Macrophage Modification Strategies for Efficient Cell Therapy


 ,
,  ,
,
Abstract

1. Development of Macrophages
2. The M1/M2 Paradigm
3. Macrophages as Perfect Cells for Reprogramming
3.1. The M1 Macrophage Applications
3.2. The M2 Macrophage Applications
4. The Role of the Monocyte-Macrophage System in Disease, or Why Should We Reprogram Macrophages?
4.1. Inflammatory Diseases
4.2. Proliferative Diseases
5. The Existing Approaches for Macrophage Reprogramming
5.1. Early Attempts at Obtaining Specific Phenotypes
5.2. Activation with Signaling Molecules
5.3. Identification of Relevant Genomic Targets
5.4. Transient Modifications
5.5. Genome Editing
5.6. Delivery of Genetic Constructs to Cells
5.7. The Induced Pluripotent Stem Cell Technologies
5.8. In Vivo Models for Genome Editing
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Steven, J.L. Metchnikoff on the Comparative Pathology of Inflammation. Glasgow Med. J. 1892, 38, 195–205. [Google Scholar]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Kosmac, K.; Peck, B.; Walton, R.; Mula, J.; Kern, P.; Bamman, M.; Dennis, R.; Jacobs, C.; Lattermann, C.; Johnson, D.; et al. Immunohistochemical Identification of Human Skeletal Muscle Macrophages. BIO-PROTOCOL 2018, 8, e2883. [Google Scholar] [CrossRef] [PubMed]
- Elchaninov, A.V.; Fatkhudinov, T.K.; Vishnyakova, P.A.; Lokhonina, A.V.; Sukhikh, G.T. Phenotypical and Functional Polymorphism of Liver Resident Macrophages. Cells 2019, 8, 1032. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.; Koniski, A.; McGrath, K.E.; Vemishetti, R.; Emerson, R.; De Mesy-Bentley, K.K.L.; Waugh, R.; Palis, J. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 2007, 109, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef]
- Bertrand, J.Y.; Jalil, A.; Klaine, M.; Jung, S.; Cumano, A.; Godin, I. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005, 106, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Geissmann, F. Development and maintainance of resident macrophages HHS Public Access Author manuscript. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M. (Ed.) Macrophages Origin, Functions and Biointervention; Springer Nature: Cham, Germany, 2017; Volume 62, ISBN 9783319540894. [Google Scholar]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. In Macrophage Activation-Biology and Disease; Bhat, K.H., Ed.; IntechOpen: London, UK, 2020; ISBN 9781789846454. [Google Scholar]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Koller, T.; Slavov, N. High-throughput single-cell proteomics quantifies the emergence of macrophage heterogeneity. bioRxiv 2019, 665307. [Google Scholar] [CrossRef]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef] [PubMed]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef]
- Yi, X.; Zhang, J.; Zhuang, R.; Wang, S.; Cheng, S.; Zhang, D.; Xie, J.; Hu, W.; Liu, X.; Zhang, Y.; et al. Silencing LAIR-1 in human THP-1 macrophage increases foam cell formation by modulating PPARγ and M2 polarization. Cytokine 2018, 111, 194–205. [Google Scholar] [CrossRef]
- Kimbrough, D.; Wang, S.H.; Wright, L.H.; Mani, S.K.; Kasiganesan, H.; LaRue, A.C.; Cheng, Q.; Nadig, S.N.; Atkinson, C.; Menick, D.R. HDAC inhibition helps post-MI healing by modulating macrophage polarization. J. Mol. Cell. Cardiol. 2018, 119, 51–63. [Google Scholar] [CrossRef]
- Cao, Q.; Rong, S.; Repa, J.J.; St Clair, R.; Parks, J.S.; Mishra, N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef]
- Mantovani, A.; Schioppa, T.; Biswas, S.K.; Marchesi, F.; Allavena, P.; Sica, A. Tumor-associated macrophages and dendritic cells as prototypic type II polarized myeloid populations. Tumori 2003, 89, 459–468. [Google Scholar] [CrossRef]
