 6912–6922. [Google Scholar] [CrossRef]
- Wu, X.; Molinaro, C.; Johnson, N.; Casiano, C.A. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: Implications for systemic autoimmunity. Arthritis Rheum. 2001, 44, 2642–2652. [Google Scholar] [CrossRef]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fülöp, T. Innate immunosenescence: Effect of aging on cells and receptors of the innate immune system in humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef]
- Locati, M.; Mantovani, A.; Sica, A. Macrophage Activation and Polarization as an Adaptive Component of Innate Immunity. In Advances in Immunology; Elsevier BV: Amsterdam, The Netherlands, 2013; Volume 120, pp. 163–184. [Google Scholar]
- Bowdish, D.M.E.; Loffredo, M.; Mukhopadhyay, S.; Mantovani, A.; Gordon, S. Macrophage receptors implicated in the “adaptive” form of innate immunity. Microbes Infect. 2007, 9, 1680–1687. [Google Scholar] [CrossRef]
- Keller, R. The macrophage response to infectious agents: Mechanisms of macrophage activation and tumour cell killing. Res. Immunol. 1993, 144, 271–273. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Lloberas, J.; Tur, J.; Vico, T.; Celada, A. Molecular and Cellular Aspects of Macrophage Aging. In Handbook of Immunosenescence; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; pp. 1631–1663. [Google Scholar]
- Takahashi, R.; Totsuka, S.; Ishigami, A.; Kobayashi, Y.; Nagata, K. Attenuated phagocytosis of secondary necrotic neutrophils by macrophages in aged and SMP30 knockout mice. Geriatr. Gerontol. Int. 2015, 16, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Villanueva, J.; Pena, J.; de la Fuente, M. Cell mediated immunity in ageing. Comp. Biochem. Physiol. Part A Physiol. 1991, 99, 1–4. [Google Scholar] [CrossRef]
- Herrero, C.; Marqués, L.; Lloberas, J.; Celada, A. IFN-γ–dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J. Clin. Investig. 2001, 107, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Mahbub, S.; Deburghgraeve, C.R.; Kovacs, E.J. Advanced Age Impairs Macrophage Polarization. J. Interf. Cytokine Res. 2012, 32, 18–26. [Google Scholar] [CrossRef]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.E.; del Proposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 2011, 187, 6208–6216. [Google Scholar] [CrossRef]
- Cao, J.J.; Wronski, T.J.; Iwaniec, U.; Phleger, L.; Kurimoto, P.; Boudignon, B.; Halloran, B.P. Aging Increases Stromal/Osteoblastic Cell-Induced Osteoclastogenesis and Alters the Osteoclast Precursor Pool in the Mouse. J. Bone Miner. Res. 2005, 20, 1659–1668. [Google Scholar] [CrossRef]
- Frei, K.; Siepl, C.; Groscurth, P.; Bodmer, S.; Schwerdel, C.; Fontana, A. Antigen presentation and tumor cytotoxicity by interferon-γ-treated microglial cells. Eur. J. Immunol. 1987, 17, 1271–1278. [Google Scholar] [CrossRef]
- Shrikant, P.; Benveniste, E.N. The central nervous system as an immunocompetent organ: Role of glial cells in antigen presentation. J. Immunol. 1996, 157, 157. [Google Scholar]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2010, 10, 263–276. [Google Scholar] [CrossRef]
- Floden, A.M.; Combs, C.K. Microglia Demonstrate Age-Dependent Interaction with Amyloid-β Fibrils. JAD 2011, 25, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2010, 33, 195. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef]
- Roubenoff, R.; Harris, T.B.; Abad, L.W.; Wilson, P.W.F.; Dallal, G.E.; Dinarello, C.A. Monocyte cytokine production in an elderly population: Effect of age and inflammation. J. Gerontol. Ser. A Biol. Sci. Med Sci. 1998, 53, M20–M26. [Google Scholar] [CrossRef] [PubMed]
- Brüünsgaard, H.; Pedersen, B.K. Age-related inflammatory cytokines, and disease. Immunol. Allergy Clin. North Am. 2003, 23, 15–39. [Google Scholar] [CrossRef]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. New York Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Roubenoff, R.; Parise, H.; Payette, H.A.; Abad, L.W.; D’Agostino, R.; Jacques, P.F.; Wilson, P.W.F.; Dinarello, C.A.; Harris, T.B. Cytokines, insulin-like growth factor 1, sarcopenia, and mortality in very old community-dwelling men and women: The Framingham Heart Study. Am. J. Med. 2003, 115, 429–435. [Google Scholar] [CrossRef]
- Pawelec, G.; Goldeck, D.; Derhovanessian, E. Inflammation, ageing and chronic disease. Curr. Opin. Immunol. 2014, 29, 23–28. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chan, C. The role of inflammasome in Alzheimer’s disease. Ageing Res. Rev. 2014, 15, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.L.; de Oliveira, E.M.L.; Pontillo, A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 29, 26–34. [Google Scholar] [CrossRef]
- Fan, Z.; Pan, Y.-T.; Zhang, Z.-Y.; Yang, H.; Yu, S.-Y.; Zheng, Y.; Ma, J.-H.; Wang, X.-M. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef]
- Aganna, E.; Martinon, F.; Hawkins, P.N.; Ross, J.B.; Swan, D.; Booth, D.; Lachmann, H.J.; Gaudet, R.; Woo, P.; Feighery, C.; et al. Association of mutations in theNALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002, 46, 2445–2452. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 55, 1977–1987. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Savage, C.D.; Lopez-Castejon, G.; Denes, A.; Brough, D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Belardo, G.; Santoro, M.G. NF-κB: A Stress-Regulated Switch for Cell Survival. Antioxid. Redox Signal. 2006, 8, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.L.; Haley, B.; Roose-Girma, M.; Phung, Q.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2009, 11, 136–140. [Google Scholar] [CrossRef]
- Niemi, K.; Teirilä, L.; Lappalainen, J.; Rajamäki, K.; Baumann, M.; Öörni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum Amyloid A Activates the NLRP3 Inflammasome via P2X7 Receptor and a Cathepsin B-Sensitive Pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: A Novel Link between Cholesterol Metabolism and Inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J.; di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Dong, X.; Vijg, J.; Suh, Y. Genetic evidence for common pathways in human age-related diseases. Aging Cell 2015, 14, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Siebold, A.P.; Sharpless, N.E. Review: A meta-analysis of GWAS and age-associated diseases. Aging Cell 2012, 11, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.V.; Deryabin, P.I.; Giukova, А.А.; Nikolsky, N.N. “Social Life” of Senescent Cells: What Is SASP and Why Study It? Acta Nat. 2018, 10, 4–14. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 2013, 14, 428–436. [Google Scholar] [CrossRef]
- De Veale, B.; Brummel, T.; Seroude, L. Immunity and aging: The enemy within? Aging Cell 2004, 3, 195–208. [Google Scholar] [CrossRef]
- Arnold, C.R.; Wolf, J.; Brunner, S.; Herndler-Brandstetter, D.; Grubeck-Loebenstein, B. Gain and Loss of T Cell Subsets in Old Age—Age-Related Reshaping of the T Cell Repertoire. J. Clin. Immunol. 2011, 31, 137–146. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J.; Li, G.; Uhrlaub, J.; Renkema, K.R.; Smithey, M. Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin. Immunol. 2012, 24, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Larbi, A.; Pawelec, G. Human T Cell Aging and the Impact of Persistent Viral Infections. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Woycicki, R.; Ilnytskyy, Y.; Metz, G.A.S.; Kovalchuk, I.; Kovalchuk, O. Immunosenescence is associated with altered gene expression and epigenetic regulation in primary and secondary immune organs. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; di Fagagna, F.D.; Jackson, S.P. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. DNA damage: A trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat. Rev. Immunol. 2006, 6, 261–270. [Google Scholar] [CrossRef]
- Liu, Z.-G.; Baskaran, R.; Lea-Chou, E.T.; Wood, L.D.; Chen, Y.; Karin, M.; Wang, J.Y.J. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 1996, 384, 273–276. [Google Scholar] [CrossRef]
- González, S.; López-Soto, A.; Suárez-Álvarez, B.; López-Vázquez, A.; López-Soto, A.; Gonzalez, S. NKG2D ligands: Key targets of the immune response. Trends Immunol. 2008, 29, 397–403. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Frontini, M.; Vijayakumar, M.; Garvin, A.; Clarke, N. A ChIP-chip approach reveals a novel role for transcription factor IRF1 in the DNA damage response. Nucleic Acids Res. 2009, 37, 1073–1085. [Google Scholar] [CrossRef]