- Bellón, T.; Martínez, V.; Lucendo, B.; del Peso, G.; Castro, M.J.; Aroeira, L.S.; Rodríguez-Sanz, A.; Ossorio, M.; Sánchez-Villanueva, R.; Selgas, R.; et al. Alternative activation of macrophages in human peritoneum: Implications for peritoneal fibrosis. Nephrol. Dial. Transplant. 2011, 26, 2995–3005. [Google Scholar] [CrossRef]
- Jiang, H.-R.; Milovanovic, M.; Allan, D.; Niedbala, W.; Besnard, A.-G.; Fukada, S.Y.; Alves-Filho, J.C.; Togbe, D.; Goodyear, C.S.; Linington, C.; et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur. J. Immunol. 2012, 42, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.; Ahn, M.; Matsumoto, Y. Mechanism of experimental autoimmune encephalomyelitis in Lewis rats: Recent insights from macrophages. Anat. Cell Biol. 2012, 45, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Maestas, D.R.; Housseau, F.; Elisseeff, J.H. Key players in the immune response to biomaterial scaffolds for regenerative medicine. Adv. Drug Deliv. Rev. 2017, 114, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7. [Google Scholar] [CrossRef]
- Fukui, S.; Iwamoto, N.; Takatani, A.; Igawa, T.; Shimizu, T.; Umeda, M.; Nishino, A.; Horai, Y.; Hirai, Y.; Koga, T.; et al. M1 and M2 Monocytes in rheumatoid arthritis: A contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front. Immunol. 2018, 8, 1958. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; PassosdosSantos, R.; Gaestel, M.; David, S. TNF and Increased Intracellular Iron Alter Macrophage Polarization to a Detrimental M1 Phenotype in the Injured Spinal Cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef]
- Qin, H.; Yan, Z.; Lee, K.S.; Gibson, S.A.; Li, X.; Harms, A.S.; Buckley, B.A.; Standaert, D.G.; Benveniste, E.N. Role of M1-polarized Macrophages in Parkinson’s Disease Models. J. Immunol. 2016, 196, 126-4. [Google Scholar]
- Mukhopadhyay, D.; Mukherjee, S.; Roy, S.; Dalton, J.E.; Kundu, S.; Sarkar, A.; Das, N.K.; Kaye, P.M.; Chatterjee, M. M2 Polarization of Monocytes-Macrophages Is a Hallmark of Indian Post Kala-Azar Dermal Leishmaniasis. PLoS Negl. Trop. Dis. 2015, 9, e0004145. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; De Lagrán, M.M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Il-13 effector Functions. Annu. Rev. Immunol. 2003, 21, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Schippers, M.; Reker-Smit, C.; Martinez, F.O.; Helming, L.; Poelstra, K.; Melgert, B.N. Hepatic localization of macrophage phenotypes during fibrogenesis and resolution of fibrosis in mice and humans. Front. Immunol. 2014, 5, 430. [Google Scholar] [CrossRef]
- Nelson, M.P.; Christmann, B.S.; Dunaway, C.W.; Morris, A.; Steele, C. Experimental Pneumocystis lung infection promotes M2a alveolar macrophage-derived MMP12 production. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L469. [Google Scholar] [CrossRef][Green Version]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- De Palma, M. Origins of Brain Tumor Macrophages. Cancer Cell 2016, 30, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef]
- Weagel, E.; Smith, C.; Liu, P.G.; Robison, R.; O’Neill, K. Macrophage Polarization and Its Role in Cancer. J. Clin. Cell. Immunol. 2015, 6, 4–11. [Google Scholar] [CrossRef]
- Yin, M.; Shen, J.; Yu, S.; Fei, J.; Zhu, X.; Zhao, J.; Zhai, L.; Sadhukhan, A.; Zhou, J. Tumor-associated macrophages (Tams): A critical activator in ovarian cancer metastasis. OncoTargets Ther. 2019, 12, 8687–8699. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. Inhibition of Pulmonary Metastasis by Intravenous Injection of Specifically Activated Macrophages. Cancer Res. 1974, 34, 1074–1078. [Google Scholar] [PubMed]