- Hayden, M.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Bonafe, M.; Storci, G.; Franceschi, C. Inflamm-aging of the stem cell niche: Breast cancer as a paradigmatic example. Bioessays 2011, 34, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Karakasilioti, I.; Kamileri, I.; Chatzinikolaou, G.; Kosteas, T.; Vergadi, E.; Robinson, A.R.; Tsamardinos, I.; Rozgaja, T.A.; Siakouli, S.; Tsatsanis, C.; et al. DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 2013, 18, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Nuñez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef]
- Rossier, M. T channels and steroid biosynthesis: In search of a link with mitochondria. Cell Calcium 2006, 40, 155–164. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Csordás, G.; das, S.; Garcia-Perez, C.; Saotome, M.; Roy, S.S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Larsson, N.-G. Somatic Mitochondrial DNA Mutations in Mammalian Aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; Pessin, J.E. Recent progress in metabolic signaling pathways regulating aging and life span. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2014, 69, S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Sevini, F.; Giuliani, C.; Vianello, D.; Giampieri, E.; Santoro, A.; Biondi, F.; Garagnani, P.; Passarino, G.; Luiselli, D.; Capri, M.; et al. mtDNA mutations in human aging and longevity: Controversies and new perspectives opened by high-throughput technologies. Exp. Gerontol. 2014, 56, 234–244. [Google Scholar] [CrossRef]
- Zamzami, N.; Hirsch, T.; Dallaporta, B.; Petit, P.X.; Kroemer, G. Mitochondrial implication in accidental and programmed cell death: Apoptosis and necrosis. J. Bioenerg. Biomembr. 1997, 29, 185–193. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Shintani, Y.; Kapoor, A.; Kaneko, M.; Smolenski, R.T.; D’Acquisto, F.; Coppen, S.R.; Harada-Shoji, N.; Lee, H.J.; Thiemermann, C.; Takashima, S.; et al. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc. Natl. Acad. Sci USA 2013, 110, 5109–5114. [Google Scholar] [CrossRef]
- Zhang, J.-Z.; Liu, Z.; Liu, J.; Ren, J.-X.; Sun, T. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int. J. Mol. Med. 2014, 33, 817–824. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, N.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Acevedo-Arozena, A.; Adachi, H.; Adams, C.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Choi, A.J.S.; Ryter, S.W. Autophagy in Inflammatory Diseases. Int. J. Cell Biol. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Yang, S.; Imamura, Y.; Jenkins, R.W.; Cañadas, I.; Kitajima, S.; Aref, A.R.; Brannon, A.L.; Oki, E.; Castoreno, A.; Zhu, Z.; et al. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol. Res. 2016, 4, 520–530. [Google Scholar] [CrossRef]
- Liu, T. Regulation of Inflammasome by Autophagy. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; Volume 1209, pp. 109–123. [Google Scholar]
- Kee, B.P.; Ng, J.G.; Ng, C.C.; Hilmi, I.; Goh, K.; Chua, K.H. Genetic polymorphisms of ATG16L1 and IRGM genes in Malaysian patients with Crohn’s disease. J. Dig. Dis. 2019, 21, 29–37. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Pai, P.-Y.; Hsueh, H.-W.; Yuan, S.-S.; Hsieh, Y.-C. Complete Induction of Autophagy Is Essential for Cardioprotection in Sepsis. Ann. Surg. 2011, 253, 1190–1200. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2007, 4, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef]
- Pantano, C.; Reynaert, N.; van der Vliet, A.; Janssen–Heininger, Y.M.W. Redox-Sensitive Kinases of the Nuclear Factor-κB Signaling Pathway. Antioxid. Redox Signal. 2006, 8, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2010, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Yoon, S.-O.; Lee, H.J.; Oh, J.Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget 2017, 8, 40817–40831. [Google Scholar] [CrossRef]
- Niida, M.; Tanaka, M.; Kamitani, T. Downregulation of active IKKβ by Ro52-mediated autophagy. Mol. Immunol. 2010, 47, 2378–2387. [Google Scholar] [CrossRef]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.S.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Jessop, F.; Hamilton, R.F.; Rhoderick, J.F.; Shaw, P.K.; Holian, A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol. Appl. Pharmacol. 2016, 309, 101–110. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Landskron, G.; de la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Bin-Lee, S.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.-M.; Dayem, A.A.; Cho, S.-G. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. Int. J. Mol. Sci. 2017, 18, 1544. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Kamp, D.W.; Shacter, E.; A Weitzman, S. Chronic inflammation, and cancer: The role of the mitochondria. Oncol. 2011, 25, 400. [Google Scholar]
- Schieber, M.; Chandel, N. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Kim, M.K.; Song, Y.S. Stress Response, Inflammaging, and Cancer. In Inflammation, Advancing Age and Nutrition; Elsevier BV: Amsterdam, The Netherlands, 2014; pp. 49–53. [Google Scholar]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; di Fusco, D.; Sica, G.S.; Sileri, P.; Macdonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2014, 34, 3493–3503. [Google Scholar] [CrossRef]
- Civenni, G.; Shinde, D.; Zoma, M.; Albino, D.; Costales, P.; Moris, F.; Carbone, G.; Catapano, C. The multi-kinase inhibitor EC-70124 delivers a double-hit to prostate cancer stem cells interfering with both STAT3 and NF-kB signaling. Eur. Urol. Suppl. 2017, 16, e1294. [Google Scholar] [CrossRef]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-κB Inducers Upregulate cFLIP, a Cycloheximide-Sensitive Inhibitor of Death Receptor Signaling. Mol. Cell. Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.-L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Bullani, R.R.; Huard, B.; Viard-Leveugle, I.; Saurat, J.-H.; French, L.; Byers, H.R.; Irmler, M.; Tschopp, J. Selective Expression of FLIP in Malignant Melanocytic Skin Lesions. J. Investig. Dermatol. 2001, 117, 360–364. [Google Scholar] [CrossRef]
- Humphreys, L.; Espona-Fiedler, M.; Longley, D.B. FLIP as a therapeutic target in cancer. FEBS J. 2018, 285, 4104–4123. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-U.; Hosotani, R.; Wada, M.; Doi, R.; Kosiba, T.; Fujimoto, K.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Nishimura, Y.; et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur. J. Cancer 1999, 35, 1374–1380. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- la Rosa, F.A.; Pierce, J.W.; Sonenshein, G.E. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol. Cell. Biol. 1994, 14, 1039–1044. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S. NF-κB Controls Cell Growth and Differentiation through Transcriptional Regulation of Cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef]
- Chen, F.; Castranova, V.; Shi, X. New Insights into the Role of Nuclear Factor-κB in Cell Growth Regulation. Am. J. Pathol. 2001, 159, 387–397. [Google Scholar] [CrossRef]
- Biliran, H.; Banerjee, S.; Thakur, A.; Sarkar, F.H.; Bollig-Fischer, A.; Ahmed, F.; Wu, J.; Sun, Y.; Liao, J.D. c-Myc Induced Chemosensitization Is Mediated by Suppression of Cyclin D1 Expression and Nuclear Factor—B Activity in Pancreatic Cancer Cells. Clin. Cancer Res. 2007, 13, 2811–2821. [Google Scholar] [CrossRef]
- Dahlman, J.M.; Wang, D.; Bakkar, N.; Guttridge, D.C. The RelA/p65 subunit of NF-κB specifically regulates cyclin D1 protein stability: Implications for cell cycle withdrawal and skeletal myogenesis. J. Cell. Biochem. 2009, 106, 42–51. [Google Scholar] [CrossRef]
- Brücher, B.L.; Lang, F.; Jamall, I.S. NF-κB signaling and crosstalk during carcinogenesis. 4open 2019, 2, 13. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Furth, P.A. STAT signaling in different breast cancer sub-types. Mol. Cell. Endocrinol. 2013, 382, 612–615. [Google Scholar] [CrossRef]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, E.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Mechanisms of apoptosis avoidance in cancer. Curr. Opin. Oncol. 1999, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Clohessy, J.G.; Zhuang, J.; de Boer, J.; Gil-Gómez, G.; Brady, H.J.M. Mcl-1 Interacts with Truncated Bid, and Inhibits Its Induction of Cytochromec Release and Its Role in Receptor-mediated Apoptosis. J. Biol. Chem. 2005, 281, 5750–5759. [Google Scholar] [CrossRef]
- Welcker, M.; Lukas, J.; Strauss, M.; Bartek, J. Enhanced protein stability: A novel mechanism of D-type cyclin over-abundance identified in human sarcoma cells. Oncogene 1996, 13, 419–425. [Google Scholar]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Bodnar, M.; Guey, B.; Hacot, S.; Lantuejoul, S. Abstract 2038: A novel role for the NLRP3 inflammasome in lung cancer. Mol. Cellul. Biol. 2015, 75, 2038. [Google Scholar] [CrossRef]