- West, M.A.; Bennet, T.; Seatter, S.C.; Clair, L.; Bellingham, J. LPS pretreatment reprograms macrophage LPS-stimulated TNF and IL-1 release without protein tyrosine kinase activation. J. Leukoc. Biol. 1997, 61, 88–95. [Google Scholar] [CrossRef]
- Andreesen, R.; Scheibenbogen, C.; Brugger, W.; Krause, S.; Meerpohl, H.G.; Leser, H.G.; Engler, H.; Löhr, G.W. Adoptive Transfer of Tumor Cytotoxic Macrophages Generated in Vitro from Circulating Blood Monocytes: A New Approach to Cancer Immunotherapy. Cancer Res. 1990, 50, 7450–7456. [Google Scholar]
- Poh, A.R.; Ernst, M. Targeting macrophages in cancer: From bench to bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef]
- Stansfield, B.K.; Ingram, D.A. Clinical significance of monocyte heterogeneity. Clin. Transl. Med. 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Passlick, B.; Flieger, D.; Loms Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Aharinejad, S.; Abraham, D.; Paulus, P.; Abri, H.; Hofmann, M.; Grossschmidt, K.; Schäfer, R.; Stanley, E.R.; Hofbauer, R. Colony-stimulating factor-1 antisense treatment suppresses growth of human tumor xenografts in mice. Cancer Res. 2002, 62, 5317–5324. [Google Scholar] [PubMed]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; Van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Invest. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Shields, C.W.; Evans, M.A.; Wang, L.L.-W.; Baugh, N.; Iyer, S.; Wu, D.; Zhao, Z.; Pusuluri, A.; Ukidve, A.; Pan, D.C.; et al. Cellular backpacks for macrophage immunotherapy. Sci. Adv. 2020, 6, eaaz6579. [Google Scholar] [CrossRef]
- Allavena, P.; Peccatori, F.; Maggioni, D.; Erroi, A.; Sironi, M.; Mantovani, A.; Colombo, N.; Lissoni, A.; Mangioni, C.; Galazka, A.; et al. Intraperitoneal Recombinant γ-Interferon in Patients with Recurrent Ascitic Ovarian Carcinoma: Modulation of Cytotoxicity and Cytokine Production in Tumor-associated Effectors and of Major Histocompatibility Antigen Expression on Tumor Cells. Cancer Res. 1990, 50, 7318–7323. [Google Scholar]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Gerrick, K.Y.; Gerrick, E.R.; Gupta, A.; Wheelan, S.J.; Yegnasubramanian, S.; Jaffee, E.M. Transcriptional profiling identifies novel regulators of macrophage polarization. PLoS ONE 2018, 13, e0208602. [Google Scholar] [CrossRef]
- Padgett, L.E.; Burg, A.R.; Lei, W.; Tse, H.M. Loss of NADPH oxidase-derived superoxide skews macrophage phenotypes to delay type 1 diabetes. Diabetes 2015, 64, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Xie, G.; Xiao, H.; Ding, F.; Bao, W.; Zhang, M. CXCR4 knockdown prevents inflammatory cytokine expression in macrophages by suppressing activation of MAPK and NF-κB signaling pathways. Cell Biosci. 2019, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, L.; Estecha, A.; Samaniego, R.; Sánchez-Ramón, S.; Vega, M.Á.; Sánchez-Mateos, P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 2011, 117, 88–97. [Google Scholar] [CrossRef]
- Gautier, E.L.; Ivanov, S.; Williams, J.W.; Huang, S.C.-C.; Marcelin, G.; Fairfax, K.; Wang, P.L.; Francis, J.S.; Leone, P.; Wilson, D.B.; et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J. Exp. Med. 2014, 211, 1525–1531. [Google Scholar] [CrossRef]
- Rosas, M.; Davies, L.C.; Giles, P.J.; Liao, C.-T.; Kharfan, B.; Stone, T.C.; O’Donnell, V.B.; Fraser, D.J.; Jones, S.A.; Taylor, P.R. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 2014, 344, 645–648. [Google Scholar] [CrossRef]
- Fainaru, O.; Woolf, E.; Lotem, J.; Yarmus, M.; Brenner, O.; Goldenberg, D.; Negreanu, V.; Bernstein, Y.; Levanon, D.; Jung, S.; et al. Runx3 regulates mouse TGF-beta-mediated dendritic cell function and its absence results in airway inflammation. EMBO J. 2004, 23, 969–979. [Google Scholar] [CrossRef]