- Pontillo, A.; Bricher, P.; Leal, V.N.C.; Lima, S.; de Souza, P.R.E.; Crovella, S. Role of inflammasome genetics in susceptibility to HPV infection and cervical cancer development. J. Med Virol. 2016, 88, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Muneer, K.M.; Tamimi, I.A.; Chang, M.E.; Ata, M.O.; Yusuf, N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol. Appl. Pharmacol. 2013, 270, 70–76. [Google Scholar] [CrossRef]
- Latz, E.; Duewell, P. NLRP3 inflammasome activation in inflammaging. Semin. Immunol. 2018, 40, 61–73. [Google Scholar] [CrossRef]
- Hotamisligil, G.; Shargill, N.; Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Weisberg, S.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Mohammadi, M.; Gozashti, M.H.; Aghadavood, M.; Mehdizadeh, M.R.; Hayatbakhsh, M.M. Clinical Significance of Serum IL-6 and TNF-α Levels in Patients with Metabolic Syndrome. Rep. Biochem. Mol. Biol. 2017, 6, 74–79. [Google Scholar]
- Di Dalmazi, G.; Pagotto, U.; Pasquali, R.; Vicennati, V. Glucocorticoids and Type 2 Diabetes: From Physiology to Pathology. J. Nutr. Metab. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2014, 58, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Leong, A.; Porneala, B.; Dupuis, J.; Florez, J.C.; Meigs, J.B. Type 2 Diabetes Genetic Predisposition, Obesity, and All-Cause Mortality Risk in the U.S.: A Multiethnic Analysis. Diabetes Care 2016, 39, 539–546. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 2008, 8, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.K.S.; Péraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M.; Morrell, V. IRS-1-Mediated Inhibition of Insulin Receptor Tyrosine Kinase Activity in TNF-alpha- and Obesity-Induced Insulin Resistance. Science 1996, 271, 665–670. [Google Scholar] [CrossRef]
- Kahn, C.R.; White, M.F. The insulin receptor and the molecular mechanism of insulin action. J. Clin. Investig. 1988, 82, 1151–1156. [Google Scholar] [CrossRef]
- Van Greevenbroek, M.M.J.; Schalkwijk, C.G.; A Stehouwer, C.D. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: Causes and consequences. Neth. J. Med. 2013, 71, 174–187. [Google Scholar]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef]
- Calder, P.C.; Dimitriadis, G.; Newsholme, P. Glucose metabolism in lymphoid and inflammatory cells and tissues. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Hisata, S.; Park, M.-A.; de Nicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Cosentino, F. Molecular mechanisms of vascular dysfunction and cardiovascular biomarkers in type 2 diabetes. Cardiovasc. Diagn. Ther. 2014, 4, 324–332. [Google Scholar]
- Prattichizzo, F.; Giuliani, A.; Ceka, A.; Rippo, M.R.; Bonfigli, A.R.; Testa, I.; Procopio, A.D.; Olivieri, F. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin. Epigenetics 2015, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Szic, K.S.V.; Declerck, K.; Vidaković, M.; Berghe, W.V. From inflammaging to healthy aging by dietary lifestyle choices: Is epigenetics the key to personalized nutrition? Clin. Epigenet. 2015, 7, 33. [Google Scholar] [CrossRef]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef]
- Bonfigli, A.R.; Spazzafumo, L.; Prattichizzo, F.; Bonafè, M.; Mensà, E.; Micolucci, L.; Giuliani, A.; Fabbietti, P.; Testa, R.; Boemi, M.; et al. Leukocyte telomere length and mortality risk in patients with type 2 diabetes. Oncotarget 2016, 7, 50835–50844. [Google Scholar] [CrossRef]
- Shalev, A.; Pise-Masison, C.A.; Radonovich, M.; Hoffmann, S.C.; Hirshberg, B.; Brady, J.N.; Harlan, D.M. Oligonucleotide Microarray Analysis of Intact Human Pancreatic Islets: Identification of Glucose-Responsive Genes and a Highly Regulated TGFβ Signaling Pathway. Endocrinology 2002, 143, 3695–3698. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef]
- Kwon, H.; Pessin, J.E. Adipokines Mediate Inflammation and Insulin Resistance. Front. Endocrinol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Yiannikouris, F.; Gupte, M.; Putnam, K.; Cassis, L.A. Adipokines, and blood pressure control. Curr. Opin. Nephrol. Hypertens. 2010, 19, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kataria, M.A.; Saini, V.; Yadav, A. Role of leptin and adiponectin in insulin resistance. Clin. Chim. Acta 2013, 417, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. 2011, 108, 15324–15329. [Google Scholar] [CrossRef]
- Jager, J.; Grémeaux, T.; Cormont, M.; le Marchand-Brustel, Y.; Tanti, J.-F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2006, 148, 241–251. [Google Scholar] [CrossRef]
- Ahmad, R.; Thomas, R.; Kochumon, S.; Sindhu, R.T.S. Increased adipose tissue expression of IL-18R and its ligand IL-18 associates with inflammation and insulin resistance in obesity. Immun. Inflamm. Dis. 2017, 5, 318–335. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.L.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Grant, R.W.; Dixit, V.D. Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Kullberg, B.J.; Tack, C.J.; van Krieken, H.; Kim, S.-H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, U.; Hall, K.; Luchsinger, J.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef]
- Neumann, H. Control of glial immune function by neurons. Glia 2001, 36, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Gimsa, U.; Bechmann, I.; Nitsch, R. Differential expression of costimulatory molecules B7-1 and B7-2 on microglial cells induced by Th1 and Th2 cells in organotypic brain tissue. Glia 2001, 36, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A. DNA microarray analysis of the aging brain. Chem. Senses 2002, 27, 299–306. [Google Scholar] [CrossRef]
- Godbout, J.P.; Moreau, M.; Lestage, J.; Chen, J.; Sparkman, N.L.; Connor, J.O.; Castanon, N.; Kelley, K.W.; Dantzer, R.; Johnson, R. Aging Exacerbates Depressive-like Behavior in Mice in Response to Activation of the Peripheral Innate Immune System. Neuropsychopharmacology 2007, 33, 2341–2351. [Google Scholar] [CrossRef]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R. Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef]
- Alzheimer’s Association 2019. Alzheimer’s Disease Facts and Figures. Alzheimers Dement 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Lane, C.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2017, 25, 59–70. [Google Scholar] [CrossRef]
- Luchsinger, J.; Gustafson, D.R. Adiposity, Type 2 Diabetes, and Alzheimer’s Disease. JAD 2009, 16, 693–704. [Google Scholar] [CrossRef]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-Analysis of Alzheimer’s Disease Risk with Obesity, Diabetes, and Related Disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Young-Pearse, T.L.; Bai, J.; Chang, R.; Zheng, J.B.; Lo-Turco, J.J.; Selkoe, D. A Critical Function for β-Amyloid Precursor Protein in Neuronal Migration Revealed by In Utero RNA Interference. J. Neurosci. 2007, 27, 14459–14469. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J. Sleep Facilitates Clearance of Metabolites from the Brain: Glymphatic Function in Aging and Neurodegenerative Diseases. Rejuvenat. Res. 2013, 16, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Ellwardt, E.; Walsh, J.T.; Kipnis, J.; Zipp, F. Understanding the Role of T Cells in CNS Homeostasis. Trends Immunol. 2016, 37, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Wagner, M.; Whitaker, N.; Grubeck-Loebenstein, B.; Jansen-Durr, P. The amyloid β peptide Aβ (25-35) induces apoptosis independent of p53. FEBS Lett. 2000, 470, 221–225. [Google Scholar] [CrossRef]
- Mudher, A.; Lovestone, S. Alzheimer’s disease—Do tauists and baptists finally shake hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Di Carlo, M.; Giacomazza, D.; Biagio, P.L.S. Alzheimer’s disease: Biological aspects, therapeutic perspectives, and diagnostic tools. J. Physics Condens. Matter 2012, 24, 244102. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.-C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and Nutritional Antioxidants in Human Diseases. Front. Physiol. 2018, 9, 477. [Google Scholar] [CrossRef]
- Ando, K.; Uemura, K.; Kuzuya, A.; Maesako, M.; Asada-Utsugi, M.; Kubota, M.; Aoyagi, N.; Yoshioka, K.; Okawa, K.; Inoue, H.; et al. N-cadherin Regulates p38 MAPK Signaling via Association with JNK-associated Leucine Zipper Protein. J. Biol. Chem. 2010, 286, 7619–7628. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2016, 101, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.; Tremblay, M.-E. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol. Stress 2018, 9, 9–21. [Google Scholar] [CrossRef]