- A-Gonzalez, N.; Guillen, J.A.; Gallardo, G.; Diaz, M.; de la Rosa, J.V.; Hernandez, I.H.; Casanova-Acebes, M.; Lopez, F.; Tabraue, C.; Beceiro, S.; et al. The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. Nat. Immunol. 2013, 14, 831–839. [Google Scholar] [CrossRef]
- Kohyama, M.; Ise, W.; Edelson, B.T.; Wilker, P.R.; Hildner, K.; Mejia, C.; Frazier, W.A.; Murphy, T.L.; Murphy, K.M. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 2009, 457, 318–321. [Google Scholar] [CrossRef]
- Gautier, E.L.; Chow, A.; Spanbroek, R.; Marcelin, G.; Greter, M.; Jakubzick, C.; Bogunovic, M.; Leboeuf, M.; van Rooijen, N.; Habenicht, A.J.; et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J. Immunol. 2012, 189, 2614–2624. [Google Scholar] [CrossRef]
- Xu, C.; Lu, Z.; Luo, Y.; Liu, Y.; Cao, Z.; Shen, S.; Li, H.; Liu, J.; Chen, K.; Chen, Z.; et al. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat. Commun. 2018, 9, 4092. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Keller, A.-A.; Maess, M.B.; Schnoor, M.; Scheiding, B.; Lorkowski, S. Transfecting Macrophages. Methods Mol. Biol. 2018, 1784, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Odaka, C.; Mizuochi, T. Macrophages are involved in DNA degradation of apoptotic cells in murine thymus after administration of hydrocortisone. Cell Death Differ. 2002, 9, 104–112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, Y.; Mout, R.; Luther, D.C.; Liu, Y.; Castellanos-García, L.; Burnside, A.S.; Ray, M.; Tonga, G.Y.; Hardie, J.; Nagaraj, H.; et al. In Vivo Editing of Macrophages through Systemic Delivery of CRISPR-Cas9-Ribonucleoprotein-Nanoparticle Nanoassemblies. Adv. Ther. 2019, 2, 1900041. [Google Scholar] [CrossRef]
- Zeng, L.; Yang, S.; Wu, C.; Ye, L.; Lu, Y. Effective transduction of primary mouse blood- and bone marrow-derived monocytes/macrophages by HIV-based defective lentiviral vectors. J. Virol. Methods 2006, 134, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Boehler, R.M.; Kuo, R.; Shin, S.; Goodman, A.G.; Pilecki, M.A.; Leonard, J.N.; Shea, L.D. Lentivirus delivery of IL-10 to promote and sustain macrophage polarization towards an anti-inflammatory phenotype. Biotechnol. Bioeng. 2014, 111, 1210–1221. [Google Scholar] [CrossRef]
- Yang, K.; Xu, C.; Zhang, Y.; He, S.; Li, D. Sestrin2 Suppresses classically activated macrophages-mediated inflammatory response in Myocardial Infarction through Inhibition of mTORC1 Signaling. Front. Immunol. 2017, 8, 728. [Google Scholar] [CrossRef]
- Jaiswal, A.; Reddy, S.S.; Maurya, M.; Maurya, P.; Barthwal, M.K. MicroRNA-99a mimics inhibit M1 macrophage phenotype and adipose tissue inflammation by targeting TNFα. Cell. Mol. Immunol. 2019, 16, 495–507. [Google Scholar] [CrossRef]
- Meng, Z.; Zhang, R.; Wang, Y.; Zhu, G.; Jin, T.; Li, C.; Zhang, S. miR-200c/PAI-2 promotes the progression of triple negative breast cancer via M1/M2 polarization induction of macrophage. Int. Immunopharmacol. 2019, 81, 106028. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tan, T.B.; Wang, L.; Zhao, X.Y.; Tan, G.J. p38MAPK/SGK1 signaling regulates macrophage polarization in experimental autoimmune encephalomyelitis. Aging (Albany NY) 2019, 11, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Do, D.C.; Mu, J.; Ke, X.; Sachdeva, K.; Qin, Z.; Wan, M.; Ishmael, F.T.; Gao, P. miR-511-3p protects against cockroach allergen–induced lung inflammation by antagonizing CCL2. JCI Insight 2019, 4, e126832. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Pan, G.; Tang, C.; Li, Z.; Zheng, D.; Wei, X.; Wu, Z. IL-34 Inhibits Acute Rejection of Rat Liver Transplantation by Inducing Kupffer Cell M2 Polarization. Transplantation 2018, 102, e265–e274. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 2, 663–676. [Google Scholar] [CrossRef]
- Gupta, R.M.; Meissner, T.B.; Cowan, C.A.; Musunuru, K. Genome-Edited Human Pluripotent Stem Cell-Derived Macrophages as a Model of Reverse Cholesterol Transport-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Cash, M.N.; Santostefano, K.E.; Nakanishi, M.; Terada, N.; Wallet, M.A. CRISPR/Cas9 knockout of USP18 enhances type I IFN responsiveness and restricts HIV-1 infection in macrophages. J. Leukoc. Biol. 2018, 103, 1225–1240. [Google Scholar] [CrossRef]
- Isogai, S.; Yamamoto, N.; Hiramatsu, N.; Goto, Y.; Hayashi, M.; Kondo, M.; Imaizumi, K. Preparation of induced pluripotent stem cells using human peripheral blood monocytes. Cell. Reprogram. 2018, 20, 347–355. [Google Scholar] [CrossRef]
- Nishimura, K.; Segawa, H.; Goto, T.; Morishita, M.; Masago, A.; Takahashi, H.; Ohmiya, Y.; Sakaguchi, T.; Asada, M.; Imamura, T.; et al. Persistent and stable gene expression by a cytoplasmic RNA replicon based on a noncytopathic variant sendai virus. J. Biol. Chem. 2007, 282, 27383–27391. [Google Scholar] [CrossRef]
- Iizuka-Koga, M.; Asashima, H.; Ando, M.; Lai, C.Y.; Mochizuki, S.; Nakanishi, M.; Nishimura, T.; Tsuboi, H.; Hirota, T.; Takahashi, H.; et al. Functional Analysis of Dendritic Cells Generated from T-iPSCs from CD4+ T Cell Clones of Sjögren’s Syndrome. Stem Cell Rep. 2017, 8, 1155–1163. [Google Scholar] [CrossRef]
- Kuhn, A.; Ackermann, M.; Mussolino, C.; Cathomen, T.; Lachmann, N.; Moritz, T. TALEN-mediated functional correction of human iPSC-derived macrophages in context of hereditary pulmonary alveolar proteinosis. Sci. Rep. 2017, 7, 15195. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.Z.; Li, P.; Weiss, D.; Fuchs, S.; Xiao, H.D.; Adams, J.A.; Williams, I.R.; Capecchi, M.R.; Taylor, W.R.; Bernstein, K.E. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am. J. Pathol. 2007, 170, 2122–2134. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Chiu, A.C.; Kanada, M.; Schaar, B.T.; Krishnan, V.; Contag, C.H.; Dorigo, O. Imaging of Tumor-Associated Macrophages in a Transgenic Mouse Model of Orthotopic Ovarian Cancer. Mol. Imaging Biol. 2017, 19, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]

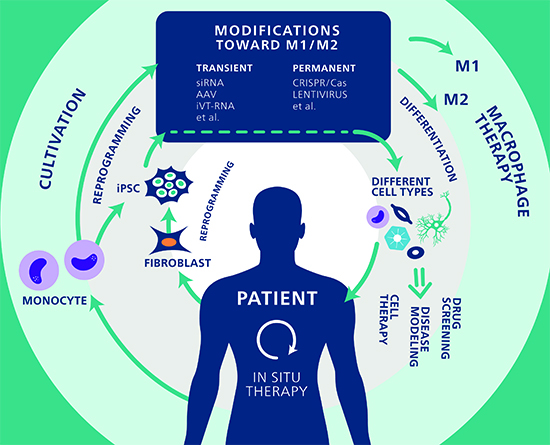{kind=link}
| Levels of Control | Key Signaling Molecules | Examples of Tactics | |
|---|---|---|---|
| M1 Polarization | M2 Polarization | ||
| Surface and matrix markers | TLR-2, TLR-4, CD80, CD86, iNOS, MHC-II | CD206, CD163, CD209, FIZZ1, Ym1/2, Arginase | search for blockers, antibodies |
| Transcription factors | NF-kB, STAT1, STAT5, IRF1, IRF5 | STAT6, IRF4, JMJD3, PPARδ, PPARγ | search for genetic blockers, search for the genes encoding proteins that regulate (activate or repress) the expression of transcription factors |
| Receptors | IFNGR1/2, CSF2Rα, IL1R, TLR4, CD163, CCR7 | FcyR, IL4Rα, IL10R, IL1R, TLR4 | search for blockers, antibodies, search for the receptors that trigger the reverse polarization process |
| Chemokines | CXCL1, CXCL3, CXCL5, CXCL8, CXCL9, CXCL10, CXCL11, CXCL13, CXCL16; CCL2, CCL3, CCL4, CCL5, CCL8, CCL15, CCL11, CCL19, CCL20; CX3CL1 | CCL1, CCL2, CCL13, CCL14, CCL17, CCL18, CCL22, CCL23, CCL24, CCL26, CCL5 | search for antagonists, search for the genes encoding proteins that enhance or weaken the binding of the inducer to the receptor |
| Cytokines | IL-1β, IL-6, IL-12, IL-18, IL-23, TNF-α, type I IFN | IL-4, IL-13, IL-10, IL-33, TGF-β | search for the genes encoding proteins that regulate (activate or repress) the expression of effector molecules |