- Gutierrez, E.R.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Diaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Lian, D.; Lai, J.; Wu, Y.; Wang, L.; Chen, Y.; Zhang, Y.; Boini, K.M.; Huang, Y.; Chen, Y. Cathepsin B-Mediated NLRP3 Inflammasome Formation and Activation in Angiotensin II -Induced Hypertensive Mice: Role of Macrophage Digestion Dysfunction. Cell. Physiol. Biochem. 2018, 50, 1585–1600. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef]
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2012, 493, 674–678. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Stutz, A.; Horvath, G.L.; Monks, B.G.; Latz, E. ASC Speck Formation as a Readout for Inflammasome Activation. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013; Volume 1040, pp. 91–101. [Google Scholar]
- Franklin, B.S.; Bossaller, L.; de Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef]
- Haque, E.; Akther, M.; Jakaria Kim, I.; Azam, S.; Choi, D. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson’s Disease. Mov. Disord. 2019, 35, 20–33. [Google Scholar] [CrossRef]
- Kim, M.-J.; Yoon, J.-H.; Ryu, J.-H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef]
- Luo, Y.; Hoffer, A.; Hoffer, B.; Qi, X. Mitochondria: A Therapeutic Target for Parkinson’s Disease? Int. J. Mol. Sci. 2015, 16, 20704–20730. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Tampellini, D.; Gerlach, M.; Riederer, P.; Fariello, R.G.; Sulzer, D. Substantia nigra neuromelanin: Structure, synthesis, and molecular behaviour. Mol. Pathol. 2001, 54, 414–418. [Google Scholar] [PubMed]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martínez-Vicente, M.; Parent, A.; Sepúlveda, M.G.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Pinho, B.R.; Reis, S.; Hartley, R.C.; Murphy, M.P.; Oliveira, J.M. Mitochondrial superoxide generation induces a parkinsonian phenotype in zebrafish and huntingtin aggregation in human cells. Free. Radic. Biol. Med. 2018, 130, 318–327. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models, and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction, and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord. 2011, 27, 8–30. [Google Scholar] [CrossRef]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into Neuroinflammation in Parkinson’s Disease: From Biomarkers to Anti-Inflammatory Based Therapies. BioMed Res. Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef]
- Beraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K. α-Synuclein Alters Toll-Like Receptor Expression. Front. Mol. Neurosci. 2011, 5. [Google Scholar] [CrossRef]
- Li, X.; Dong, C.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; Petersen, N.O.; Woodside, M.T. Early stages of aggregation of engineered α-synuclein monomers and oligomers in solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Riggs, B.L.; Wahner, H.W.; Mazess, R.B.; Seeman, E.; Offord, K.P.; Dunn, W.L.; Johnson, K.A.; Melton, L.J. Changes in Bone Mineral Density of the Proximal Femur and Spine with Aging. J. Clin. Investig. 1982, 70, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.A.; Pietschmann, P.; Savastano, S. Osteoporosis and Sarcopenia Increase Frailty Syndrome in the Elderly. Front. Endocrinol. 2019, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Colón-Emeric, C.S. Recent Advances: Osteoporosis in the “Oldest Old”. Curr. Osteoporos. Rep. 2013, 11, 270–275. [Google Scholar] [CrossRef]
- Al-Saedi, A.; Stupka, N.; Duque, G. Pathogenesis of Osteoporosis. In Drug Delivery; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2020; pp. 1–15. [Google Scholar]
- Teitelbaum, S. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Udagawa, N. The mechanism of osteoclast differentiation from macrophages: Possible roles of T lymphocytes in osteoclastogenesis. J. Bone Miner. Metab. 2003, 21, 337–343. [Google Scholar] [CrossRef]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen, and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef]
- Hill, S.C.; Namde, M.; Dwyer, A.; Poznanski, A.; Canna, S.W.; Goldbach-Mansky, R. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA). Pediatr. Radiol. 2006, 37, 145–152. [Google Scholar] [CrossRef]