| Direction of the Shift | Disease |
|---|---|
| Multiple sclerosis [38] | |
| Encephalomyelitis [38] | |
| M1 | Rheumatoid arthritis [39] |
| Alzheimer’s disease [41] Parkinson’s disease [42] | |
| M2 | Skin leishmaniasis [43] |
| Duchenne muscular dystrophy [44] | |
| Asthma-associated fibrosis [45] | |
| Hepatic cirrhosis [46] | |
| Pneumocystis murina infections [47] | |
| Various types of cancer (breast cancers, thyroid cancers, head and neck cancers, etc.) [50,51,52] |
| Disease | Phenotype | Technology | Delivery | In Vitro/In Vivo | Results |
|---|---|---|---|---|---|
| Inflammatory diseases Acute lung injury | M2 to M1 | Knockdown CXCR4 | liposomes | In vitro | Inhibition of the inflammatory cytokine expression [76] |
| Atherosclerosis | M1 to M2 | Knockdown LAIR-1 | lentiviral vectors | In vitro | Increased foam cell formation and cholesterol uptake [29] |
| Vessel stroke | M1 to M2 | Knockout HDAC9 | transgenesis | In vivo | Upregulation of the lipid homeostasis genes, downregulation of the pro-inflammatory genes [31] |
| Myocardial infarction | M1 to M2 | Overexpression of Sestrin2 | lentiviral vectors | In vitro In vivo | Cardiac tissue repair [91] |
| Inflammation | M1 to M2 | Knockout NLRP3 (inflammasomes) | nanoparticles | In vivo | Mitigation of the acute inflammation [84] |
| M1 to M2 | IL-10 | lentiviral vectors | In vitro | Reduction in the amounts of TNF-α, sustained macrophage polarization towards an M2 phenotype promoting local immune responses [90] | |
| Rejection of of transplants | M1 to M2 | Overexpression of Il34 | AAV | In vivo | Anti-inflammatory polarization of Kupffer cells and alleviated liver transplant rejection [96] |
| Proliferative diseases Ovarian cancer, lung cancer, glioma | M2 to M1 | IVT-RNA | nanoparticles | In vivo | High anti-tumor activity [23] |
| Melanoma, hepato- carcinoma | M2 to M1 | Chloroquine | i/v | In vivo | Decreased immunosuppressive infiltration of the myeloid-derived suppressor cells and Treg cells, enhancement of the antitumor T cell-mediated immunity [24] |
| Ovarian cancer | M2 to M1 | IFN-γ therapy | i/v | In vivo | Significant tumor regression [70] |
| Breast cancer, melanoma | M2 to M1 | Paclitaxel | i/p | In vivo | Antitumor effect [25] |
| Breast cancer | block M2 | CCL2 inhibitors | i/v | In vivo | Suppression of metastasis and prolongation of the survival [68] |
| Lewis lung carcinoma | block M2 | MEL-dKLA | i/p | In vivo | Lower rates of tumor growth and angiogenesis, decreased tumor weights [27] |
| Viral diseases Anti-HIV response | block M1 | iPSCT Knockout USP18 | nucleofection | In vitro | Inhibition of replication of the virus [99] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poltavets, A.S.; Vishnyakova, P.A.; Elchaninov, A.V.; Sukhikh, G.T.; Fatkhudinov, T.K. Macrophage Modification Strategies for Efficient Cell Therapy. Cells 2020, 9, 1535. https://doi.org/10.3390/cells9061535
Poltavets AS, Vishnyakova PA, Elchaninov AV, Sukhikh GT, Fatkhudinov TK. Macrophage Modification Strategies for Efficient Cell Therapy. Cells. 2020; 9(6):1535. https://doi.org/10.3390/cells9061535
Chicago/Turabian StylePoltavets, Anastasiya S., Polina A. Vishnyakova, Andrey V. Elchaninov, Gennady T. Sukhikh, and Timur Kh. Fatkhudinov. 2020. "Macrophage Modification Strategies for Efficient Cell Therapy" Cells 9, no. 6: 1535. https://doi.org/10.3390/cells9061535
APA StylePoltavets, A. S., Vishnyakova, P. A., Elchaninov, A. V., Sukhikh, G. T., & Fatkhudinov, T. K. (2020). Macrophage Modification Strategies for Efficient Cell Therapy. Cells, 9(6), 1535. https://doi.org/10.3390/cells9061535