- Bonar, S.L.; Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.; Chen, D.; Grimston, S.K.; Hickman-Brecks, C.L.; Ravindran, S.; McAlinden, A.; et al. Constitutively Activated NLRP3 Inflammasome Causes Inflammation and Abnormal Skeletal Development in Mice. PLoS ONE 2012, 7, e35979. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Kreider, J.; Galecki, A.; Goldstein, S.A. Preservation of femoral bone thickness in middle age predicts survival in genetically heterogeneous mice. Aging Cell 2011, 10, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, M.G.H.; de Hair, M.J.H.; Schuller, Y.; van de Sande, G.P.M.; Wijbrandts, C.A.; Dinant, H.J.; Gerlag, D.M.; Tak, P.-P. The Features of the Synovium in Early Rheumatoid Arthritis According to the 2010 ACR/EULAR Classification Criteria. PLoS ONE 2012, 7, e36668. [Google Scholar] [CrossRef]
- Nalbant, S.; Birlik, A.M. Cytokines in Rheumatoid Arthritis (RA). In New Developments in the Pathogenesis of Rheumatoid Arthritis; IntechOpen: London, UK, 2017. [Google Scholar]
- Noack, M.; Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef]
- Mathews, R.J.; Robinson, J.I.; Battellino, M.; Wong, C.; Taylor, J.; Eyre, S.; Churchman, S.; Wilson, A.G.; Isaacs, J.D.; Hyrich, K.L.; et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann. Rheum. Dis. 2013, 73, 1202–1210. [Google Scholar] [CrossRef]
- Choulaki, C.; Papadaki, G.; Repa, A.; Kampouraki, E.; Kambas, K.; Ritis, K.; Bertsias, G.; Boumpas, D.T.; Sidiropoulos, P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. 2015, 17, 257. [Google Scholar] [CrossRef]
- Xie, Q.; Wei, M.; Zhang, B.; Kang, X.; Liu, D.; Zheng, W.; Pan, X.; Quan, Y.; Liao, D.; Shen, J. MicroRNA-33 regulates the NLRP3 inflammasome signaling pathway in macrophages. Mol. Med. Rep. 2017, 17, 3318–3327. [Google Scholar] [CrossRef]
- Ruscitti, P.; Cipriani, P.; di Benedetto, P.; Liakouli, V.; Berardicurti, O.; Carubbi, F.; Ciccia, F.; Alvaro, S.; Triolo, G.; Giacomelli, R. Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1β via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: A pos. Clin. Exp. Immunol. 2015, 182, 35–44. [Google Scholar] [CrossRef]
- Kolly, L.; Busso, N.; Palmer, G.; Talabot-Ayer, D.; Chobaz, V.; So, A. Expression and function of the NALP3 inflammasome in rheumatoid synovium. Immunology 2010, 129, 178–185. [Google Scholar] [CrossRef]
- Ab-Joosten, L.; Radstake, T.R.; Lubberts, E.; Bersselaar, L.A.M.V.D.; van Riel, P.L.C.M.; van Lent, P.L.E.M.; Barrera, P.; Berg, W.B.V.D. Association of interleukin-18 expression with enhanced levels of both interleukin-1? And tumor necrosis factor? In knee synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2003, 48, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.-P.; Zhou, L.-J.; Lu, S.-Y.; Liang, Y.-E.; Chen, X.-Y.; Liu, L.; Lin, J. Association of IL-18 promoter gene polymorphisms with rheumatoid arthritis: A meta-analysis. Mol. Biol. Rep. 2014, 41, 8211–8217. [Google Scholar] [CrossRef] [PubMed]
- Chalan, P.; Berg, A.V.D.; Kroesen, B.-J.; Brouwer, L.; Boots, A. Rheumatoid Arthritis, Immunosenescence and the Hallmarks of Aging. Curr. Aging Sci. 2015, 8, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, R.F.A.G.D.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130. [Google Scholar] [CrossRef]
- Dhingra, R.; Vasan, R.S. Age as a Risk Factor. Med Clin. North Am. 2011, 96, 87–91. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1098. [Google Scholar] [CrossRef]
- Moslehi, J.; Depinho, R.A.; Sahin, E. Telomeres and mitochondria in the aging heart. Circ. Res. 2012, 110, 1226–1237. [Google Scholar] [CrossRef]
- Sasaki, Y.; Ikeda, Y.; Iwabayashi, M.; Akasaki, Y.; Ohishi, M. The Impact of Autophagy on Cardiovascular Senescence and Diseases. Int. Hear. J. 2017, 58, 666–673. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013, 1–12. [Google Scholar] [CrossRef]
- Garcia, K.C.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebastian-Valverde, M.; Pasinetti, G.M. The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process. Cells 2020, 9, 1552. https://doi.org/10.3390/cells9061552
Sebastian-Valverde M, Pasinetti GM. The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process. Cells. 2020; 9(6):1552. https://doi.org/10.3390/cells9061552
Chicago/Turabian StyleSebastian-Valverde, Maria, and Giulio M. Pasinetti. 2020. "The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process" Cells 9, no. 6: 1552. https://doi.org/10.3390/cells9061552
APA StyleSebastian-Valverde, M., & Pasinetti, G. M. (2020). The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process. Cells, 9(6), 1552. https://doi.org/10.3390/cells9061552